Synthesis and Biological Evaluation of RGD–Cryptophycin Conjugates for Targeted Drug Delivery

 , , , , , , , , and
, , , , , , , , and
Abstract
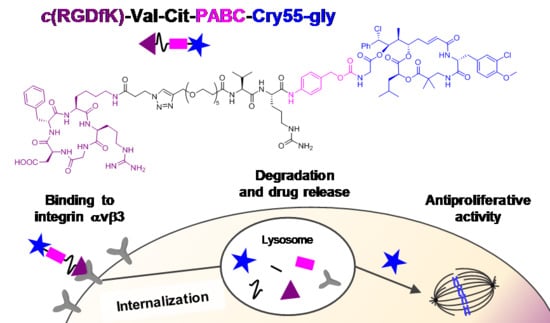
1. Introduction
2. Materials and Methods
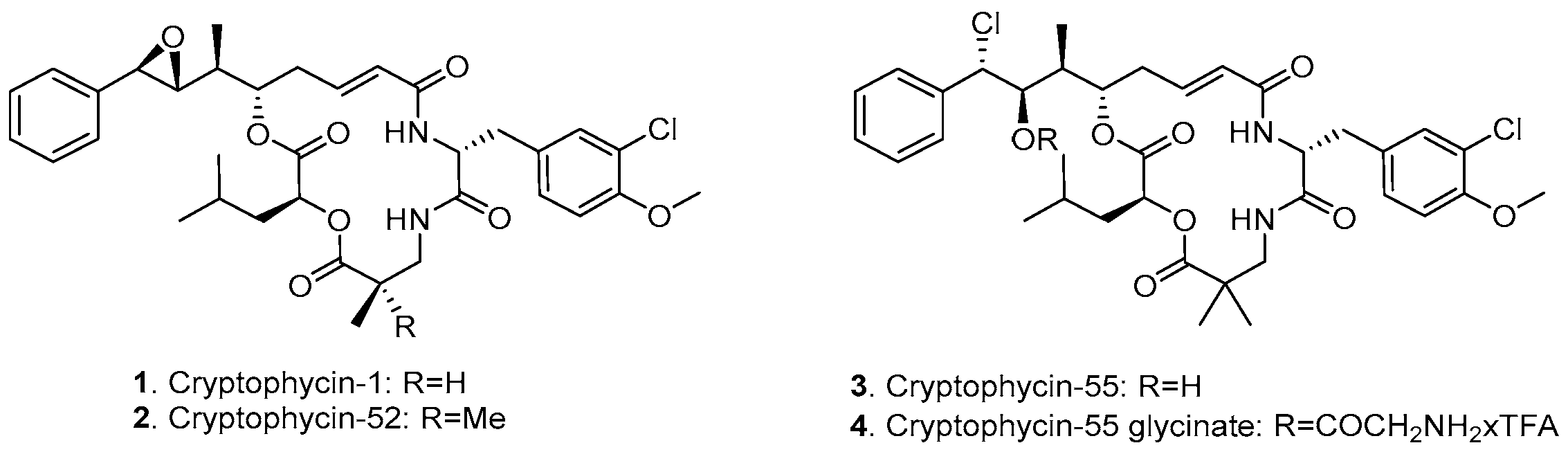
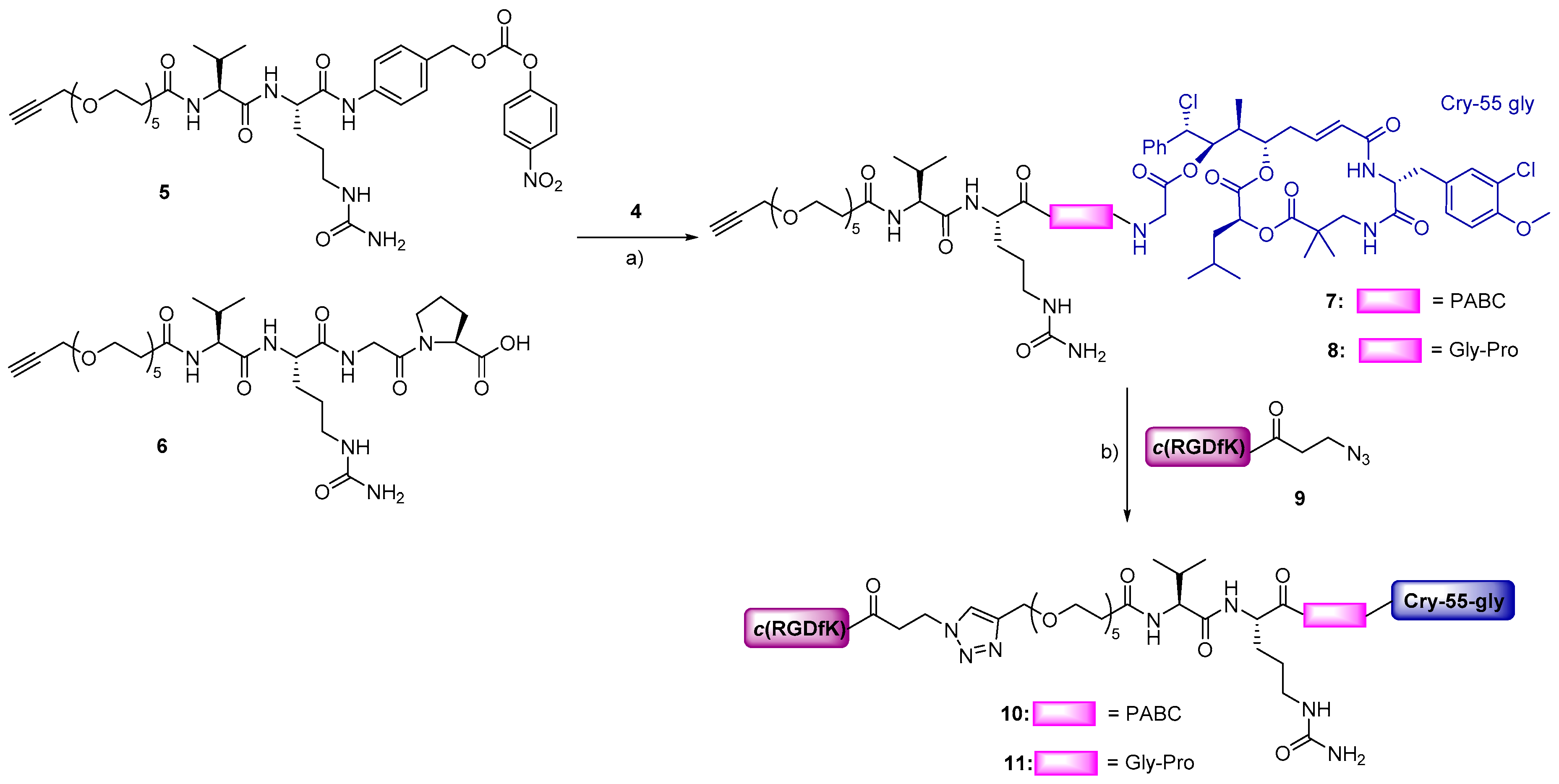
2.1. Synthesis of Cryptophycin Conjugates
2.2. Integrin Binding Assay
2.3. Plasma Metabolic Stability Assay
2.4. Cathepsin B Cleavage Assay
2.5. Degradation in Human Liver Lysosomal Homogenate
2.6. In Vitro Cytotoxicity Assay
2.7. Flow Cytometry Analysis
2.8. Confocal Microscopy
3. Results and Discussion
3.1. Design and Synthesis
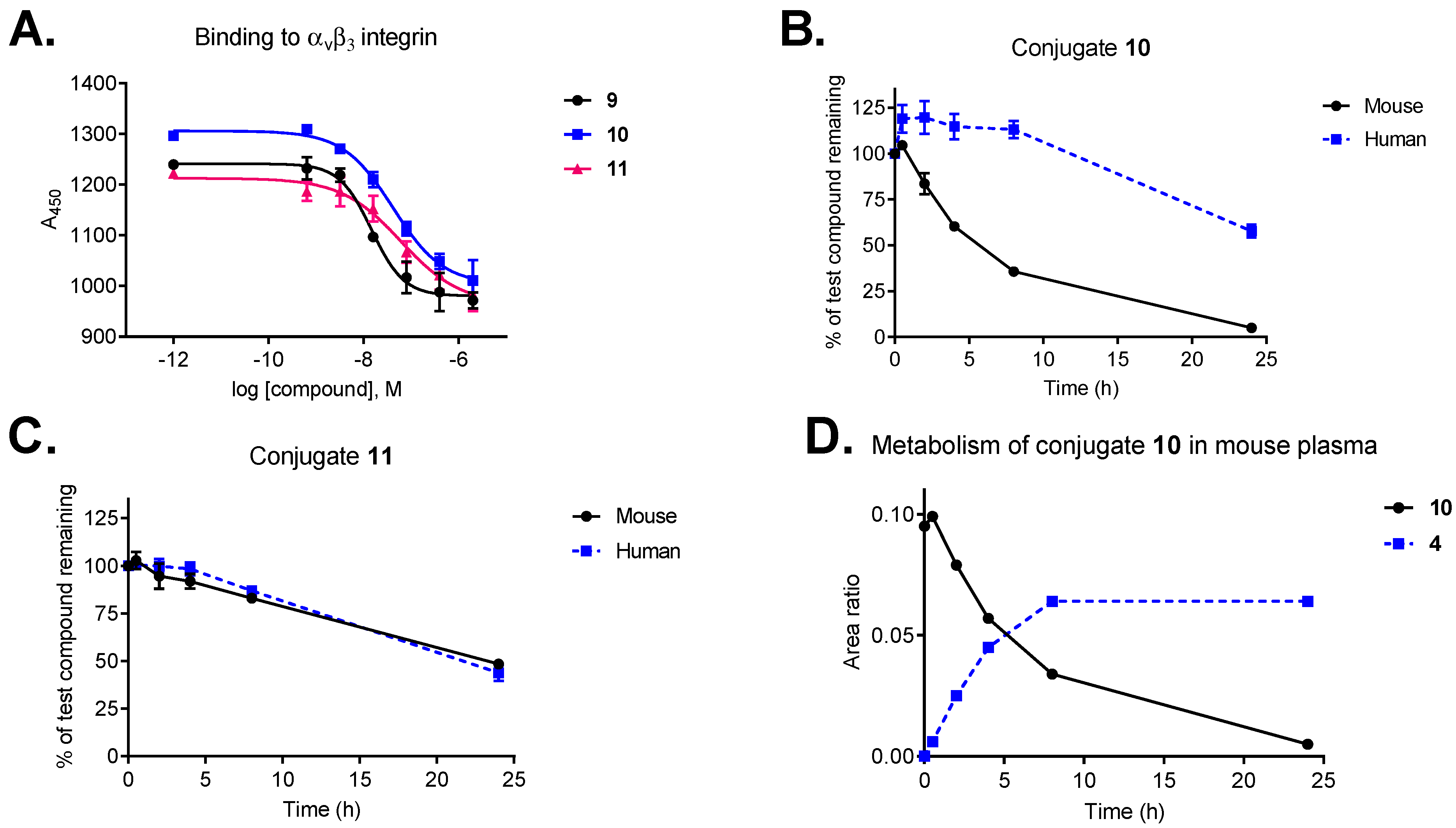
3.2. Integrin Binding Affinity
3.3. Plasma Metabolic Stability
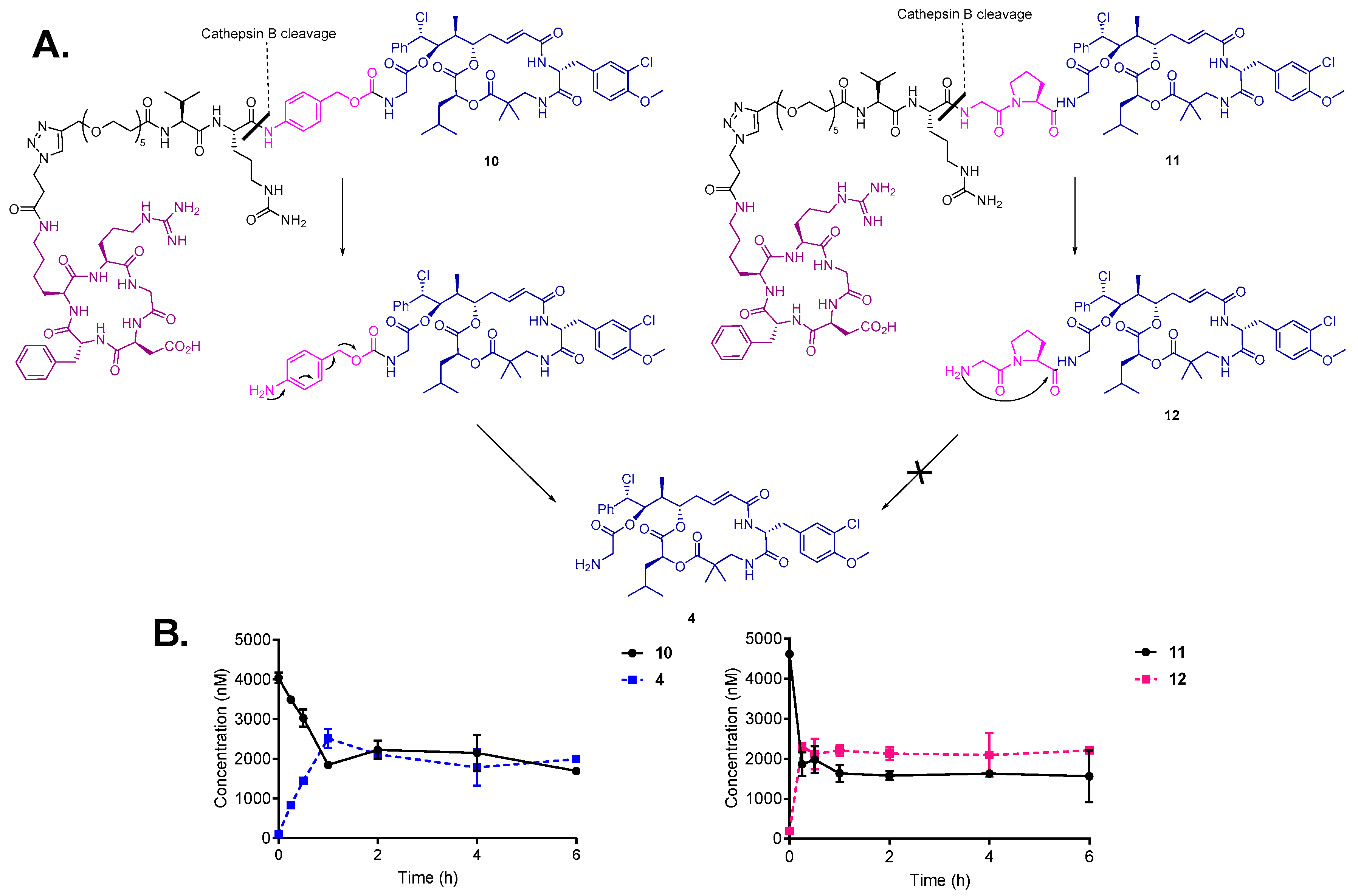
3.4. Drug Release by Cathepsin B and Degradation in Human Liver Lysosomes
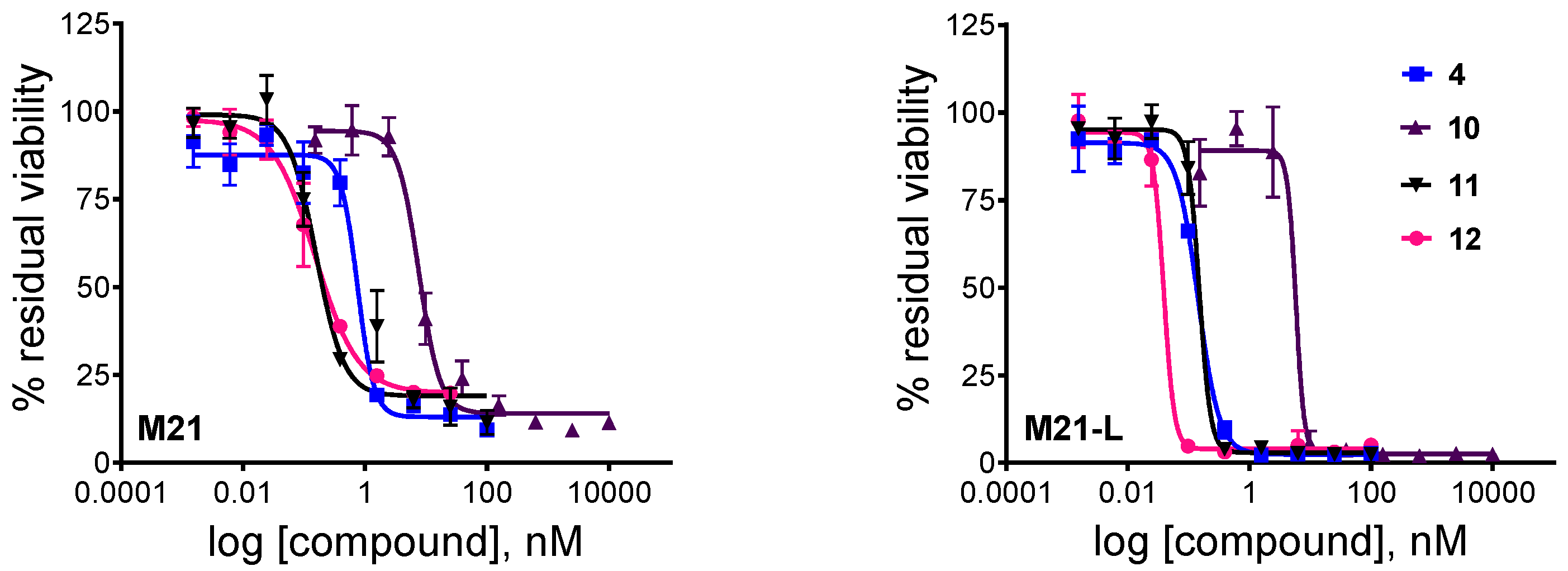
3.5. In Vitro Cytotoxicity Assay
3.6. Intracellular Uptake Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chari, R.V.J.; Miller, M.L.; Widdison, W.C. Antibody-Drug Conjugates: An Emerging Concept in Cancer Therapy. Angew. Chem. Int. Ed. 2014, 53, 3796–3827. [Google Scholar] [CrossRef]
- Srinivasarao, M.; Galliford, C.V.; Low, P.S. Principles in the Design of Ligand-Targeted Cancer Therapeutics and Imaging Agents. Nat. Rev. Drug Discov. 2015, 14, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Vrettos, E.I.; Mező, G.; Tzakos, A.G. On the Design Principles of Peptide–drug Conjugates for Targeted Drug Delivery to the Malignant Tumor Site. Beilstein J. Org. Chem. 2018, 14, 930–954. [Google Scholar] [CrossRef] [PubMed]
- Krall, N.; Scheuermann, J.; Neri, D. Small Targeted Cytotoxics: Current State and Promises from DNA-Encoded Chemical Libraries. Angew. Chem. Int. Ed. 2013, 52, 1384–1402. [Google Scholar] [CrossRef]
- William, D.H.; Tamer, E.F.; Hossam, M.A.; Hongbing, W.; Hanzem, E.H. Antibody-Drug Conjugates: Pharmacokinetic/Pharmacodynamic Modeling, Preclinical Characterization, Clinical Studies, and Lessons Learned. Clin. Pharmacokinet. 2018, 57, 687–703. [Google Scholar]
