Spironolactone-Loaded LeciPlexes as Potential Topical Delivery Systems for Female Acne: In Vitro Appraisal and Ex Vivo Skin Permeability Studies



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Drug-Loaded LeciPlexes
2.3. Effect of Drug/Lipid Ratio on LeciPlex Properties
2.4. Effect of CTAB/PL-90G Molar Ratio on LeciPlex Properties
2.5. Evaluation of SP-Loaded LeciPlexes
2.5.1. Determination of Drug Entrapment Efficiency
2.5.2. HPLC Assay of SP
2.5.3. Determination of Particle Size and Zeta Potential
2.5.4. FT-IR Studies
2.5.5. Differential Scanning Calorimetry (DSC) Studies
2.5.6. TEM Measurements
2.6. Formulation of LeciPlex Gel
2.7. In Vitro Drug Release Studies
2.8. Kinetic Treatment of the Release Data
2.9. Ex Vivo Rat Skin Penetration Study
2.9.1. Preparation of Rat Skin
2.9.2. Ex Vivo Permeation Experiments
2.10. Statistical Analysis
3. Results and Discussion
3.1. Preparation of SP-Loaded LeciPlexes
3.1.1. Effect of SP Concentration on LeciPlex Properties
3.1.2. Effect of CTAB/PL-90G Ratio on LeciPlex Properties
3.2. FT-IR Studies
3.3. DSC Studies
3.4. TEM Measurements
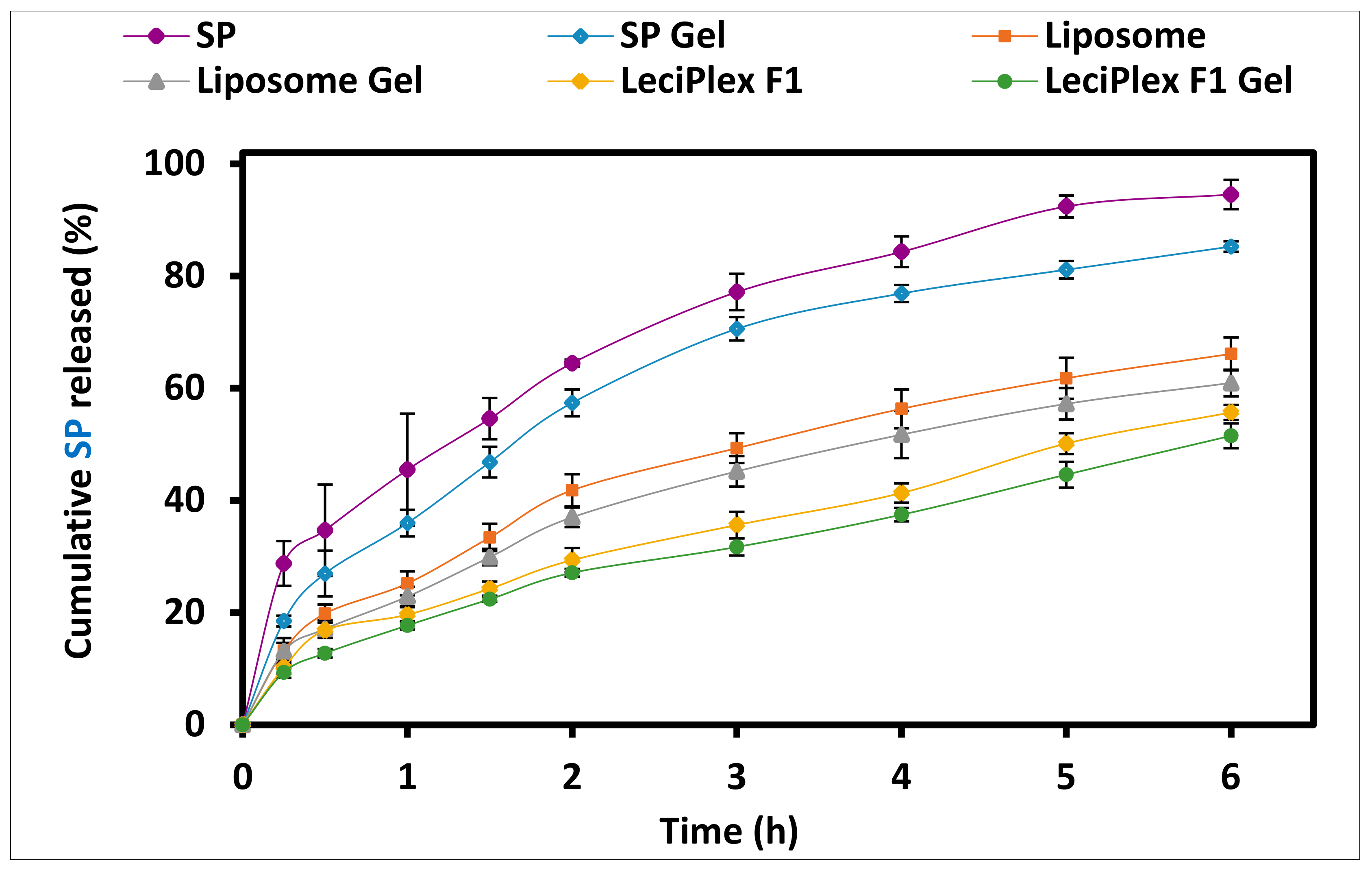
3.5. In Vitro Drug Release Studies
3.6. Kinetics of Drug Release
3.7. Ex Vivo Skin Permeation Studies
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cong, T.X.; Hao, D.; Wen, X.; Li, X.H.; He, G.; Jiang, X. From pathogenesis of acne vulgaris to anti-acne agents. Arch. Dermatol. Res. 2019, 311, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.A.; Bagatin, E. Adult-onset acne: Prevalence, impact, and management challenges. Clin. Cosmet. Investig. Dermatol. 2018, 11, 59–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhate, K.; Williams, H.C. Epidemiology of acne vulgaris. Br. J. Dermatol. 2013, 168, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Hay, R.J.; Johns, N.E.; Williams, H.C.; Bolliger, I.W.; Dellavalle, R.P.; Margolis, D.J.; Marks, R.; Naldi, L.; Weinstock, M.A.; Wulf, S.K.; et al. The global burden of skin disease in 2010: An analysis of the prevalence and impact of skin conditions. J. Investig. Dermatol. 2014, 134, 1527–1534. [Google Scholar] [CrossRef] [Green Version]
- Omer, H.; McDowell, A.; Alexeyev, O.A. Understanding the role of Propionibacterium acnes in acne vulgaris: The critical importance of skin sampling methodologies. Clin. Dermatol. 2017, 35, 118–129. [Google Scholar] [CrossRef]
- Hosthota, A.; Bondade, S.; Basavaraja, V. Impact of acne vulgaris on quality of life and self-esteem. Cutis 2016, 98, 121–124. [Google Scholar]
- Perkins, A.C.; Maglione, J.; Hillebrand, G.G.; Miyamoto, K.; Kimball, A.B. Acne vulgaris in women: Prevalence across the life span. J. Womens Health 2012, 21, 223–230. [Google Scholar] [CrossRef]
- Rocha, M.A.; Bagatin, E. Skin barrier and microbiome in acne. Arch. Dermatol. Res. 2018, 310, 181–185. [Google Scholar] [CrossRef]
