1. Introduction
Poor drug solubility is a major challenge of many new and emerging therapeutic agents. It is frequently cited that 40% of recently marketed drug compounds have limited solubility in aqueous media [
1], with this figure higher again for drugs in the developmental stage (>70%) [
2]. This is problematic for several reasons, with one of the major issues being unacceptably poor drug bioavailability. As a result, many poorly soluble drugs are delivered using invasive hypodermic injections [
3], or as high-dose oral preparations [
4]. These approaches often result in manufacturing complexities and adverse effects, ultimately causing poorer treatment outcomes. To capitalise on the therapeutic potential of drugs that are promising in developmental stages but hampered by formulation issues due to their solubility profile, a variety of solubility enhancement strategies can be used [
4]. These techniques are most commonly used for medications delivered orally but are not limited to this route of administration.
Transdermal drug delivery is an attractive alternative route to oral and parenteral preparations. This non-invasive method of delivery offers many benefits including avoidance of first-pass metabolism, increased patient compliance and the ability to terminate drug delivery rapidly [
5,
6]. Unfortunately, however, it is limited in terms of the number of compounds that can permeate across the resilient barrier posed by the outermost layer of the skin, the
stratum corneum. Various passive and active technologies have been developed that reduce the barrier effect of the
stratum corneum, increasing the number of drugs amenable to transdermal delivery [
7]. Microneedle (MN) technology is one of these that is rapidly advancing towards large-scale manufacture and commercialisation [
8].
MNs are minimally invasive devices consisting of numerous micron-sized projections assembled on a baseplate. MNs penetrate the
stratum corneum sufficiently to enable a port of access to the skin’s rich microcirculation yet are short enough to avoid painful stimulation of dermal nerves [
5]. They can be subdivided into numerous different categories, each with their own distinct characteristics [
9]. In this work, hydrogel-forming MNs have been used due to the vast number of advantages they confer [
10]. When inserted into the skin, they are hard and sharp. Following insertion, they rapidly take-up interstitial fluid, swelling and forming a continuous hydrogel pathway through which drugs can diffuse [
11]. To date, there have been relatively few reports of MNs being employed in transdermal delivery of hydrophobic molecules. This is most likely due to the inherent mechanism of action of MNs as they create aqueous channels within the skin. To facilitate MN-mediated delivery of poorly soluble drugs, alternative approaches to MN or MN-reservoir formulation need to be adopted. Ma and Gill (2014) demonstrated that they could coat solid MNs with a solid dispersion, whereby a poorly soluble compound (lidocaine) was dispersed within a hydrophilic matrix [
12]. In vitro testing of this system demonstrated increased lidocaine delivery, in comparison with the marketed Emla
® cream. In a similar approach, Boehm et al. (2016) coated the poorly soluble antifungal agent, itraconazole onto poly(glycolic acid) MNs and demonstrated enhanced delivery [
13]. Others have investigated dissolving MNs [
14,
15]. For example, dissolving MNs fabricated from oligomeric sodium hyaluronate and containing the poorly soluble and lipophilic compound artemether have been described [
14]. Vora et al. (2018) have more recently used dissolving MNs for delivery of the hydrophobic compound, cholecalciferol [
16]. While these examples provide considerable advancement to the transdermal delivery of poorly soluble compounds, both the coated and dissolving systems have limitations. Drug loading is limited to what can be coated onto the surface of the device with coated MNs, therefore, most amenable to highly potent molecules, rendering this a rather niche product. Although dissolving MNs have been developed to enable delivery of low potency substances at higher doses, delivery of hydrophobic drugs using dissolving MNs is typically limited to what can be loaded into the needles themselves, and so dosing capacity may again be limited. Coated and dissolving MNs have a part to play within advanced drug delivery, but alternative MN technologies such as hydrogel-forming MNs have been shown to overcome several of the previously described challenges [
9]. Through the adoption of solubility enhancement strategies, there is potential to expand the MN drug delivery portfolio to include poorly soluble drugs.
For hydrogel-forming MNs to be used to deliver poorly soluble drug compounds, novel drug containing reservoirs displaying water miscibility as well as solubilising ability are required. Considering the various solubility enhancement techniques [
2], a convenient and readily adaptable method is co-solvency. Co-solvency is a simple, popular approach used to enhance drug solubility. Drug substances are formulated in a liquid medium in which they display high solubility. Addition of a non-polar solvent reduces the polarity of the entire system, encouraging dissolution of hydrophobic molecules in aqueous media. It is important that the solvents chosen are biocompatible, non-toxic and relatively inexpensive [
17].
This novel study presents proof of principle for the transdermal delivery of poorly soluble drugs using hydrogel-forming MNs, specifically focusing on the use of water-miscible, liquid co-solvents as drug reservoirs. As this is the first report of a liquid reservoir with hydrogel-forming MNs, development of an appropriate in vitro Franz cell setup is also described with Nile red (NR) dye (Log P = 3.8) used as a model hydrophobic compound in these studies. Delivery of the therapeutic compounds olanzapine (OLP) (Log P = 3.6), an antipsychotic compound and atorvastatin calcium trihydrate (ATR) (Log P = 5.4), a lipid regulating statin, were subsequently investigated using the developed in vitro method.
2. Materials and Methods
2.1. Materials
Poly(ethylene glycol) (PEG; MW 10,000, 600, 400 and 200 Da), methylene blue and HPLC grade methanol and acetonitrile were purchased from Sigma–Aldrich (Dorset, UK). ATR, NR, OLP and propylene glycol were purchased from Tokyo Chemical Industry UK Ltd. (Oxford, UK). Gantrez® S-97, a copolymer of methyl vinyl ether and maleic acid (PMVE/MA, molecular mass: 1,500,000 Da) was a gift from Ashland (Kidderminster, UK). Anhydrous sodium carbonate was obtained from VWR International Ltd., (Poole, UK). Oxoid™ phosphate buffered saline (PBS) pH 7.4 tablets were obtained from Thermo Scientific (Sheffield, UK). Poly(ester) film, one-side siliconised, release liner, FL2000 PET 75 was purchased from Rexam Release B.V (Apeldoom, The Netherlands) Glisseal®N vacuum grease was purchased from Borer Chemie (Zuchwil, Switzerland). All other chemicals used were of analytical reagent grade.
2.2. Fabrication of Hydrogel-Forming Microneedle Arrays
Hydrogel-forming MNs were fabricated as described previously [
11]. Briefly, polymer blends containing 20%
w/
w Gantrez
® S-97, 7.5%
w/
w PEG 10,000 and 3%
w/
w anhydrous sodium carbonate were cast into laser engineered silicone moulds. Cast moulds were centrifuged at 2205 g for 15 min using an Eppendorf
® 5804 series centrifuge (Eppendorf
® UK Ltd., Stevenage, UK) and the formulation dried under ambient conditions for 48 h. Crosslinking was induced by storing MNs at 80 °C for 24 h. Two array designs were fabricated, 11 × 11 (conical shaped; 600 µm height, 300 µm width at base and 150 µm interspacing) arrays and 19 × 19 (conical shaped; 600 μm height, 300 μm width at base and 50 μm interspacing). Mechanical properties of hydrogel-forming MN arrays were determined as previously reported [
10].