- Casi, G.; Neri, D. Antibody-Drug Conjugates and Small Molecule-Drug Conjugates: Opportunities and Challenges for the Development of Selective Anticancer Cytotoxic Agents. J. Med. Chem. 2015, 58, 8751–8761. [Google Scholar] [CrossRef]
- Cazzamalli, S.; Corso, A.D.; Neri, D. Targeted Delivery of Cytotoxic Drugs: Challenges, Opportunities and New Developments. Chim. Int. J. Chem. 2017, 71, 712–715. [Google Scholar] [CrossRef]
- Deonarain, M.; Yahioglu, G.; Stamati, I.; Pomowski, A.; Clarke, J.; Edwards, B.; Diez-Posada, S.; Stewart, A. Small-Format Drug Conjugates: A Viable Alternative to ADCs for Solid Tumours? Antibodies 2018, 7, 16. [Google Scholar] [CrossRef]
- Schwartz, R.E.; Hirsch, C.F.; Sesin, D.F.; Flor, J.E.; Chartrain, M.; Fromtling, R.E.; Harris, G.H.; Salvatore, M.J.; Liesch, J.M.; Yudin, K. Pharmaceuticals from Cultured Algae. J. Ind. Microbiol. 1990, 5, 113–123. [Google Scholar] [CrossRef]
- Panda, D.; Himes, R.H.; Moore, R.E.; Wilson, L.; Jordan, M.A. Mechanism of Action of the Unusually Potent Microtubule Inhibitor Cryptophycin 1. Biochemistry 1997, 36, 12948–12953. [Google Scholar] [CrossRef] [PubMed]
- Barbier, P.; Gregoire, C.; Devred, F.; Sarrazin, M.; Peyrot, V. In Vitro Effect of Cryptophycin 52 on Microtubule Assembly and Tubulin: Molecular Modeling of the Mechanism of Action of a New Antimitotic Drug. Biochemistry 2001, 40, 13510–13519. [Google Scholar] [CrossRef]
- Smith, C.D.; Zhang, X.; Mooberry, S.L.; Patterson, G.M.L.; Moore, R.E. Cryptophycin: A New Antimicrotubule Agent Active against Drug-Resistant Cells. Cancer Res. 1994, 54, 3779–3784. [Google Scholar] [PubMed]
- Edelman, M.J.; Gandara, D.R.; Hausner, P.; Israel, V.; Thornton, D.; DeSanto, J.; Doyle, L.A. Phase 2 Study of Cryptophycin 52 (LY355703) in Patients Previously Treated with Platinum Based Chemotherapy for Advanced Non-Small Cell Lung Cancer. Lung Cancer 2003, 39, 197–199. [Google Scholar] [CrossRef]
- D’Agostino, G.; del Campo, J.; Mellado, B.; Izquierdo, M.A.; Minarik, T.; Cirri, L.; Marini, L.; Perez-Gracia, J.L.; Scambia, G. A Multicenter Phase II Study of the Cryptophycin Analog LY355703 in Patients with Platinum-Resistant Ovarian Cancer. Int. J. Gynecol. Cancer 2006, 16, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Sammet, B.; Bogner, T.; Nahrwold, M.; Weiss, C.; Sewald, N. Approaches for the Synthesis of Functionalized Cryptophycins. J. Org. Chem. 2010, 75, 6953–6960. [Google Scholar] [CrossRef]