- Ju, Q.; Tao, T.; Hu, T.; Karadag, A.S.; Al-Khuzaei, S.; Chen, W. Sex hormones and acne. Clin. Dermatol. 2017, 35, 130–137. [Google Scholar] [CrossRef]
- Cunliffe, W.J. The sebaceous gland and acne-40 years on. Dermatology 1998, 196, 9–15. [Google Scholar] [CrossRef]
- Dunn, L.K.; O’Neill, J.L.; Feldman, S.R. Acne in adolescents: Quality of life, self-esteem, mood, and psychological disorders. Dermatol. Online J. 2011, 17, 1. [Google Scholar] [PubMed]
- Dreno, B.; Bagatin, E.; Blume-Peytavi, U.; Rocha, M.; Gollnick, H. Female type of adult acne: Physiological and psychological considerations and management. J. Dtsch. Dermatol. Ges. 2018, 16, 1185–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, J.C. Acne: Effect of hormones on pathogenesis and management. Am. J. Clin. Dermatol. 2002, 3, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Rathnayake, D.; Sinclair, R. Use of spironolactone in dermatology. Skinmed 2010, 8, 328–332. [Google Scholar] [PubMed]
- Kim, G.K.; Del Rosso, J.Q. Oral spironolactone in post-teenage female patients with acne vulgaris: Practical considerations for the clinician based on current data and clinical experience. J. Clin. Aesthet. Dermatol. 2012, 5, 37–50. [Google Scholar] [PubMed]
- Boisselle, A.; Dionne, F.T.; Tremblay, R.R. Interaction of spironolactone with rat skin androgen receptor. Can. J. Biochem. 1979, 57, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- George, R.; Clarke, S.; Thiboutot, D. Hormonal therapy for acne. Semin. Cutan. Med. Surg. 2008, 27, 188–196. [Google Scholar] [CrossRef]
- Shaw, J.C.; White, L.E. Long-term safety of spironolactone in acne: Results of an 8-year followup study. J. Cutan. Med. Surg. 2002, 6, 541–545. [Google Scholar] [CrossRef]
- Lai, J.-J.; Chang, P.; Lai, K.-P.; Chen, L.; Chang, C. The role of androgen and androgen receptor in skin-related disorders. Arch. Dermatol. Res. 2012, 304, 499–510. [Google Scholar] [CrossRef]
- Krunic, A.; Ciurea, A.; Scheman, A. Efficacy and tolerance of acne treatment using both spironolactone and a combined contraceptive containing drospirenone. J. Am. Acad. Dermatol. 2008, 58, 60–62. [Google Scholar] [CrossRef]
- Schäfer-Korting, M.; Mehnert, W.; Korting, H.-C. Lipid nanoparticles for improved topical application of drugs for skin diseases. Adv. Drug Deliv. Rev. 2007, 59, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.S.; Mohammed, Y.; Pastore, M.N.; Namjoshi, S.; Yousef, S.; Alinaghi, A.; Haridass, I.N.; Abd, E.; Leite-Silva, V.R.; Benson, H.A.E.; et al. Topical and cutaneous delivery using nanosystems. J. Control. Release 2017, 247, 86–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afzali, B.M.; Yaghoobi, E.; Yaghoobi, R.; Bagherani, N.; Dabbagh, M.A. Comparison of the efficacy of 5% topical spironolactone gel and placebo in the treatment of mild and moderate acne vulgaris: A randomized controlled trial. J. Dermatol. Treat. 2012, 23, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Carita, A.C.; Eloy, J.O.; Chorilli, M.; Lee, R.J.; Leonardi, G.R. Recent advances and perspectives in liposomes for cutaneous drug delivery. Curr. Med. Chem. 2018, 25, 606–635. [Google Scholar] [CrossRef]
