Design and In Vivo Pharmacokinetic Evaluation of Triamcinolone Acetonide Microcrystals-Loaded PLGA Microsphere for Increased Drug Retention in Knees after Intra-Articular Injection

Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of TA Microcrystals Using an Ultra-Sonication Method
2.3. Preparation of TA Microcrystals-Loaded MSs Using a Spray-Drying Technique
2.4. Morphological Features of TA Microcrystals and Microcrystals-Loaded MSs
2.4.1. Appearance f TA Microcrystals and Microcrystals-Loaded MSs
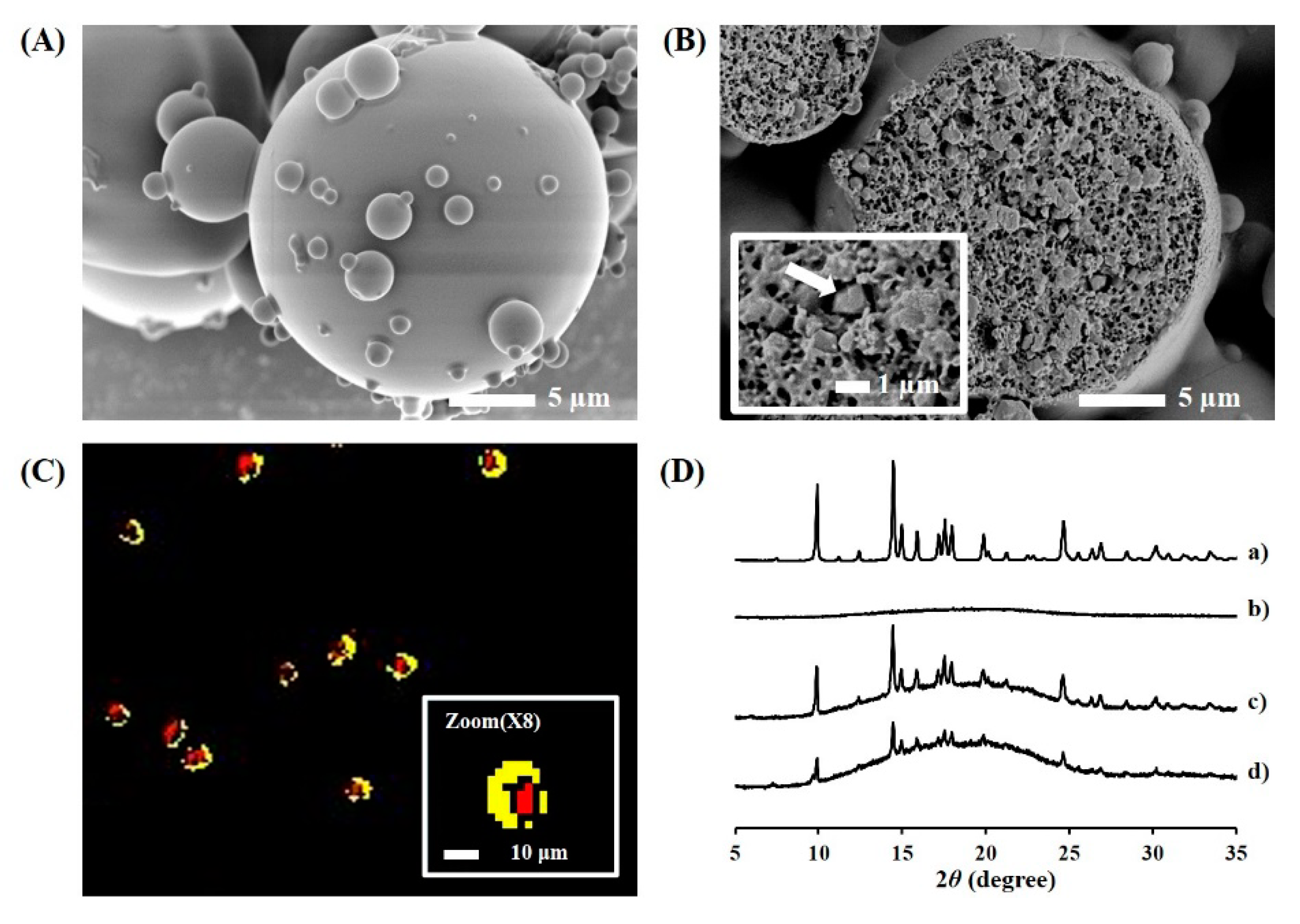
2.4.2. Cross-Sectional Image of TA Microcrystals-Loaded MSs
2.4.3. Hyperspectral Mapping Images of TA Microcrystals-Loaded MSs
2.5. Physicochemical Characterization TA Microcrystals and Microcrystals-Loaded MSs
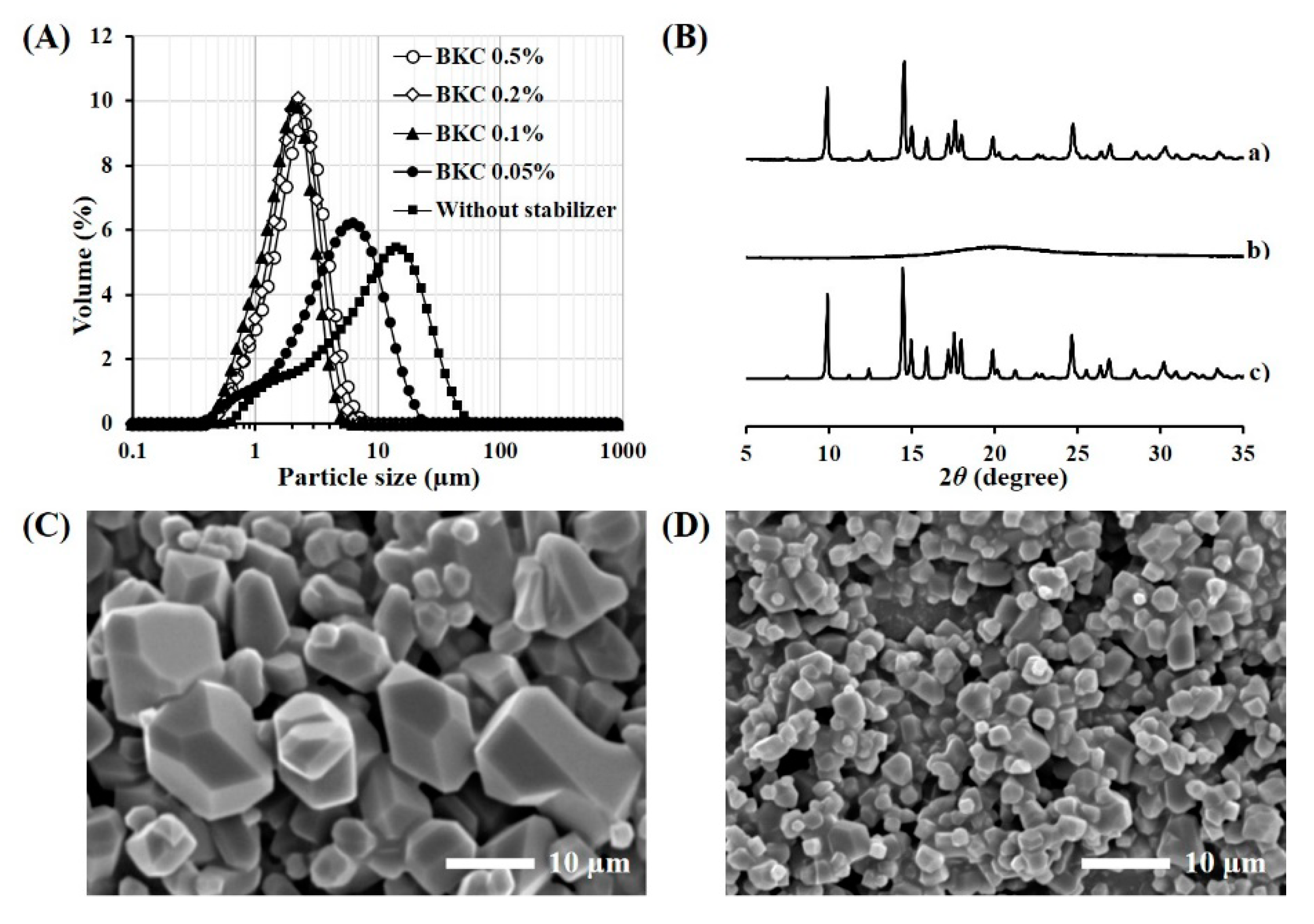
2.5.1. Crystallinity Analysis
2.5.2. Particle Size Analysis
2.5.3. Determination of Loading Amount and Efficiency of TA Microcrystals in MSs
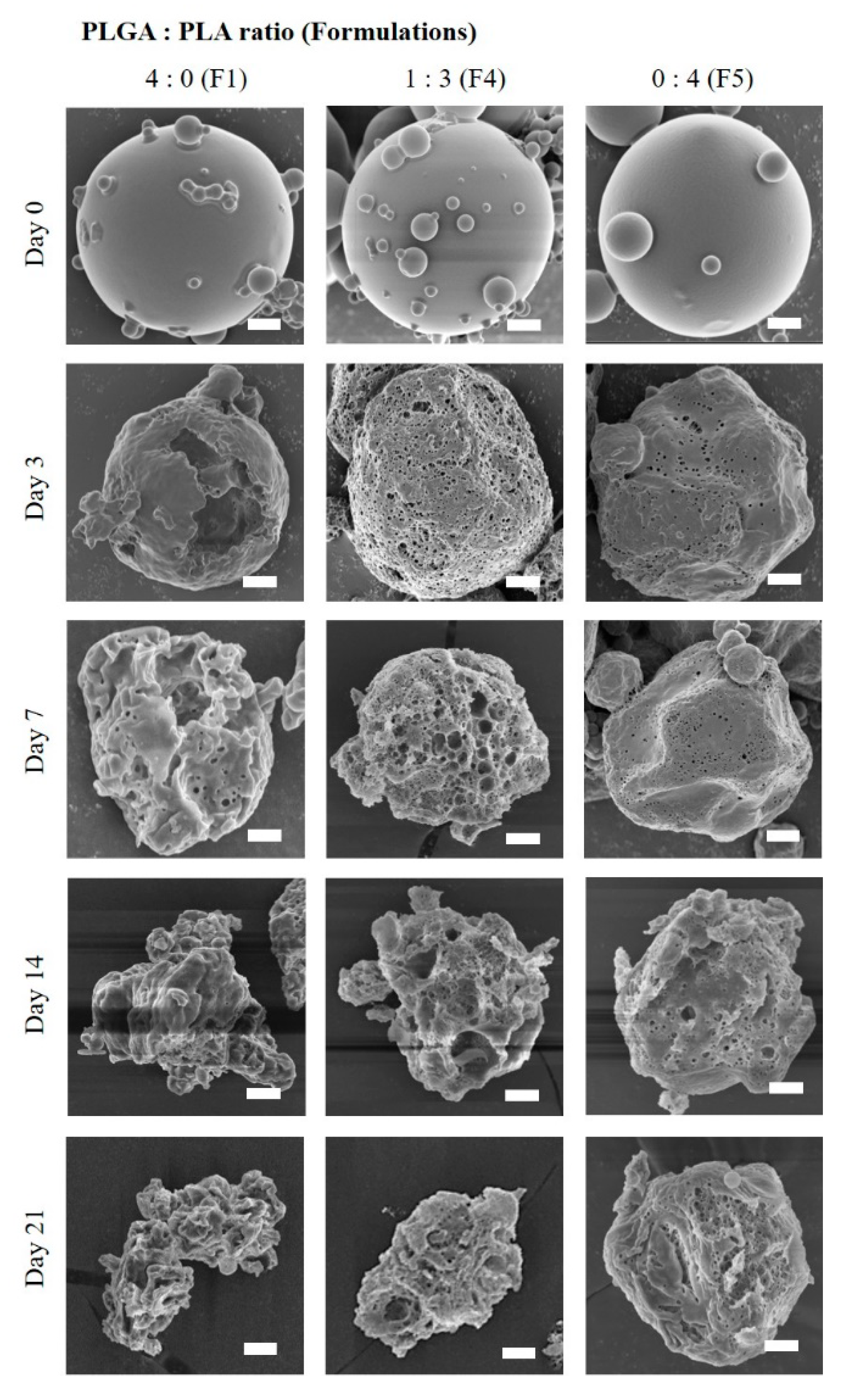
2.6. In Vitro Drug Release Profiles and Morphological Changes of Microcrystals-Loaded MSs
2.7. In Vivo Systemic Exposure and Joint Retention of TA after IA Injection in Rats
2.7.1. Animals and Experimental Protocols
2.7.2. LC-MS/MS Analysis of TA Concentration in Plasma and Knee Tissues
2.7.3. Pharmacokinetic Parameters from TA Concentration Profile in Plasma
2.8. Physicochemical Stability of TA Microcrystals-Loaded MSs
2.9. Statistical Analysis
3. Results and Discussion
3.1. Formulation and Physical Characteristics of TA Microcrystals
3.2. Formulation and Physical Characteristics of TA Microcrystals-Loaded MSs
3.3. In Vitro Drug Release and Degradation Profiles of TA Microcrystals-Loaded MSs
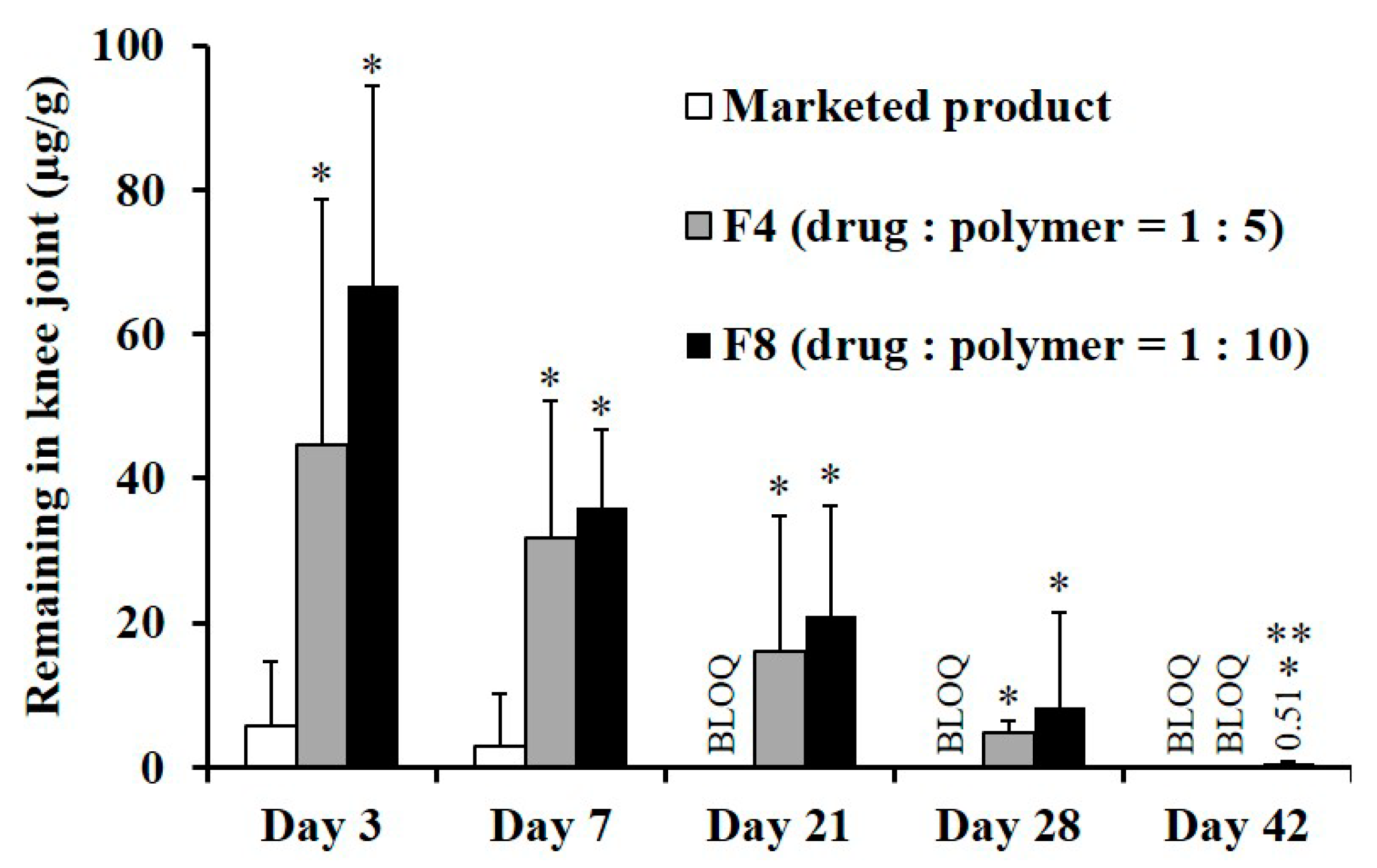
3.4. In Vivo Systemic Exposure and Ioint Retention of TA after IA Injection in Rats
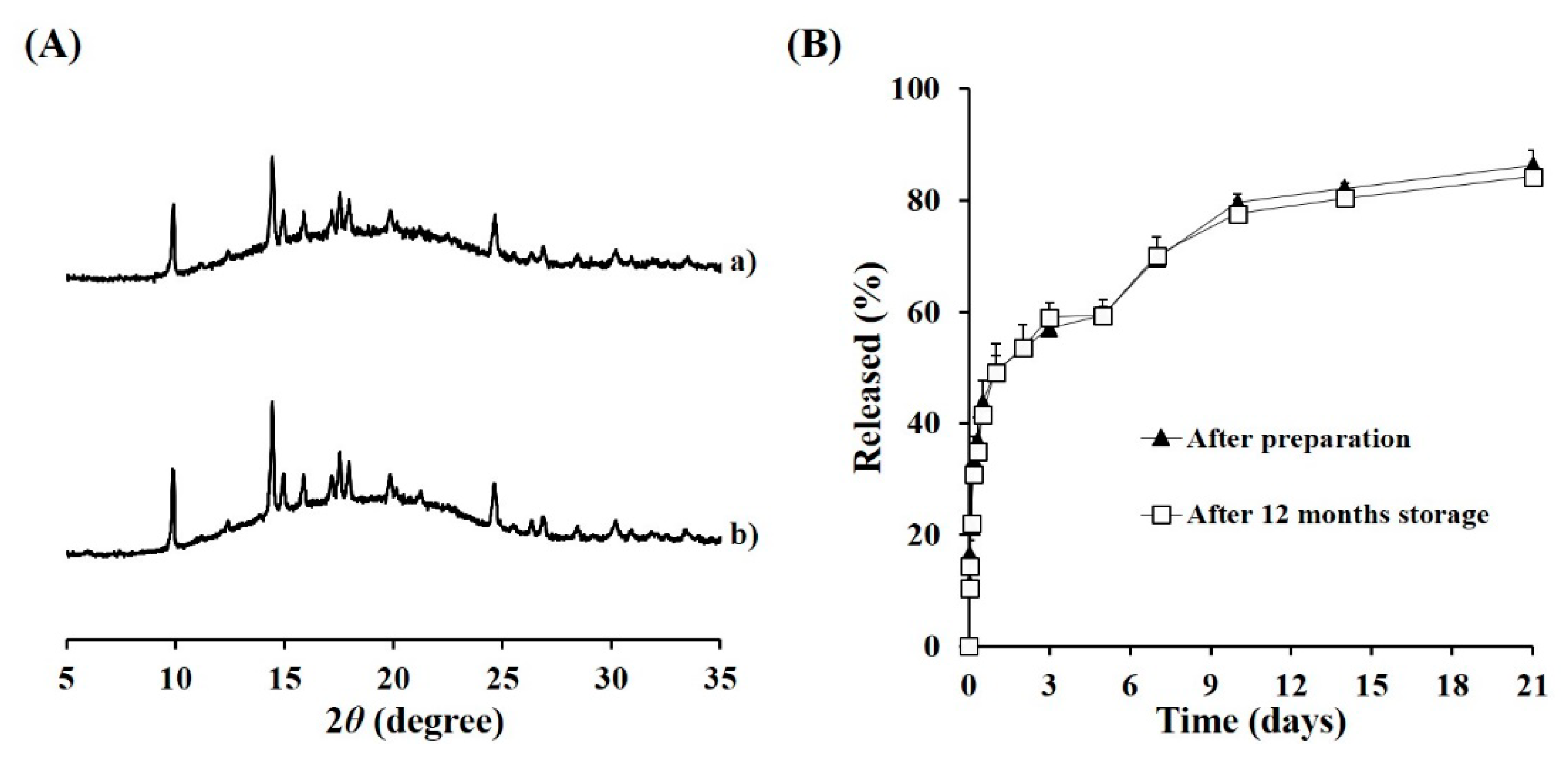