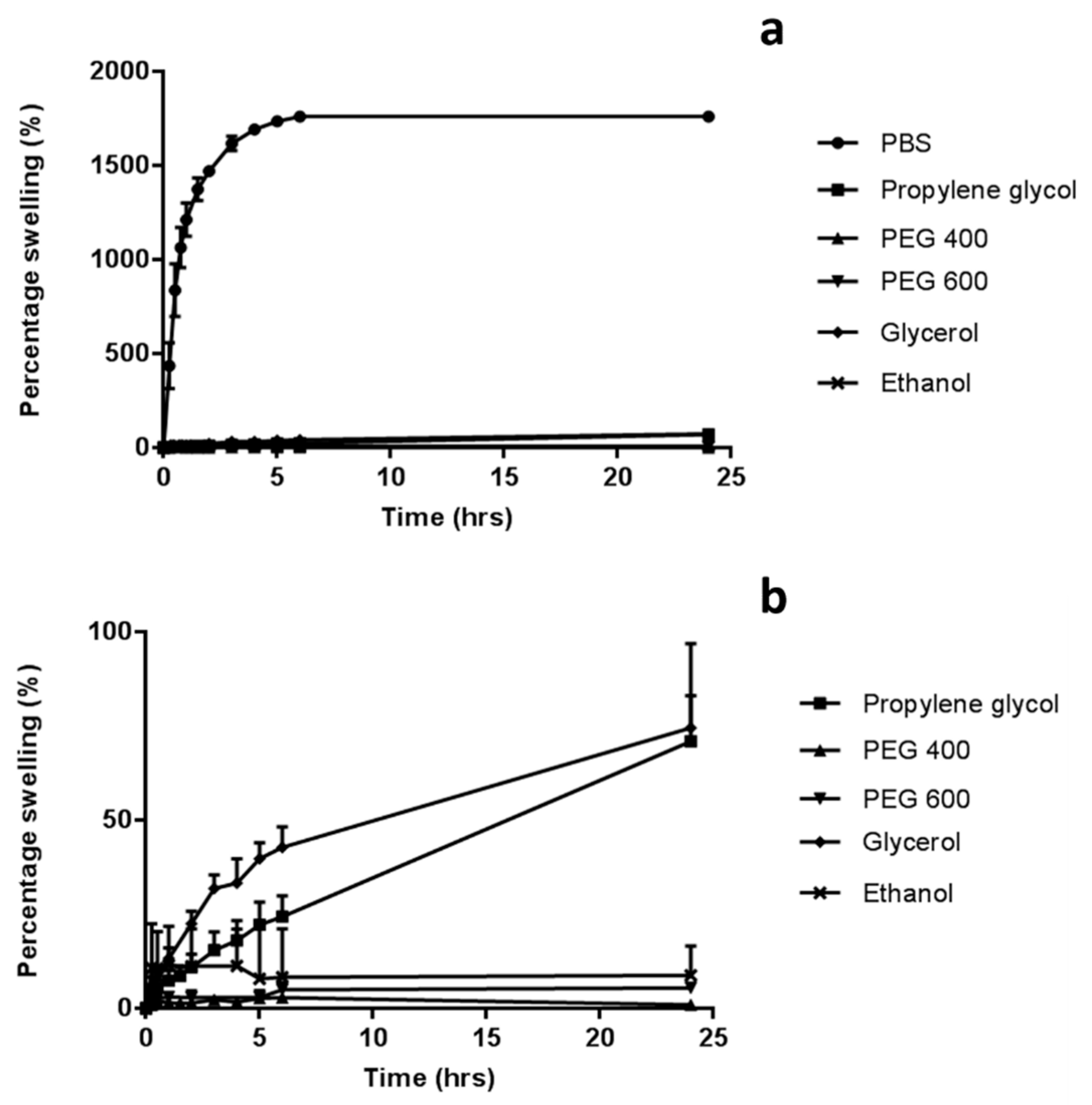
2.3. Swelling Studies
The swelling of the hydrogel formulation was investigated using 1 cm
2 needle-free baseplates, over 24 h at room temperature. Baseplates were prepared following the same method employed to fabricate MN arrays, except using silicone moulds containing no laser-engineered template for needle formation. The dry weight of each of these samples was recorded (
mo). These were subsequently placed in disposable polystyrene weigh boats containing 30 mL of either phosphate buffered saline (PBS) (pH 7.4), glycerol, propylene glycol, PEG 600 or PEG 400. At regular intervals samples were removed from the solvents, blotted with tissue paper to remove excess solvent and weighed (
mt). The same experiment protocol was employed, using ethanol as a co-solvent, with the weighing boats replaced by glass screw top jars to prevent ethanol evaporation. The percentage swelling was calculated using Equation (1):
2.4. Saturation Solubility
The saturation solubility of NR, OLP and ATR in various solvents was determined using the shake-flask method [
18]. Briefly, an excess of drug was added to solvent in snap-top glass vials. Vials were sonicated for 10 min and placed in a rotary incubator (GFL Shaking Incubator 3033, Burgwedel, Germany) at 37 °C for 24 h at 40 rpm. Following incubation, samples were filtered through a 0.22 µm syringe filter (Chromacol, Herts, UK), diluted as appropriate and analysed using ultraviolet–visible (UV–vis) spectroscopy. Solvents tested included ethanol, PEG 200, PEG 400, PEG 600 and propylene glycol.
2.5. Development of In Vitro Liquid Delivery System
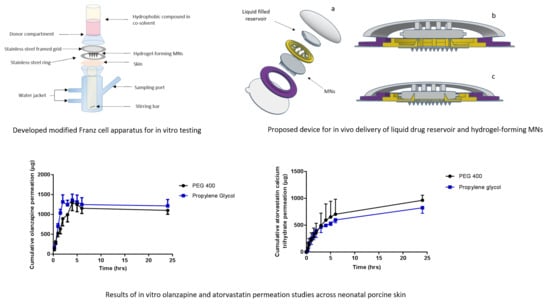
2.5.1. Franz Cell Apparatus Modification
The conventional Franz cell setup, as described previously [
19], is not suitable for testing in vitro delivery of a liquid formulation due to the presence of a steel weight which occludes the surface available for movement of liquid into, and through the MNs. This necessitated an alternative approach to be developed. Various modifications to conventional Franz diffusion cells (PermeGear Inc., Hellertown, PA, USA), were implemented to obtain a final set-up that provides a reliable and reproducible method of testing the in vitro permeation of liquids via hydrogel-forming MNs.
Shaved dermatomed (Integra Life Sciences, Padgett Instruments, NJ, USA) neonatal porcine skin (thickness ~350 µm) was attached to the flat flange of a custom-made Franz cell donor compartment (15 mm orifice diameter, 40 mm height) using cyanoacrylate glue;
stratum corneum side orientated upwards. The skin was placed on dental wax to provide support and to facilitate confirmation of MN penetration. MNs were inserted into the skin by applying gentle pressure to a syringe plunger for 30 s, with the flat end pressing on the baseplate. A stainless steel 40 Mesh Mini Basket (Quality Lab Accessories, LLC, Telford, PA, USA) was placed on top of the inserted MNs with Blu Tack
® (Bostik Ltd, Stafford, UK) attached to provide a mass of 8 g. Liquid drug formulation (1 mL NR dye in PEG 400, 50 µg/mL) was pipetted into the donor compartment, sealed using ParafilmM
® and mounted onto the receiving compartment. Receiving compartment contained 20%
v/v PEG 400 in PBS (pH 7.4) stirred continuously at 600 rpm and thermostated at 37 ± 1 °C (
Figure 1). Samples (200 µL) were removed at pre-defined time intervals and the receiver fluid replenished with an equivalent volume of degassed, pre-warmed receptor medium. Samples were centrifuged for 5 min at 12,225 g using an Eppendorf Minispin centrifuge (Eppendorf UK Ltd., Stevenage, UK) and then analysed using UV–vis spectroscopy.