- Sala, M.; Diab, R.; Elaissari, A.; Fessi, H. Lipid nanocarriers as skin drug delivery systems: Properties, mechanisms of skin interactions and medical applications. Int. J. Pharm. 2018, 535, 1–17. [Google Scholar] [CrossRef]
- Date, A.A.; Srivastava, D.; Nagarsenker, M.S.; Mulherkar, R.; Panicker, L.; Aswal, V.; Hassan, P.A.; Steiniger, F.; Thamm, J.; Fahr, A. Lecithin-based novel cationic nanocarriers (LeciPlex) I: Fabrication, characterization and evaluation. Nanomedicine Lond. Engl. 2011, 6, 1309–1325. [Google Scholar] [CrossRef]
- Shah, S.M.; Ashtikar, M.; Jain, A.S.; Makhija, D.T.; Nikam, Y.; Gude, R.P.; Steiniger, F.; Jagtap, A.A.; Nagarsenker, M.S.; Fahr, A. LeciPlex, invasomes, and liposomes: A skin penetration study. Int. J. Pharm. 2015, 490, 391–403. [Google Scholar] [CrossRef]
- Wang, S.; Lu, W.; Tovmachenko, O.; Rai, U.S.; Yu, H.; Ray, P.C. Challenge in understanding size and shape dependent toxicity of gold nanomaterials in human skin keratinocytes. Chem. Phys. Lett. 2008, 463, 145–149. [Google Scholar] [CrossRef] [Green Version]
- Cui, Z.; Qiu, F.; Sloat, B.R. Lecithin-based cationic nanoparticles as a potential DNA delivery system. Int. J. Pharm. 2006, 313, 206–213. [Google Scholar] [CrossRef]
- Hassan, D.H.; Abdelmonem, R.; Abdellatif, M.M. Formulation and characterization of carvedilol leciplex for glaucoma treatment: In-vitro, ex-vivo and in-vivo study. Pharmaceutics 2018, 10, 197. [Google Scholar] [CrossRef] [Green Version]
- Date, A.A.; Nagarsenker, M.S.; Patere, S.; Dhawan, V.; Gude, R.P.; Hassan, P.A.; Aswal, V.; Steiniger, F.; Thamm, J.; Fahr, A. Lecithin-based novel cationic nanocarriers (Leciplex) II: Improving therapeutic efficacy of quercetin on oral administration. Mol. Pharm. 2011, 8, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Kelidari, H.R.; Saeedi, M.; Akbari, J.; Morteza-Semnani, K.; Gill, P.; Valizadeh, H.; Nokhodchi, A. Formulation optimization and in vitro skin penetration of spironolactone loaded solid lipid nanoparticles. Coll. Surf. B Biointerfaces 2015, 128, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Kelidari, H.R.; Saeedi, M.; Hajheydari, Z.; Akbari, J.; Morteza-Semnani, K.; Akhtari, J.; Valizadeh, H.; Asare-Addo, K.; Nokhodchi, A. Spironolactone loaded nanostructured lipid carrier gel for effective treatment of mild and moderate acne vulgaris: A randomized, double-blind, prospective trial. Coll. Surf. B Biointerfaces 2016, 146, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Fathalla, D.; Soliman, G.; Fouad, E. Development and in vitro/in vivo evaluation of liposomal gels for the sustained ocular delivery of latanoprost. J. Clin. Exp. Ophthalmol. 2015, 6, 2. [Google Scholar]