- Weiss, C.; Sammet, B.; Sewald, N. Recent Approaches for the Synthesis of Modified Cryptophycins. Nat. Prod. Rep. 2013, 30, 924–940. [Google Scholar] [CrossRef]
- Figueras, E.; Borbély, A.; Ismail, M.; Frese, M.; Sewald, N. Novel Unit B Cryptophycin Analogues as Payloads for Targeted Therapy. Beilstein J. Org. Chem. 2018, 14, 1281–1286. [Google Scholar] [CrossRef]
- Nahrwold, M.; Weiß, C.; Bogner, T.; Mertink, F.; Conradi, J.; Sammet, B.; Palmisano, R.; Royo Gracia, S.; Preuße, T.; Sewald, N. Conjugates of Modified Cryptophycins and RGD-Peptides Enter Target Cells by Endocytosis. J. Med. Chem. 2013, 56, 1853–1864. [Google Scholar] [CrossRef]
- Bouchard, H.; Brun, M.-P.; Commercon, A.; Zhang, J. Conjugates, Preparation Thereof, and Therapeutic Use Thereof. U.S. Patent 8952147B2, 10 February 2015. [Google Scholar]
- Verma, V.A.; Pillow, T.H.; DePalatis, L.; Li, G.; Phillips, G.L.; Polson, A.G.; Raab, H.E.; Spencer, S.; Zheng, B. The Cryptophycins as Potent Payloads for Antibody Drug Conjugates. Bioorg. Med. Chem. Lett. 2015, 25, 864–868. [Google Scholar] [CrossRef]
- Weiss, C.; Figueras, E.; Borbely, A.N.; Sewald, N. Cryptophycins: Cytotoxic Cyclodepsipeptides with Potential for Tumor Targeting. J. Pept. Sci. 2017, 23, 514–531. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, H.; Brun, M.-P.; Hubert, P. Novel Peptidic Linkers and Cryptophycin Conjugates, their Preparation and their Therapeutic Use. US2018/0369401 A1, 27 December 2018. [Google Scholar]
- Liang, J.; Moore, R.E.; Moher, E.D.; Munroe, J.E.; Al-awar, R.S.; Hay, D.A.; Varie, D.L.; Zhang, T.Y.; Aikins, J.A.; Martinelli, M.J.; et al. Cryptophycins-309, 249 and Other Cryptophycin Analogs: Preclinical Efficacy Studies with Mouse and Human Tumors. Investig. New Drugs 2005, 23, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Steinkühler, C.; Gallinari, P.; Osswald, B.; Sewald, N.; Ritzefeld, M.; Frese, M.; Figueras, E.; Pethö, L. Cryptophycin-Based Antibody-Drug Conjugates with Novel Self-Immolative Linkers. WO2016/146638 A1, 22 September 2016. [Google Scholar]