A variation of the above approach was adopted with the basket and Blu Tack® replaced with an adhesive foam (prototype sample from TG Eakin, Comber, UK) with a central opening (outer diameter = 12 mm and inner diameter = 8 mm). The MN array was attached to the adhesive foam prior to insertion. The arrays were assembled with only the needle-free border of the array attached to the foam. All other aspects of the setup were the same as previously described, with the same sampling intervals and analytic methods adopted.
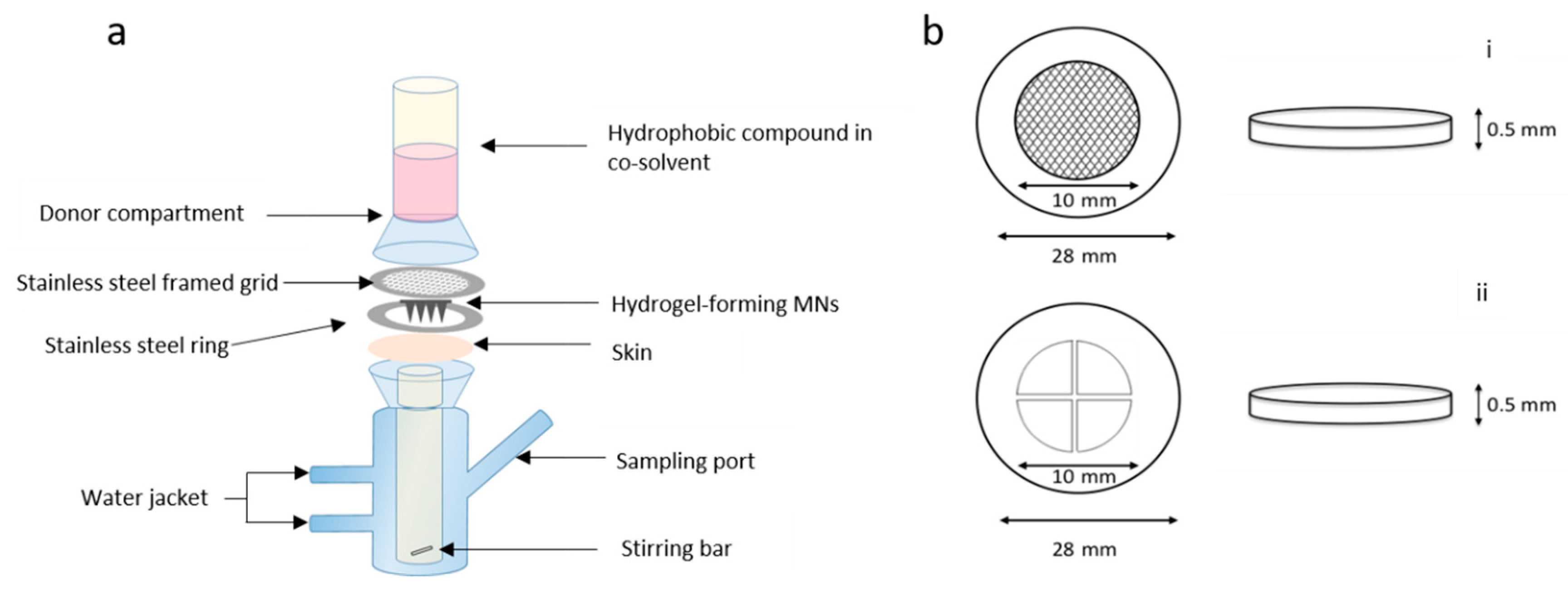
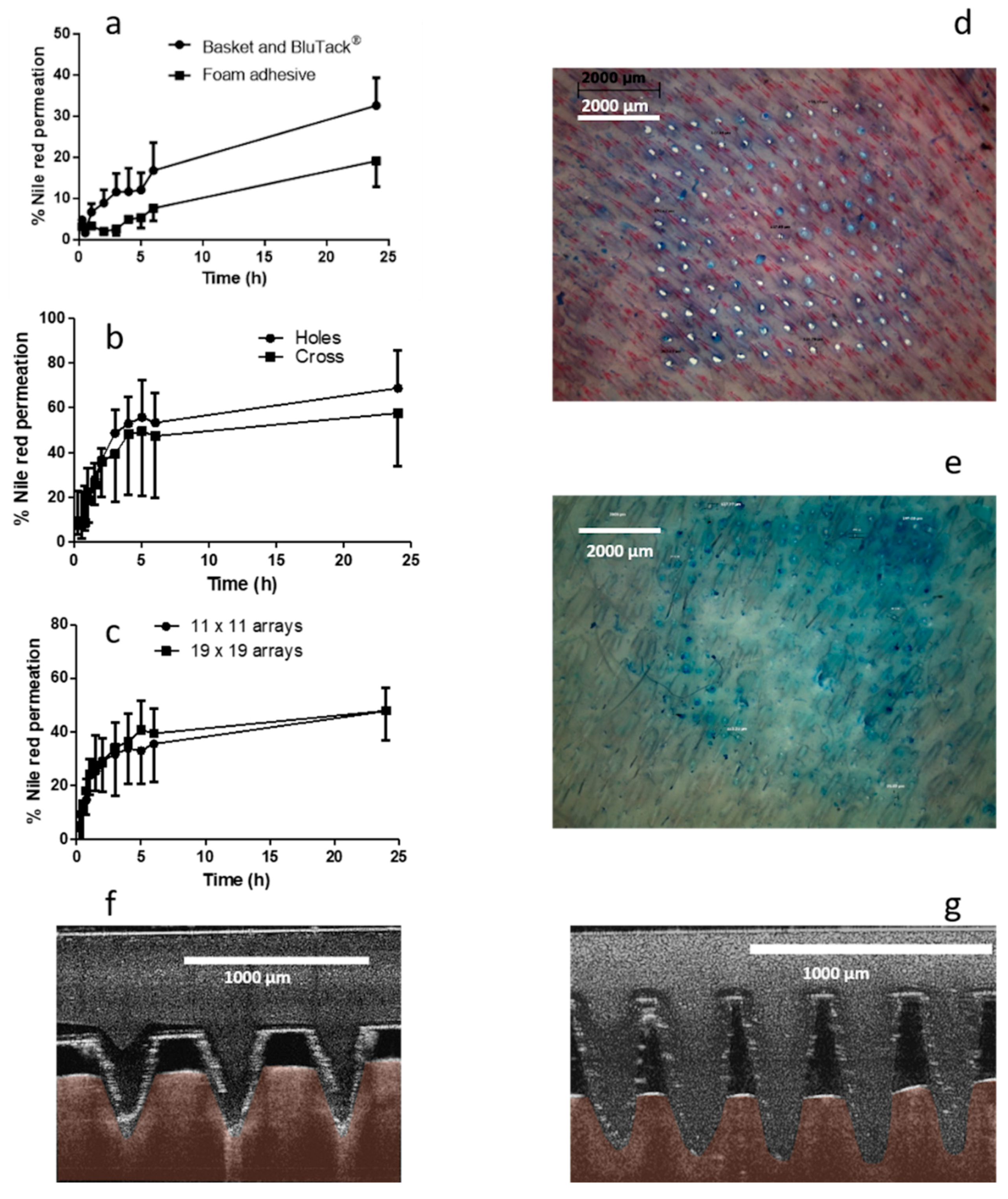
2.5.2. Effect of Stainless Steel Grid Design on In Vitro NR Permeation
Franz cell assembly was further modified to facilitate more efficient delivery of the drug from a liquid reservoir (
Figure 2a). MNs were inserted into dermatomed neonatal porcine skin attached to a custom-designed stainless steel ring. To secure MNs in place, a stainless steel-framed grid (Currie Engineering, Coleraine, UK) was placed between a modified glass donor compartment and skin, which was secured in place with a clamp. Two grid designs were tested; both grids were laser cut into cylindrical stainless steel disks, 0.5 mm in thickness and an outer diameter of 28 mm (
Figure 2b). The inner grid was either circular holes or cross shaped. The open area of the grid represented in
Figure 2bi is 60 mm
2 and 60.54 mm
2 in
Figure 2bii. All other aspects of the setup were the same as previously described above, with the same sampling intervals and analytical methods adopted.