3.5. Long-Term Stability of TA Microcrystals-Loaded MS
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

References
- Gerwin, N.; Hops, C.; Lucke, A. Intraarticular drug delivery in osteoarthritis. Adv. Drug Deliv. Rev. 2006, 58, 226–242. [Google Scholar] [CrossRef] [PubMed]
- Luzardo-Álvarez, A.; Lamela-Gómez, I.; Otero-Espinar, F.; Blanco-Méndez, J. Development, characterization, and in vitro evaluation of resveratrol-loaded poly-(ε-caprolactone) microcapsules prepared by ultrasonic atomization for intra-articular administration. Pharmaceutics 2019, 11, 249. [Google Scholar] [CrossRef] [PubMed]
- Creamer, P. Intra-articular corticosteroid injections in osteoarthritis: Do they work and if so, how? Ann. Rheum. Dis. 1997, 56, 634–636. [Google Scholar] [CrossRef] [PubMed]
- Godwin, M.; Dawes, M. Intra-articular steroid injections for painful knees. Systematic review with meta-analysis. Can. Fam. Physician 2004, 50, 241–248. [Google Scholar] [PubMed]
- Bjordal, J.M.; Johnson, M.I.; Lopes-Martins, R.A.; Bogen, B.; Chow, R.; Ljunggren, A.E. Short-term efficacy of physical interventions in osteoarthritic knee pain. A systematic review and meta-analysis of randomised placebo-controlled trials. BMC Musculoskelet. Disord. 2007, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Ayhan, E.; Kesmezacar, H.; Akgun, I. Intraarticular injections (corticosteroid, hyaluronic acid, platelet rich plasma) for the knee osteoarthritis. World J. Orthop. 2014, 5, 351. [Google Scholar] [CrossRef]
- Knight, A.D.; Levick, J.R. Morphometry of the ultrastructure of the blood-joint barrier in the rabbit knee. Q. J. Exp. Physiol. 1984, 69, 271–288. [Google Scholar] [CrossRef]
- Ayral, X. Injections in the treatment of osteoarthritis. Best Pract. Res. Clin. Rheumatol. 2001, 15, 609–626. [Google Scholar] [CrossRef]