- Cazzamalli, S.; Figueras, E.; Pethő, L.; Borbély, A.; Steinkühler, C.; Neri, D.; Sewald, N. In Vivo Antitumor Activity of a Novel Acetazolamide–Cryptophycin Conjugate for the Treatment of Renal Cell Carcinomas. ACS Omega 2018, 3, 14726–14731. [Google Scholar] [CrossRef] [PubMed]
- Figueras, E.; Martins, A.; Borbély, A.; Le Joncour, V.; Cordella, P.; Perego, R.; Modena, D.; Pagani, P.; Esposito, S.; Auciello, G.; et al. Octreotide Conjugates for Tumor Targeting and Imaging. manuscript in preparation.
- Hamidi, H.; Ivaska, J. Every Step of the Way: Integrins in Cancer Progression and Metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in Cancer: Biological Implications and Therapeutic Opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Le Breton, A.; Préat, V. RGD-Based Strategies To Target Alpha(v) Beta(3) Integrin in Cancer Therapy and Diagnosis. Mol. Pharm. 2012, 9, 2961–2973. [Google Scholar] [CrossRef]
- Nieberler, M.; Reuning, U.; Reichart, F.; Notni, J.; Wester, H.-J.; Schwaiger, M.; Weinmüller, M.; Räder, A.; Steiger, K.; Kessler, H. Exploring the Role of RGD-Recognizing Integrins in Cancer. Cancers 2017, 9, 116. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E.; Pierschbacher, M. New Perspectives in Cell Adhesion: RGD and Integrins. Science 1987, 238, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Da Ressurreição, A.S.M.; Vidu, A.; Civera, M.; Belvisi, L.; Potenza, D.; Manzoni, L.; Ongeri, S.; Gennari, C.; Piarulli, U. Cyclic RGD-Peptidomimetics Containing Bifunctional Diketopiperazine Scaffolds as New Potent Integrin Ligands. Chem. Eur. J. 2009, 15, 12184–12188. [Google Scholar] [CrossRef] [PubMed]
- Urman, S.; Gaus, K.; Yang, Y.; Strijowski, U.; Sewald, N.; De Pol, S.; Reiser, O. The Constrained Amino Acid β-Acc Confers Potency and Selectivity to Integrin Ligands. Angew. Chem. Int. Ed. 2007, 46, 3976–3978. [Google Scholar] [CrossRef] [PubMed]