2.5.3. Effect of Microneedle Density on NR Permeation
MN arrays in an 11 × 11 configuration were initially used to create a pathway through the skin that enabled the permeation of NR from an organic solvent-based liquid formulation. Previous work completed by Garland et al. (2012) has investigated drug delivery from arrays containing 361 needles [
20], approximately three times the number of needles in an 11 × 11 array. As such, it was hypothesised that by using 19 × 19 arrays, the permeation of NR could be increased by up to three-fold. MNs were tested using the same modified Franz cell set-up described previously with two designs tested, i.e., 11 × 11 arrays and 19 × 19 arrays. An aliquot (1 mL) of PEG 400 containing NR at a concentration of 50 µg/mL was filled into the Franz cell donor compartment. Samples (200 µL) were removed at regular intervals and replaced with fresh receiver media (PBS containing 20%
v/v PEG 400). Samples were analysed using UV–vis spectroscopy. Following MN removal, skin samples were stained with methylene blue dye and visualised using a digital light microscope to assess MN insertion (Leica EZ4 D stereo microscope, Leica Microsystems, Milton Keynes, UK). Optical coherence tomography (OCT) (EX1301 OCT Microscope (Michelson Diagnostics Ltd., Kent, UK)) was used to visualise MN insertion of both array geometries, immediately post-insertion, in neonatal porcine skin, as previously described [
21].
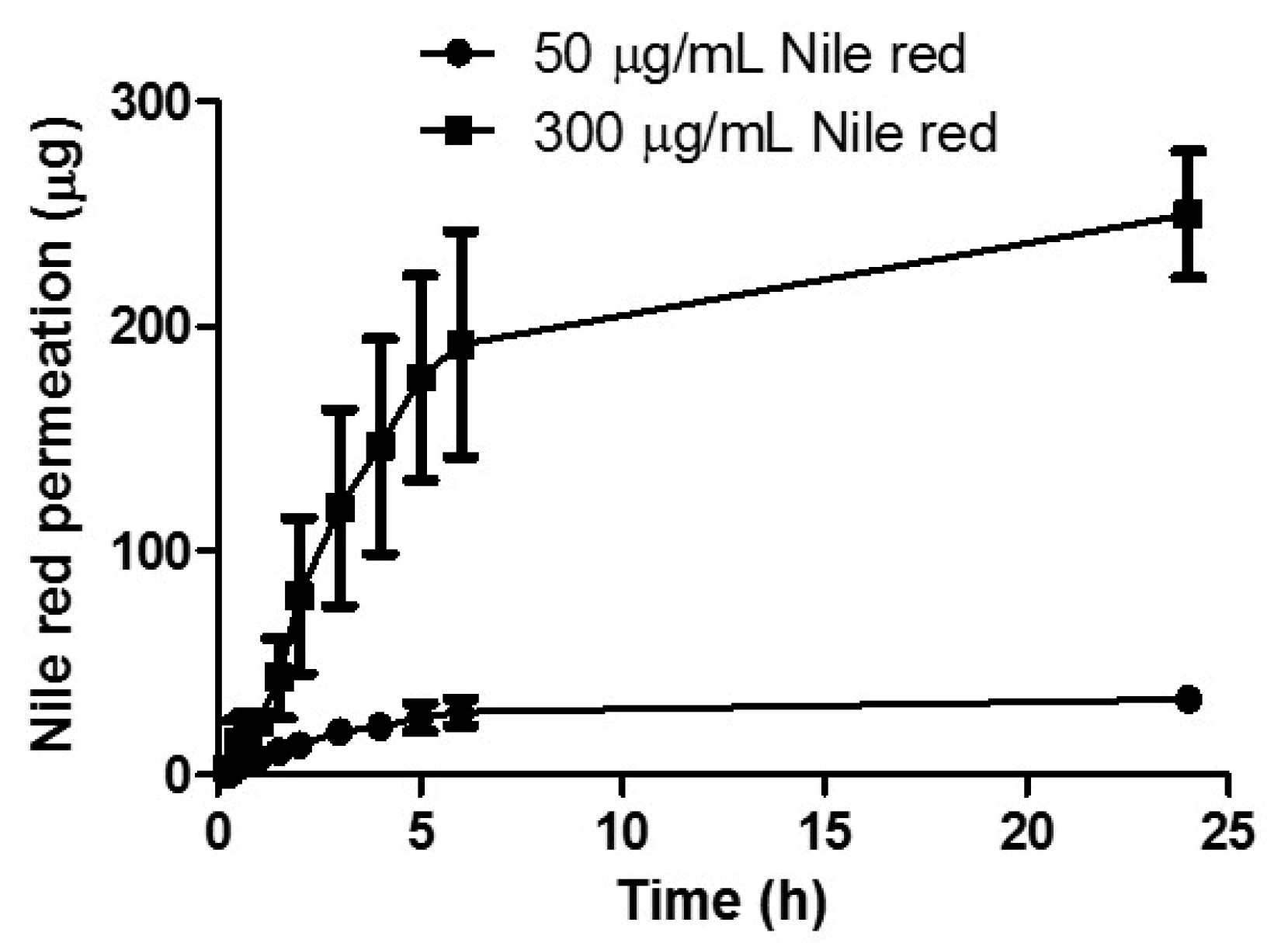
2.5.4. Effect of NR Concentration on In Vitro Permeation
Two concentrations of NR dissolved in PEG 400 were tested. Aliquots (1 mL) of 50 µg/mL or 300 µg/mL NR in PEG 400 were filled into the donor compartment following assembly of the developed Franz cell apparatus. Receiver compartment composition was 20% v/v PEG 400 in PBS and 30% v/v PEG 400 in PBS for the 50 µg/mL and 300 µg/mL test solutions respectively. The remainder of the experimental set-up was as stated above.
2.6. Investigation of OLP Stability
OLP is recognised to display varying degrees of instability in both aqueous and biological matrices [
22]. The extent of this instability is controversial, with contradictory reports on OLP behaviour in various environments [
23,
24,
25]. As a result, OLP stability studies were conducted. The stability of OLP standards was assessed over 24 h to determine sample stability for analysis purposes. A further stability study of OLP reservoirs was conducted over 28 days to inform storage conditions of potential OLP reservoirs.
2.6.1. OLP Stability over 24 h
The stability of OLP dissolved in a solution of 50:50 methanol:PBS was assessed across three concentrations. Working standards of 20 µg/mL, 10 µg/mL and 5 µg/mL were prepared by diluting the appropriate volume of a stock solution of olanzapine (0.5 mg/mL) with 50:50 methanol:PBS diluent solution. Solutions were stored at 37 °C for 24 h with 1 mL of the solution removed at pre-defined time intervals and analysed using reverse-phase high-performance liquid chromatography (RP-HPLC).