- Dong, Y.; Ng, W.K.; Shen, S.; Kim, S.; Tan, R.B.H. Preparation and characterization of spironolactone nanoparticles by antisolvent precipitation. Int. J. Pharm. 2009, 375, 84–88. [Google Scholar] [CrossRef]
- Clogston, J.D.; Patri, A.K. Zeta potential measurement. In Characterization of Nanoparticles Intended for Drug Delivery; McNeil, S.E., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 63–70. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Hixson, A.W.; Crowell, J.H. Dependence of reaction Vvelocity upon surface and agitation. Ind. Eng. Chem. 1931, 23, 923–931. [Google Scholar] [CrossRef]
- Baker, R.W.; Lonsdale, H. Controlled release: Mechanisms and rates. In Controlled Release of Biologically Active Agents; Plenum Press: New York, NY, USA, 1974; pp. 15–71. [Google Scholar]
- Alomrani, A.H.; Al-Agamy, M.H.; Badran, M.M. In vitro skin penetration and antimycotic activity of itraconazole loaded niosomes: Various non-ionic surfactants. J. Drug Deliv. Sci. Technol. 2015, 28, 37–45. [Google Scholar] [CrossRef]
- Sloan, K.B.; Beall, H.D.; Weimar, W.R.; Villanueva, R. The effect of receptor phase composition on the permeability of hairless mouse skin in diffusion cell experiments. Int. J. Pharm. 1991, 73, 97–104. [Google Scholar] [CrossRef]
- Dhawan, V.V.; Joshi, G.V.; Jain, A.S.; Nikam, Y.P.; Gude, R.P.; Mulherkar, R.; Nagarsenker, M.S. Apoptosis induction and anti-cancer activity of LeciPlex formulations. Cell. Oncol. 2014, 37, 339–351. [Google Scholar] [CrossRef]
- Aggarwal, N.; Goindi, S. Preparation and evaluation of antifungal efficacy of griseofulvin loaded deformable membrane vesicles in optimized guinea pig model of Microsporum canis—Dermatophytosis. Int. J. Pharm. 2012, 437, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S. DLS and zeta potential—What they are and what they are not? J. Control. Release 2016, 235, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Chandarana, C.V.; Prajapati, P.R.; Jani, G.K. Quantification of spironolactone by Fourier transform infrared spectrophotometry in bulk and tablet dosage form. Vib. Spectrosc. 2019, 100, 185–190. [Google Scholar] [CrossRef]
- Su, G.; Yang, C.; Zhu, J.-J. Fabrication of gold nanorods with tunable longitudinal surface plasmon resonance peaks by reductive dopamine. Langmuir 2015, 31, 817–823. [Google Scholar] [CrossRef]
- Al-Hadiya, B.M.H.; Belal, F.; Asiri, Y.A.; Gubara, O.A. Spironolactone. In Analytical Profiles of Drug Substances and Excipients; Brittain, H.G., Ed.; Academic Press: Cambridge, MA, USA, 2002; Volume 29, pp. 261–320. [Google Scholar]
- Das Neves, J.; Amiji, M.; Bahia, M.F.; Sarmento, B. Assessing the physical–chemical properties and stability of dapivirine-loaded polymeric nanoparticles. Int. J. Pharm. 2013, 456, 307–314. [Google Scholar] [CrossRef]
- Tawfeek, H.M.; Faisal, W.; Soliman, G.M. Enalapril maleate orally disintegrating tablets: Tableting and in vivo evaluation in hypertensive rats. Pharm. Dev. Technol. 2018, 23, 496–503. [Google Scholar] [CrossRef]