- Kambhampati, S.P.; Mishra, M.K.; Mastorakos, P.; Oh, Y.; Lutty, G.A.; Kannan, R.M. Intracellular delivery of dendrimer triamcinolone acetonide conjugates into microglial and human retinal pigment epithelial cells. Eur. J. Pharm. Biopharm. 2015, 95, 239–249. [Google Scholar] [CrossRef]
- Rudnik-Jansen, I.; Colen, S.; Berard, J.; Plomp, S.; Que, I.; van Rijen, M.; Woike, N.; Egas, A.; van Osch, G.; van Maarseveen, E.; et al. Prolonged inhibition of inflammation in osteoarthritis by triamcinolone acetonide released from a polyester amide microsphere platform. J. Control. Release 2017, 253, 64–72. [Google Scholar] [CrossRef]
- Elron-Gross, I.; Glucksam, Y.; Margalit, R. Liposomal dexamethasone–diclofenac combinations for local osteoarthritis treatment. Int. J. Pharm. 2009, 376, 84–91. [Google Scholar] [CrossRef]
- Rudnik-Jansen, I.; Woike, N.; de Jong, S.; Versteeg, S.; Kik, M.; Emans, P.; Mihov, G.; Thies, J.; Eijkelkamp, N.; Tryfonidou, M.; et al. Applicability of a modified rat model of acute arthritis for long-term testing of drug delivery systems. Pharmaceutics 2019, 11, 70. [Google Scholar] [CrossRef]
- He, Z.; Wang, B.; Hu, C.; Zhao, J. An overview of hydrogel-based intra-articular drug delivery for the treatment of osteoarthritis. Colloids Surf. B Biointerfaces 2017, 154, 33–39. [Google Scholar] [CrossRef]
- Zhang, Z.; Wei, X.; Gao, J.; Zhao, Y.; Zhao, Y.; Guo, L.; Chen, C.; Duan, Z.; Li, P.; Wei, L.; et al. Intra-articular injection of cross-linked hyaluronic acid-dexamethasone hydrogel attenuates osteoarthritis: An experimental study in a rat model of osteoarthritis. Int. J. Mol. Sci. 2016, 17, 411. [Google Scholar] [CrossRef]
- Ho, M.J.; Kim, S.R.; Choi, Y.W.; Kang, M.J. Recent advances in intra-articular drug delivery systems to extend drug retention in joint. J. Pharm. Investig. 2019, 49, 9–15. [Google Scholar] [CrossRef]