2.6.2. OLP Stability in Organic Solvents over 28 days
The stability of OLP in three organic solvents, methanol, PEG 400 and propylene glycol, was assessed over 28 days. OLP solutions with three drug loadings were prepared (20 µg/mL, 10 µg/mL and 1 µg/mL) in each of the respective solvents. OLP solutions were divided into two cohorts with one group stored in light at ambient temperatures and the second group protected from light and stored in a refrigerator at a temperature between 2 and 8 °C. A sample was removed at pre-defined time intervals, filtered through a 0.22 µm syringe filter, and analysed using RP-HPLC.
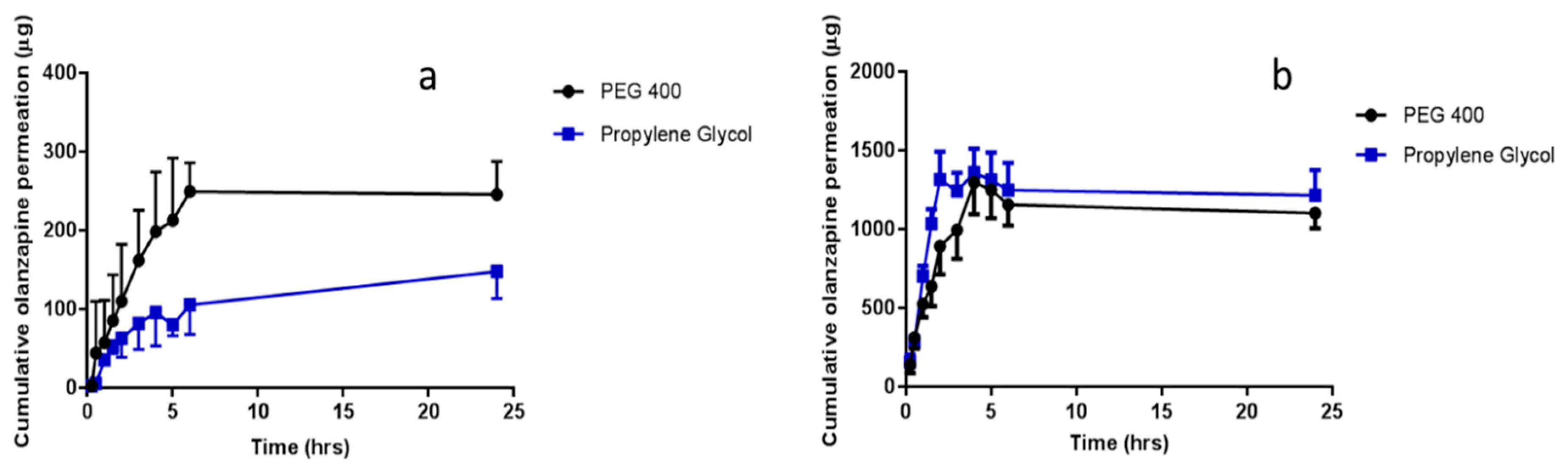
2.7. In Vitro Permeation of OLP and ATR
To determine whether hydrogel-forming MNs could be used to enhance the delivery of therapeutic compounds, liquid reservoirs containing either OLP or ATR were prepared. In vitro permeation was investigated using the custom-designed Franz cell set-up described previously. Following saturation solubility and swelling testing, PEG 400 and propylene glycol were selected as the most appropriate solvents to prepare both OLP-containing and ATR-containing liquid reservoirs. The saturation solubility of OLP in PEG 400 and propylene glycol was 31.59 mg/mL ± 3.15 mg/mL and 24.10 mg/mL ± 2.51 mg/mL, respectively. The saturation solubility of ATR in PEG 400 was 47.09 mg/mL ± 10.08 mg/mL and 48.43 mg/mL ± 4.55 mg/mL in propylene glycol. The receiver compartment contained PBS (pH 7.4) with 5%
v/v of the solvent used in the drug formulation to maintain the solubility of the drugs in the receiver compartment and ensure sink conditions [
26,
27]. Sampling was conducted as described above for NR. For each organic solvent, two OLP concentrations were tested, i.e., 0.5 mg/mL and 2.5 mg/mL, and one ATR concentration was tested, i.e., 2 mg/mL. Samples were analysed using RP-HPLC.
2.8. Pharmaceutical Analysis
NR was quantified using UV–vis spectroscopy (PowerWave XS microplate spectrophotometer, BioTek®, Swindon, UK) with absorbance read at 570 nm. The saturation solubility analysis of OLP and ATR was determined using UV–vis spectroscopy, with absorbance read at 225 nm and 250 nm, respectively.
RP-HPLC (Agilent 1200® Binary Pump, Agilent 1200®, Standard Autosampler, Agilent 1200® Variable Wavelength Detector; Agilent Technologies UK Ltd., Stockport, UK) methods were developed and validated for both OLP and ATR. The chromatographic analysis of OLP in 50:50 methanol:PBS was achieved using a Waters™ XSelect CSH C18 column (75 mm × 3 mm internal diameter, 2.5 μm packing) with UV detection at 225 nm (run time of 5 min). Mobile phase consisted of 10:90 acetonitrile:0.025 M potassium dihydrogen phosphate buffer (pH 3.0) at a flow rate of 0.7 mL/min. The injection volume was 20 µL and column temperature was maintained at 20 °C. OLP samples were diluted in 50% v/v methanol in PBS (pH 7.4) prior to analysis, where necessary. The chromatograms obtained were analysed using Agilent ChemStation® Software B.02.01.
The chromatographic analysis of ATR in PBS (pH 7.4) was completed as described previously [
28]. Briefly, gradient separation was obtained using a Luna C18 (ODS1) column (150 mm × 4.6 mm i.d. with 5 µm packing; Phenomenex, Macclesfield, UK) with UV detection at 250 nm (run time of 20 minutes with post-run time set at 2 min). The mobile phase was a mixture of 25mM potassium dihydrogen phosphate buffer, pH 2.5, and methanol–acetonitrile (50:50,
v/
v), adjusted in composition over time. The injection volume was 20 µL and column temperature was maintained at 20 °C.
The described methods for the quantification of OLP and ATR were validated according to guidelines from the International Conference on Harmonisation (ICH) [
29]. Least-squares linear regression analysis and correlation analysis were performed on the triplicate calibration curves produced on each day of the validation period. This enabled the generation of the equation of each line and their respective coefficients of determination. To determine the individual limits of detection (LoD) and quantification (LoQ), an approach based on the standard deviation of the response and the slope of the representative calibration curve was employed.
2.9. Statistical Analysis
GraphPad Prism® version 5.0 (GraphPad Software Inc., San Diego, CA, USA) was used to perform statistical analysis. Where appropriate, a Kruskal–Wallis test with post-hoc Dunn’s test was used for comparison of multiple groups, with a Mann–Whitney U test performed for comparison of two groups when n < 5. All data were expressed as the mean ± standard deviation. Statistical significance was denoted by p < 0.05 in all cases.
4. Discussion
Water-miscible organics solvents, termed co-solvents, can be used to enhance the solubility of poorly soluble substances [
30]. Relatively simple to use in a formulation and generally very effective, co-solvents are a popular pharmaceutical method used to increase solubility. Co-solvents in conventional transdermal systems often have a dual function; they act to increase drug solubility in the vehicle, whilst also acting as a permeation enhancer by increasing permeant partitioning into, and solubility within, the
stratum corneum, thereby improving percutaneous absorption [
31].