- Faisal, W.; Soliman, G.M.; Hamdan, A.M. Enhanced skin deposition and delivery of voriconazole using ethosomal preparations. J. Liposome Res. 2018, 28, 14–21. [Google Scholar] [CrossRef]
- Akbari, J.; Saeedi, M.; Morteza-Semnani, K.; Sadeghi Ghadi, Z.; Hosseini, S.S. Improving the dissolution properties of spironolactone using liquisolid technique. Pharm. Biomed. Res. 2015, 1, 59–70. [Google Scholar] [CrossRef]
- Nounou, M.M.; El-Khordagui, L.K.; Khalafallah, N.A.; Khalil, S.A. In vitro release of hydrophilic and hydrophobic drugs from liposomal dispersions and gels. Acta Pharm. 2006, 56, 311–324. [Google Scholar]
- El-Sayed, M.M.; Hussein, A.K.; Sarhan, H.A.; Mansour, H.F. Flurbiprofen-loaded niosomes-in-gel system improves the ocular bioavailability of flurbiprofen in the aqueous humor. Drug Dev. Ind. Pharm. 2017, 43, 902–910. [Google Scholar] [CrossRef]
- Li, M.; Zheng, Y.; Shan, F.-y.; Zhou, J.; Gong, T.; Zhang, Z.-R. Development of ionic-complex-based nanostructured lipid carriers to improve the pharmacokinetic profiles of breviscapine. Acta Pharmacol. Sin. 2013, 34, 1108–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Tsui, C.P.; Tang, C.Y.; Gu, L. Effects of compositional tailoring on drug delivery behaviours of silica xerogel/polymer core-shell composite nanoparticles. Sci. Rep. 2018, 8, 13002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, P.; Sousa Lobo, J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Siepmann, J.; Siepmann, F. Mathematical modeling of drug delivery. Int. J. Pharm. 2008, 364, 328–343. [Google Scholar] [CrossRef] [PubMed]
- Tomić, I.; Juretić, M.; Jug, M.; Pepić, I.; Cetina Čižmek, B.; Filipović-Grčić, J. Preparation of in situ hydrogels loaded with azelaic acid nanocrystals and their dermal application performance study. Int. J. Pharm. 2019, 563, 249–258. [Google Scholar] [CrossRef]
- Ruela, A.L.M.; Perissinato, A.G.; Lino, M.E.d.S.; Mudrik, P.S.; Pereira, G.R. Evaluation of skin absorption of drugs from topical and transdermal formulations. Braz. J. Pharm. Sci. 2016, 52, 527–544. [Google Scholar] [CrossRef] [Green Version]
- Shamma, R.N.; Sayed, S.; Sabry, N.A.; El-Samanoudy, S.I. Enhanced skin targeting of retinoic acid spanlastics: In vitro characterization and clinical evaluation in acne patients. J. Liposome Res. 2019, 29, 283–290. [Google Scholar] [CrossRef]
- Goyal, G.; Garg, T.; Malik, B.; Chauhan, G.; Rath, G.; Goyal, A.K. Development and characterization of niosomal gel for topical delivery of benzoyl peroxide. Drug Deliv. 2015, 22, 1027–1042. [Google Scholar] [CrossRef]
- Patel, M.R.; Patel, R.B.; Parikh, J.R.; Patel, B.G. Formulation consideration and skin retention study of microemulsion containing tazarotene for targeted therapy of acne. J. Pharm. Investig. 2016, 46, 55–66. [Google Scholar] [CrossRef]