- Evans, C.H.; Kraus, V.B.; Setton, L.A. Progress in intra-articular therapy. Nat. Rev. Rheumatol. 2014, 10, 11–22. [Google Scholar] [CrossRef]
- Bodick, N.; Lufkin, J.; Willwerth, C.; Kumar, A.; Bolognese, J.; Schoonmaker, C.; Ballal, R.; Hunter, D.; Clayman, M. An intra-articular, extended-release formulation of triamcinolone acetonide prolongs and amplifies analgesic effect in patients with osteoarthritis of the knee: A randomized clinical trial. J. Bone Jt. Surg. Am. 2015, 97, 877–888. [Google Scholar] [CrossRef]
- Ho, M.J.; Kim, S.R.; Choi, Y.W.; Kang, M.J. A novel stable crystalline triamcinolone acetonide-loaded PLGA microsphere for prolonged release after intra-articular injection. Bull. Korean Chem. Soc. 2016, 37, 1496–1500. [Google Scholar] [CrossRef]
- Ige, P.P.; Baria, R.K.; Gattani, S.G. Fabrication of fenofibrate nanocrystals by probe sonication method for enhancement of dissolution rate and oral bioavailability. Colloids Surf. B Biointerfaces 2013, 108, 366–373. [Google Scholar] [CrossRef]
- Xiao, C.-D.; Shen, X.-C.; Tao, L. Modified emulsion solvent evaporation method for fabricating core–shell microspheres. Int. J. Pharm. 2013, 452, 227–232. [Google Scholar] [CrossRef]
- Ouyang, H.; Zheng, A.; Heng, P.; Chan, L. Effect of lipid additives and drug on the rheological properties of molten paraffin wax, degree of surface drug coating, and drug release in spray-congealed microparticles. Pharmaceutics 2018, 10, 75. [Google Scholar] [CrossRef]
- Naik, J.B.; Waghulde, M.R. Development of vildagliptin loaded Eudragit® microspheres by screening design: In Vitro evaluation. J. Pharm. Investig. 2018, 48, 627–637. [Google Scholar] [CrossRef]