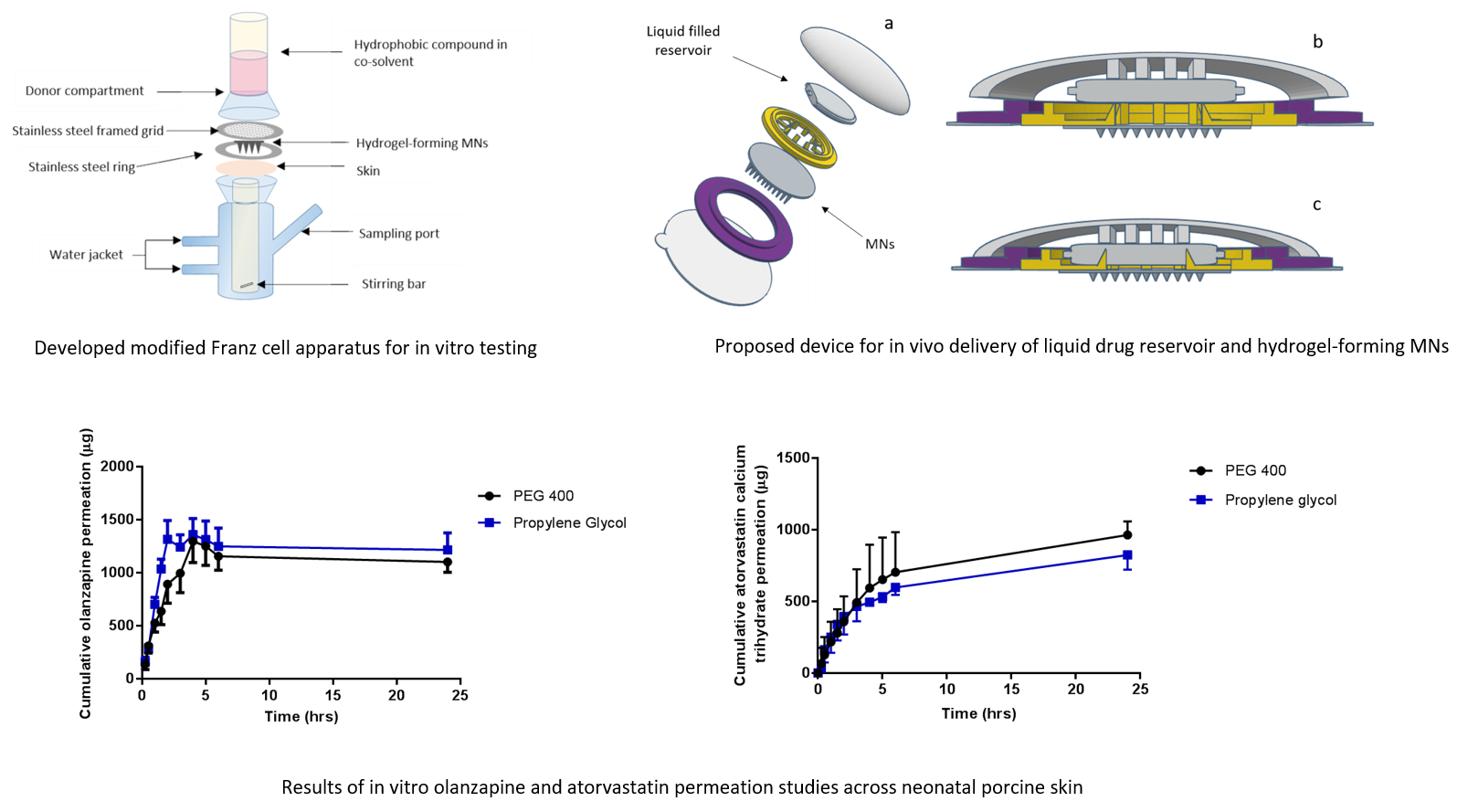
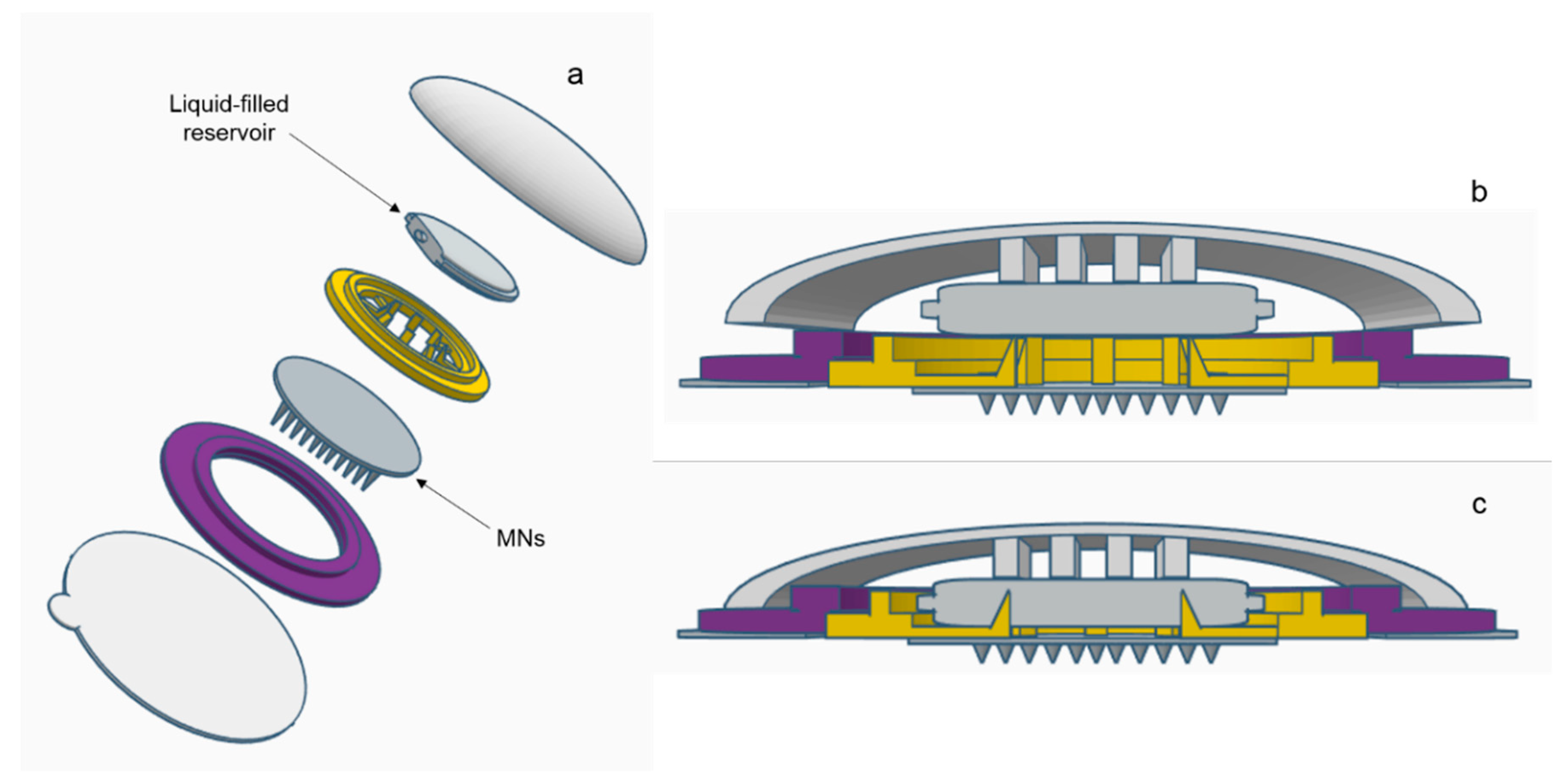
A range of water-miscible organic co-solvents were tested to determine those most appropriate for use with hydrogel-forming MNs in terms of hydrogel swelling. As expected, the highest degree of swelling was observed in 100% PBS. Contrastingly, when placed in a 100% organic solvent, the extent of swelling was significantly reduced for all solvents. This ensures that the MNs will not swell in the absence of physiological fluid. When considering an integrated MN patch consisting of hydrogel-forming MNs and a liquid reservoir, it would be imagined that the liquid reservoir would be maintained completely intact and separated from the array itself, yet contained within a singular patch, until the point of application. This is important to ensure the structural and mechanical integrity of the MN array itself as even a small degree of swelling may disturb the rigid hydrogel matrix in the dry state. Presented in
Figure 9 is a schematic representation of a device that could be adapted to facilitate the combination of hydrogel-forming MNs and liquid reservoirs. Such a device would overcome the practical issue of liquid reservoirs such as those reported by Martanto et al. (2004) who reported the adhering of a ‘flanged glass chamber’ to the back of rats to retain liquid in place [
32].