- Kapp, T.G.; Rechenmacher, F.; Neubauer, S.; Maltsev, O.V.; Cavalcanti-Adam, E.A.; Zarka, R.; Reuning, U.; Notni, J.; Wester, H.-J.; Mas-Moruno, C.; et al. A Comprehensive Evaluation of the Activity and Selectivity Profile of Ligands for RGD-Binding Integrins. Sci. Rep. 2017, 7, 39805. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide Combined with Standard Treatment for Patients with Newly Diagnosed Glioblastoma with Methylated MGMT Promoter (CENTRIC EORTC 26071-22072 Study): A Multicentre, Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef]
- Garanger, E.; Boturyn, D.; Dumy, P. Tumor Targeting with RGD Peptide Ligands-Design of New Molecular Conjugates for Imaging and Therapy of Cancers. Anticancer Agents Med. Chem. 2007, 7, 552–558. [Google Scholar] [CrossRef]
- Chen, H.; Niu, G.; Wu, H.; Chen, X. Clinical Application of Radiolabeled RGD Peptides for PET Imaging of Integrin αvβ3. Theranostics 2016, 6, 78–92. [Google Scholar] [CrossRef]
- Dal Corso, A.; Pignataro, L.; Belvisi, L.; Gennari, C. αvβ3 Integrin-Targeted Peptide/Peptidomimetic-Drug Conjugates: In-Depth Analysis of the Linker Technology. Curr. Top. Med. Chem. 2016, 16, 314–329. [Google Scholar] [CrossRef] [PubMed]
- Katsamakas, S.; Chatzisideri, T.; Thysiadis, S.; Sarli, V. RGD-Mediated Delivery of Small-Molecule Drugs. Future Med. Chem. 2017, 9, 579–604. [Google Scholar] [CrossRef]
- Chatzisideri, T.; Leonidis, G.; Sarli, V. Cancer-Targeted Delivery Systems Based on Peptides. Future Med. Chem. 2018, 10, 2201–2226. [Google Scholar] [CrossRef] [PubMed]
- Araste, F.; Abnous, K.; Hashemi, M.; Taghdisi, S.M.; Ramezani, M.; Alibolandi, M. Peptide-Based Targeted Therapeutics: Focus on Cancer Treatment. J. Control. Release 2018, 292, 141–162. [Google Scholar] [CrossRef]
- Esposito, S.; Mele, R.; Ingenito, R.; Bianchi, E.; Bonelli, F.; Monteagudo, E.; Orsatti, L. An Efficient Liquid Chromatography-High Resolution Mass Spectrometry Approach for the Optimization of the Metabolic Stability of Therapeutic Peptides. Anal. Bioanal. Chem. 2017, 409, 2685–2696. [Google Scholar] [CrossRef]