- César, I.C.; Byrro, R.M.D.; de Santana e Silva Cardoso, F.F.; Mundim, I.M.; de Souza Teixeira, L.; de Sousa, W.C.; Gomes, S.A.; Bellorio, K.B.; Brêtas, J.M.; Pianetti, G.A. Determination of triamcinolone in human plasma by a sensitive HPLC-ESI-MS/MS method: Application for a pharmacokinetic study using nasal spray formulation. J. Mass Spectrom. 2011, 46, 320–326. [Google Scholar] [CrossRef]
- Zulian, F.; Martini, G.; Gobber, D.; Plebani, M.; Zacchello, F.; Manners, P. Triamcinolone acetonide and hexacetonide intra-articular treatment of symmetrical joints in juvenile idiopathic arthritis: A double-blind trial. Rheumatology 2004, 43, 1288–1291. [Google Scholar] [CrossRef]
- Zhang, Z.; Bi, X.; Li, H.; Huang, G. Enhanced targeting efficiency of PLGA microspheres loaded with Lornoxicam for intra-articular administration. Drug Deliv. 2011, 18, 536–544. [Google Scholar] [CrossRef]
- Zolnik, B.S.; Leary, P.E.; Burgess, D.J. Elevated temperature accelerated release testing of PLGA microspheres. J. Control. Release 2006, 112, 293–300. [Google Scholar] [CrossRef]
- Shen, J.; Burgess, D.J. Accelerated in vitro release testing of implantable PLGA microsphere/PVA hydrogel composite coatings. Int. J. Pharm. 2012, 422, 341–348. [Google Scholar] [CrossRef]
- Tomic, I.; Vidis-Millward, A.; Mueller-Zsigmondy, M.; Cardot, J.-M. Setting accelerated dissolution test for PLGA microspheres containing peptide, investigation of critical parameters affecting drug release rate and mechanism. Int. J. Pharm. 2016, 505, 42–51. [Google Scholar] [CrossRef]
- Frank, A.; Rath, S.K.; Venkatraman, S.S. Controlled release from bioerodible polymers: Effect of drug type and polymer composition. J. Control. Release 2005, 102, 333–344. [Google Scholar] [CrossRef]
- Stewart, S.; Domínguez-Robles, J.; Donnelly, R.; Larrañeta, E.; Stewart, S.A.; Domínguez-Robles, J.; Donnelly, R.F.; Larrañeta, E. Implantable polymeric drug delivery devices: Classification, manufacture, materials, and clinical applications. Polymers 2018, 10, 1379. [Google Scholar] [CrossRef]