The saturation solubility of each test hydrophobic substance in various co-solvents was investigated to identify a suitable reservoir medium. The optimum solvent in terms of NR solubility was found to be PEG 400. However, OLP and ATR both displayed high solubility in PEG 400 and propylene glycol and so both co-solvents were further investigated. The concentration of each drug in the liquid reservoir in subsequent permeation studies was chosen to be notably lower than the experimental values obtained, to minimise the risk of drug precipitation.
Co-solvents as drug reservoir media are beneficial in terms of drug solubility but can create challenges for experimental design. The Franz cell apparatus is the standard equipment used for in vitro testing of drug delivery via MN arrays [
33,
34,
35,
36]. To date, in vitro testing of hydrogel-forming MNs typically involves the use of a weight placed on top of the solid drug-containing reservoir to retain MNs in place. Liquid reservoirs have not been previously described in combination with hydrogel-forming MNs and, therefore, required a custom-designed donor compartment. This apparatus was designed to facilitate storage of a liquid reservoir, as well as preventing MN expulsion, while allowing movement of the liquid into the MN array. Following extensive preliminary work, an effective and reproducible experimental setup was established, as described above. Grids, of two different designs, were designed and fabricated to hold the MNs in place. Additionally, a stainless steel spacer ring of 0.5 mm thickness was used to facilitate MN swelling. Grid and spacer design was key to successful experimental set-up.
Initial in vitro development studies investigated the effect of MN array density on drug permeation. It was assumed that if the number of individual MNs were increased by 3-fold, that there would be an equivalent 3-fold increase in NR permeation. However, this was not found to be the case. Microscope images obtained following removal of MNs indicated that MN insertion efficiency was superior for 11 × 11 arrays in comparison to 19 × 19. Post-experiment skin analysis showed differences in MN insertion efficiency. The design of the glass donor compartment prohibited manual thumb pressure being applied, which has previously been shown to be suitable for the insertion of 19 × 19 MN arrays into rats [
37]. It has been shown that MN insertion depth is lower with 19 × 19 arrays when compared to 11 × 11 arrays, when an equivalent force is applied [
38]. This may help explain the difference in the skin samples post MN removal.
To establish the effect of drug loading on total permeation of NR, two donor solution concentrations were tested. As expected, increasing drug concentration caused a correlating increase in permeation, suggesting that concentration is a major influence on the transdermal delivery of NR using a liquid reservoir. Studying the profile in more detail, it is evident that there is no lag time with the linear period continuing for up to 5 hours. When MN arrays are inserted into the skin, they rapidly imbibe interstitial fluid. This fluid causes the MN array to swell, opening the hydrogel matrix and facilitating permeation of drug into the skin. As interstitial fluid is assimilated, it mixes with the liquid reservoir compartment and contributes to even more extensive swelling as the MNs swell from both upper and lower surfaces of the array. The rapid rate of hydrogel swelling, coupled with a liquid reservoir medium resulted in prompt and, and indeed rapid delivery of the hydrophobic compound.
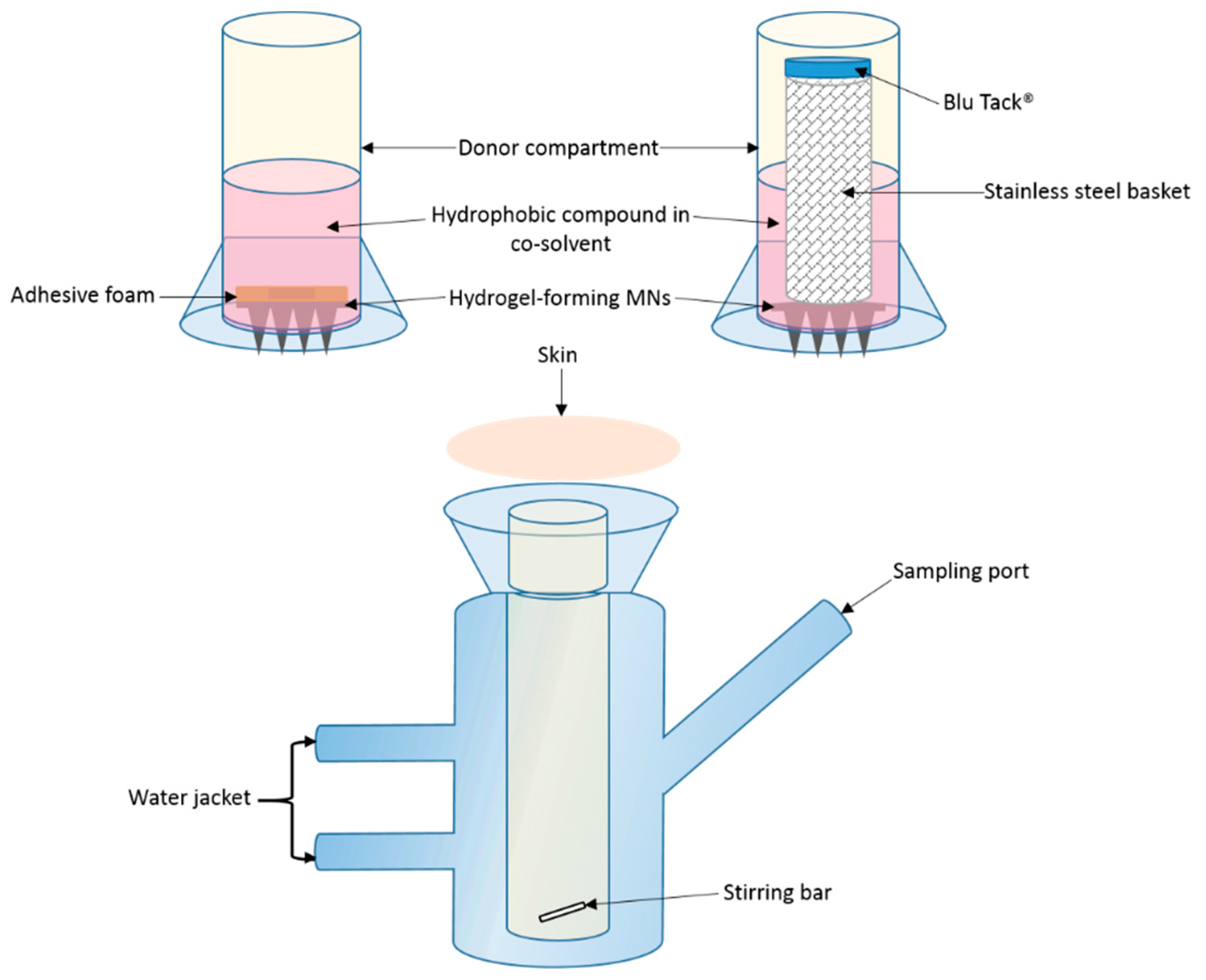
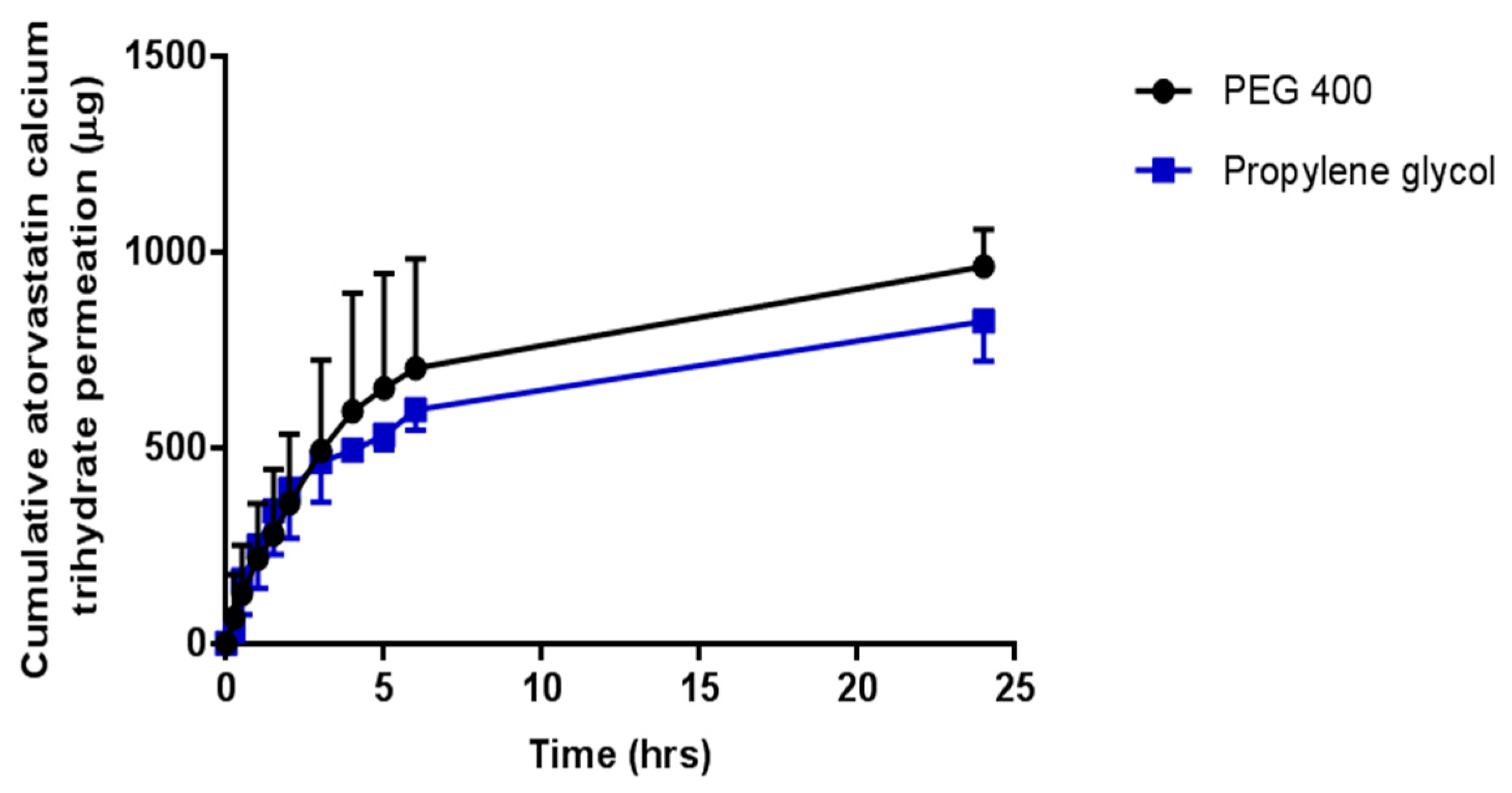