- Veronese, F.M.; Schiavon, O.; Pasut, G.; Mendichi, R.; Andersson, L.; Tsirk, A.; Ford, J.; Wu, G.; Kneller, S.; Davies, J.; et al. PEG-Doxorubicin Conjugates: Influence of Polymer Structure on Drug Release, in Vitro Cytotoxicity, Biodistribution, and Antitumor Activity. Bioconj. Chem. 2005, 16, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and Challenges for the next Generation of Antibody-Drug Conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin B-Labile Dipeptide Linkers for Lysosomal Release of Doxorubicin from Internalizing Immunoconjugates: Model Studies of Enzymatic Drug Release and Antigen-Specific in Vitro Anticancer Activity. Bioconj. Chem. 2002, 13, 855–869. [Google Scholar] [CrossRef]
- Caculitan, N.G.; dela Cruz Chuh, J.; Ma, Y.; Zhang, D.; Kozak, K.R.; Liu, Y.; Pillow, T.H.; Sadowsky, J.; Cheung, T.K.; Phung, Q.; et al. Cathepsin B Is Dispensable for Cellular Processing of Cathepsin B-Cleavable Antibody-Drug Conjugates. Cancer Res. 2017, 77, 7027–7037. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-C.; Chalouni, C.; Doll, S.; Nalle, S.C.; Darwish, M.; Tsai, S.P.; Kozak, K.R.; Del-Rosario, G.; Yu, S.-F.; Erickson, H.; et al. FRET Reagent Reveals the Intracellular Processing of Peptide-Linked Antibody-Drug Conjugates. Bioconj. Chem. 2018, 29, 2468–2477. [Google Scholar] [CrossRef] [PubMed]
- Battersby, J.E.; Hancock, W.S.; Canova-Davis, E.; Oeswein, J.; O’Onnor, B. Diketopiperazine Formation and N-Terminal Degradation in Recombinant Human Growth Hormone. Int. J. Pept. Protein Res. 1994, 44, 215–222. [Google Scholar] [CrossRef]
- DiMarchi, R.D.; Brooke, G.S. Selective Chemical Removal of a Protein Amino-Terminal Residue. US4782139, 1 November 1988. [Google Scholar]
- Su, D.; Kozak, K.R.; Sadowsky, J.; Yu, S.-F.; Fourie-O’Donohue, A.; Nelson, C.; Vandlen, R.; Ohri, R.; Liu, L.; Ng, C.; et al. Modulating Antibody–Drug Conjugate Payload Metabolism by Conjugation Site and Linker Modification. Bioconj. Chem. 2018, 29, 1155–1167. [Google Scholar] [CrossRef]
- Dorywalska, M.; Dushin, R.; Moine, L.; Farias, S.E.; Zhou, D.; Navaratnam, T.; Lui, V.; Hasa-Moreno, A.; Casas, M.G.; Tran, T.-T.; et al. Molecular Basis of Valine-Citrulline-PABC Linker Instability in Site-Specific ADCs and Its Mitigation by Linker Design. Mol. Cancer Ther. 2016, 15, 958–970. [Google Scholar] [CrossRef]