- Witt, C.; Kissel, T. Morphological characterization of microspheres, films and implants prepared from poly(lactide-co-glycolide) and ABA triblock copolymers: Is the erosion controlled by degradation, swelling or diffusion? Eur. J. Pharm. Biopharm. 2001, 51, 171–181. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J.; Makadia, H.K.; Siegel, S.J. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Hopkins, R.L.; Leinung, M.C. Exogenous Cushing’s syndrome and glucocorticoid withdrawal. Endocrinol. Metab. Clin. N. Am. 2005, 34, 371–384. [Google Scholar] [CrossRef]
- Wernecke, C.; Braun, H.J.; Dragoo, J.L. The Effect of Intra-articular corticosteroids on articular cartilage. Orthop. J. Sport. Med. 2015, 3, 232596711558116. [Google Scholar] [CrossRef]
- Jüni, P.; Hari, R.; Rutjes, A.W.; Fischer, R.; Silletta, M.G.; Reichenbach, S.; da Costa, B.R. Intra-articular corticosteroid for knee osteoarthritis. Cochrane Database Syst. Rev. 2015, 10, CD005328. [Google Scholar] [CrossRef]
- Hepper, C.T.; Halvorson, J.J.; Duncan, S.T.; Gregory, A.J.M.; Dunn, W.R.; Spindler, K.P. The efficacy and duration of intra-articular corticosteroid injection for knee osteoarthritis: A systematic review of level I studies. J. Am. Acad. Orthop. Surg. 2009, 17, 638–646. [Google Scholar] [CrossRef]
- Kraus, V.B.; Conaghan, P.G.; Aazami, H.A.; Mehra, P.; Kivitz, A.J.; Lufkin, J.; Hauben, J.; Johnson, J.R.; Bodick, N. Synovial and systemic pharmacokinetics (PK) of triamcinolone acetonide (TA) following intra-articular (IA) injection of an extended-release microsphere-based formulation (FX006) or standard crystalline suspension in patients with knee osteoarthritis (OA). Osteoarthr. Cartil. 2018, 26, 34–42. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stabilizer (w/v %) 1 | Crystal Size (d0.5, µm) 2,3 | Homogeneity (SPAN) 2,4 | Dispersibility 5 |