In vitro OLP permeation and ATR permeation from liquid reservoirs in combination with hydrogel-forming MNs was tested using the newly developed Franz cell model. At both a high (2.5 mg/reservoir) and low (0.5 mg/reservoir) OLP loading there was no significant difference between the two co-solvents in terms of cumulative drug permeation. It was taken into consideration that there was possible OLP degradation occurring in the receiver compartment between 6 and 24 h. Based on stability studies conducted it is known that OLP loss over this time duration is less than 10%. The slight reduction in OLP observed between these two points can, therefore, be accounted for. Results from ATR-containing reservoirs were similar to OLP with no difference found between propylene glycol and PEG 400. As the drugs were already in solution, the onset of permeation was rapid, with both drugs detected in their respective receiver compartments within 15 min of MN insertion. With respect to the drug not accounted for at 24 h, from a practical perspective, some of the liquid added to the donor compartment remained adhered to the glass sides of the vessel and on the surface of the skin. Some drug may also have been retained within the skin and the MN array itself.
Comparing percentage permeation of OLP with ATR, using the same hydrogel-forming MNs, confirms co-solvency is a suitable method for facilitating MN-mediated delivery of hydrophobic drugs. If the permeant is soluble in the water-miscible solvent, diffusion through the hydrogel matrix can be facilitated. Solute-solvent interaction will also bear an effect on interaction of the solvent with the hydrogel matrix and, therefore, influence permeation, as demonstrated by the higher percentage permeation of NR. Further to this, as the MN system swells, the co-solvent is diluted and it is known from saturation solubility studies that increasing solvent polarity, readily reduces the solubility of the hydrophobic compound and could potentially cause precipitation of the drug within the hydrogel. This further highlights the importance of selecting a solvent in which the drug displays optimum solubility to minimise the likelihood of this occurring. The delivery of the drug in solution also increased the volume of co-solvent in the receiver compartment of the Franz diffusion cell, further aiding drug solubility in this medium. In vivo, it is thought that the rich dermal microcirculation may rapidly absorb the delivered drug, hastening its removal from the local area, thereby reducing the risk of drug precipitation and deposition in the predominantly aqueous environment of the viable skin. However, the exact drug permeation profile through the skin will have to be explored in greater detail.
Further improvements to solubility could be achieved through a combination of co-solvents and surfactants. However, the main challenge for delivery of the drug from a liquid reservoir in combination with MNs is appropriate device design. This work presented an adapted in vitro model which provided a robust method for assessing permeation. It would be envisaged that to progress this system, a suitable patch design would be required for in vivo application. An alternative approach could be to solidify the co-solvent system through the inclusion of higher molecular weight polymers, thereby having the solubilising enhancement effects of co-solvents but a more convenient formulation.
Future work will include further investigation of the parameters governing drug permeation, considering in more depth the distribution profile of these drugs within the skin. Other hydrophobic compounds, as well as additional co-solvents will be investigated. Further to this, attempts will be made to solidify the system through the addition of higher molecular weight polymers, yet maintain the solubility enhancing effect provided by co-solvents.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