- Cazzamalli, S.; Dal Corso, A.; Neri, D. Linker Stability Influences the Anti-Tumor Activity of Acetazolamide-Drug Conjugates for the Therapy of Renal Cell Carcinoma. J. Control. Release 2017, 246, 39–45. [Google Scholar] [CrossRef]
- Gikanga, B.; Adeniji, N.S.; Patapoff, T.W.; Chih, H.W.; Yi, L. Cathepsin B Cleavage of VcMMAE-Based Antibody-Drug Conjugate Is Not Drug Location or Monoclonal Antibody Carrier Specific. Bioconj. Chem. 2016, 27, 1040–1049. [Google Scholar] [CrossRef]
- Manabe, S.; Machida, H.; Aihara, Y.; Yasunaga, M.; Ito, Y.; Matsumura, Y. Development of a Diketopiperazine-Forming Dipeptidyl Gly-Pro Spacer for Preparation of an Antibody-Drug Conjugate. Medchemcomm 2013, 4, 792–796. [Google Scholar] [CrossRef]
- Cheresh, D.A.; Spiro, R.C. Biosynthetic and Functional Properties of an Arg-Gly-Asp-Directed Receptor Involved in Human Melanoma Cell Attachment to Vitronectin, Fibrinogen, and von Willebrand Factor. J. Biol. Chem. 1987, 262, 17703–17711. [Google Scholar]
- Felding-Habermann, B.; Mueller, B.M.; Romerdahl, C.A.; Cheresh, D.A. Involvement of Integrin αv Gene Expression in Human Melanoma Tumorigenicity. J. Clin. Investig. 1992, 89, 2018–2022. [Google Scholar] [CrossRef] [PubMed]
- Bodero, L.; López Rivas, P.; Korsak, B.; Hechler, T.; Pahl, A.; Müller, C.; Arosio, D.; Pignataro, L.; Gennari, C.; Piarulli, U. Synthesis and Biological Evaluation of RGD and isoDGR Peptidomimetic-α-Amanitin Conjugates for Tumor-Targeting. Beilstein J. Org. Chem. 2018, 14, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Castel, S.; Pagan, R.; Mitjans, F.; Piulats, J.; Goodman, S.; Jonczyk, A.; Huber, F.; Vilaró, S.; Reina, M. RGD Peptides and Monoclonal Antibodies, Antagonists of αv-Integrin, Enter the Cells by Independent Endocytic Pathways. Lab. Investig. 2001, 81, 1615–1626. [Google Scholar] [CrossRef]
- Lucie, S.; Elisabeth, G.; Stéphanie, F.; Guy, S.; Amandine, H.; Corinne, A.R.; Didier, B.; Catherine, S.; Alexeï, G.; Pascal, D.; et al. Clustering and Internalization of Integrin αvβ3 with a Tetrameric RGD-Synthetic Peptide. Mol. Ther. 2009, 17, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.D.C.; Nakeff, A.N.; Valeriote, F.V. Cellular Uptake of a Novel Cytotoxic Agent, Cryptophycin-52, by Human THP-1 Leukemia Cells and H-125 Lung Tumor Cells. Int. J. Cancer 1998, 77, 869–873. [Google Scholar] [CrossRef]