|---|---|---|---|
| - 6 | 7.21 ± 1.02 | 2.14 ± 0.13 | Aggregated |
| Polysorbate 20 0.5% | 6.63 ± 0.82 | 2.11 ± 0.07 | Aggregated |
| Polysorbate 80 0.5% | 7.35 ± 0.91 | 1.99 ± 0.04 | Aggregated |
| Span 20 0.5% | 17.7 ± 6.27 | 1.81 ± 0.11 | Aggregated |
| PEG 4000 0.5% | 7.15 ± 3.07 | 2.10 ± 0.88 | Aggregated |
| Poloxamer 188 0.5% | 5.44 ± 1.84 | 2.00 ± 0.52 | Aggregated |
| Cholesterol 0.5% | 8.72 ± 3.49 | 1.81 ± 0.60 | Aggregated |
| BKC 0.5% | 2.11 ± 0.05 | 1.32 ± 0.03 | Re-dispersible |
| BKC 0.2% | 1.94 ± 0.06 | 1.20 ± 0.03 | Re-dispersible |
| BKC 0.1% | 1.73 ± 0.02 | 1.21 ± 0.01 | Re-dispersible |
| BKC 0.05% | 4.75 ± 0.23 | 1.99 ± 0.02 | Aggregated |
| Compositions | Characteristics | ||||||
|---|---|---|---|---|---|---|---|
| TA crystal Size (µm) 1, 5 | PLGA:PLA Ratio (w:w) | Drug:polymer Ratio (w:w) | Particle Size (µm) 1, 5 | SPAN 2, 5 | Loading Amount 3, 5 | Loading Efficiency (%) 4, 5 | |
| F0 | 7.21 ± 1.02 | 1:3 | 1:5 | 15.9 ± 0.95 | 2.03 ± 0.04 | 0.16 ± 0.02 | 97.2 ± 2.35 |
| F1 | 1.73 ± 0.02 | 4:0 | 1:5 | 16.2 ± 1.35 | 1.89 ± 0.05 | 0.15 ± 0.01 | 93.5 ± 0.87 |
| F2 | 1.73 ± 0.02 | 3:1 | 1:5 | 17.2 ± 0.43 | 1.89 ± 0.02 | 0.15 ± 0.04 | 92.7 ± 5.76 |
| F3 | 1.73 ± 0.02 | 2:2 | 1:5 | 16.1 ± 0.56 | 2.06 ± 0.02 | 0.14 ± 0.01 | 90.9 ± 1.01 |
| F4 | 1.73 ± 0.02 | 1:3 | 1:5 | 16.9 ± 0.35 | 1.80 ± 0.01 | 0.16 ± 0.01 | 98.1 ± 1.84 |
| F5 | 1.73 ± 0.02 | 0:4 | 1:5 | 15.9 ± 0.06 | 2.11 ± 0.01 | 0.15 ± 0.02 | 96.3 ± 2.48 |
| F6 | 1.73 ± 0.02 | 1:3 | 1:2 | 18.9 ± 0.25 | 1.70 ± 0.01 | 0.31 ± 0.00 | 96.0 ± 0.18 |
| F7 | 1.73 ± 0.02 | 1:3 | 1:3 | 15.8 ± 1.11 | 2.04 ± 0.07 | 0.22 ± 0.01 | 93.7 ± 1.33 |
| F8 | 1.73 ± 0.02 | 1:3 | 1:10 | 16.9 ± 0.42 | 1.20 ± 0.01 | 0.09 ± 0.02 | 99.5 ± 2.55 |
| Parameters | Marketed Product | F4 | F8 |
|---|---|---|---|
| AUC0–7day (ng·h/mL) | 2787.4 ± 187.4 | 1500.4 ± 218.9 * | 1022.2 ± 161.9 *,** |