- Panda, D.; DeLuca, K.; Williams, D.; Jordan, M.A.; Wilson, L. Antiproliferative Mechanism of Action of Cryptophycin-52: Kinetic Stabilization of Microtubule Dynamics by High-Affinity Binding to Microtubule Ends. Proc. Natl. Acad. Sci. USA 1998, 95, 9313–9318. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | IC50 (nM) | |
|---|---|---|---|
| M21 (αvβ3+) | M21-L (αv-, αvβ3-) | ||
| 1 | Cry-55gly (4) | 0.75 ± 0.11 | 0.14 ± 0.08 |
| 2 | Gly-Pro-Cry-55gly (12) | 0.15 ± 0.03 | 0.039 ± 0.01 |
| 3 | c(RGDfK)-Val-Cit-PABC-Cry-55gly (10) | 7.63 ± 0.76 | 5.80 ± 4.30 |
| 4 | c(RGDfK)-Val-Cit-Gly-Pro-Cry-55gly (11) | 0.16 ± 0.02 | 0.15 ± 0.02 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borbély, A.; Figueras, E.; Martins, A.; Esposito, S.; Auciello, G.; Monteagudo, E.; Di Marco, A.; Summa, V.; Cordella, P.; Perego, R.; et al. Synthesis and Biological Evaluation of RGD–Cryptophycin Conjugates for Targeted Drug Delivery. Pharmaceutics 2019, 11, 151. https://doi.org/10.3390/pharmaceutics11040151
Borbély A, Figueras E, Martins A, Esposito S, Auciello G, Monteagudo E, Di Marco A, Summa V, Cordella P, Perego R, et al. Synthesis and Biological Evaluation of RGD–Cryptophycin Conjugates for Targeted Drug Delivery. Pharmaceutics. 2019; 11(4):151. https://doi.org/10.3390/pharmaceutics11040151
Chicago/Turabian StyleBorbély, Adina, Eduard Figueras, Ana Martins, Simone Esposito, Giulio Auciello, Edith Monteagudo, Annalise Di Marco, Vincenzo Summa, Paola Cordella, Raffaella Perego, and et al. 2019. "Synthesis and Biological Evaluation of RGD–Cryptophycin Conjugates for Targeted Drug Delivery" Pharmaceutics 11, no. 4: 151. https://doi.org/10.3390/pharmaceutics11040151
APA StyleBorbély, A., Figueras, E., Martins, A., Esposito, S., Auciello, G., Monteagudo, E., Di Marco, A., Summa, V., Cordella, P., Perego, R., Kemker, I., Frese, M., Gallinari, P., Steinkühler, C., & Sewald, N. (2019). Synthesis and Biological Evaluation of RGD–Cryptophycin Conjugates for Targeted Drug Delivery. Pharmaceutics, 11(4), 151. https://doi.org/10.3390/pharmaceutics11040151