| Cmax (ng/mL) | 218.7 ± 26.6 | 75.6 ± 17.2 * | 32.2 ± 12.0 *,** |
| Tmax (h) | 3.7 ± 0.8 | 1.8 ± 0.4 * | 1.4 ± 0.5 * |
| T1/2 (h) 1 | 5.3 ± 0.1 | 9.0 ± 0.5 * | 13.3 ± 2.7 *,** |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ho, M.J.; Jeong, H.T.; Im, S.H.; Kim, H.T.; Lee, J.E.; Park, J.S.; Cho, H.R.; Kim, D.Y.; Choi, Y.W.; Lee, J.W.; et al. Design and In Vivo Pharmacokinetic Evaluation of Triamcinolone Acetonide Microcrystals-Loaded PLGA Microsphere for Increased Drug Retention in Knees after Intra-Articular Injection. Pharmaceutics 2019, 11, 419. https://doi.org/10.3390/pharmaceutics11080419
Ho MJ, Jeong HT, Im SH, Kim HT, Lee JE, Park JS, Cho HR, Kim DY, Choi YW, Lee JW, et al. Design and In Vivo Pharmacokinetic Evaluation of Triamcinolone Acetonide Microcrystals-Loaded PLGA Microsphere for Increased Drug Retention in Knees after Intra-Articular Injection. Pharmaceutics. 2019; 11(8):419. https://doi.org/10.3390/pharmaceutics11080419
Chicago/Turabian StyleHo, Myoung Jin, Hoe Taek Jeong, Sung Hyun Im, Hyung Tae Kim, Jeong Eun Lee, Jun Soo Park, Ha Ra Cho, Dong Yoon Kim, Young Wook Choi, Joon Woo Lee, and et al. 2019. "Design and In Vivo Pharmacokinetic Evaluation of Triamcinolone Acetonide Microcrystals-Loaded PLGA Microsphere for Increased Drug Retention in Knees after Intra-Articular Injection" Pharmaceutics 11, no. 8: 419. https://doi.org/10.3390/pharmaceutics11080419
APA StyleHo, M. J., Jeong, H. T., Im, S. H., Kim, H. T., Lee, J. E., Park, J. S., Cho, H. R., Kim, D. Y., Choi, Y. W., Lee, J. W., Choi, Y. S., & Kang, M. J. (2019). Design and In Vivo Pharmacokinetic Evaluation of Triamcinolone Acetonide Microcrystals-Loaded PLGA Microsphere for Increased Drug Retention in Knees after Intra-Articular Injection. Pharmaceutics, 11(8), 419. https://doi.org/10.3390/pharmaceutics11080419