- Abd, E.; Yousef, S.A.; Pastore, M.N.; Telaprolu, K.; Mohammed, Y.H.; Namjoshi, S.; Grice, J.E.; Roberts, M.S. Skin models for the testing of transdermal drugs. Clin. Pharmacol. 2016, 8, 163–176. [Google Scholar] [CrossRef] [Green Version]
- Safwat, M.A.; Soliman, G.M.; Sayed, D.; Attia, M.A. Gold nanoparticles capped with benzalkonium chloride and poly (ethylene imine) for enhanced loading and skin permeability of 5-fluorouracil. Drug Dev. Ind. Pharm. 2017, 43, 1780–1791. [Google Scholar] [CrossRef] [PubMed]
- Ryman-Rasmussen, J.P.; Riviere, J.E.; Monteiro-Riviere, N.A. Penetration of intact skin by quantum dots with diverse physicochemical properties. Toxicol. Sci. 2006, 91, 159–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safwat, M.A.; Soliman, G.M.; Sayed, D.; Attia, M.A. Fluorouracil-loaded gold nanoparticles for the treatment of skin cancer: Development, in vitro characterization, and in vivo evaluation in a mouse skin cancer xenograft model. Mol. Pharm. 2018, 15, 2194–2205. [Google Scholar] [CrossRef]
- Belubbi, T.; Shevade, S.; Dhawan, V.; Sridhar, V.; Majumdar, A.; Nunes, R.; Araujo, F.; Sarmento, B.; Nagarsenker, K.; Steiniger, F.; et al. Lipid architectonics for superior oral bioavailability of nelfinavir mesylate: Comparative in vitro and in vivo assessment. AAPS PharmSciTech 2018, 19, 3584–3598. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredients | Formula | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 | F10 | |
| SP (mg) | 10 | 20 | 30 | 40 | 50 | 10 | 10 | 10 | 10 | 10 |
| PL-90G (mM) | 24 | 24 | 24 | 24 | 24 | 24 | 24 | 24 | 24 | 24 |
| CTAB (mM) | 24 | 24 | 24 | 24 | 24 | 6 | 12 | 36 | 48 | 72 |
| Transcutol®P (mL) | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| Distilled water (mL) | 9.5 | 9.5 | 9.5 | 9.5 | 9.5 | 9.5 | 9.5 | 9.5 | 9.5 | 9.5 |
| Formula | Particle Size (nm) | PDI a | Zeta Potential (mV) | Encapsulation Efficiency (%) b |
|---|---|---|---|---|
| F1 | 337.0 ± 11.1 | 0.3 | 49.3 ± 3.5 | 88.7 ± 3.4 |
| F2 | 668.5 ± 120.3 | 0.7 | 57.6 ± 2.1 | 92.7 ± 5.7 |
| F3 | 591.0 ± 13.7 | 0.9 | 56.9 ± 1.0 | 93.6 ± 6.9 |
| F4 | 487.9 ± 14.5 | 0.5 | 57.7 ± 3.3 | 86.6 ± 2.3 |
| F5 | 439.6 ± 25.1 | 0.8 | 54.3 ± 0.2 | 79.5 ± 7.6 |
| F6 | 381.5 ± 8.8 | 0.3 | 55.2 ± 1.4 | 85.3 ± 1.9 |
| F7 | 355.3 ± 9 | 0.4 | 51.9 ± 1.2 | 87.7 ± 5.7 |
| F8 | 321.5 ± 40.5 | 0.9 | 52.0 ± 1.4 | 92.8 ± 5.7 |
| F9 | 206.5 ± 100 | 0.9 | 54.5 ± 3.7 | 72.6 ± 7.1 |
| F10 | 108.0 ± 25.3 | 0.6 | 51.6 ± 2.1 | 65.8 ± 4.8 |
| Formulation | Zero Order | First Order | Higuchi Model | Hixson–Crowell Model | Baker–Lonsdale Model | Korsemeyer–Peppas Equation | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R2 | K | R2 | K | R2 | K | R2 | K | R2 | K | R2 | K | n | |
| SP suspension | 0.9662 | 11.64 | −0.9965 | 0.46 | 0.9947 | 36.04 | 0.9968 | 0.43 | 0.9965 | 0.06 | 0.9922 | 0.46 | 0.40 |
| SP gel | 0.9535 | 11.50 | −0.9936 | 0.30 | 0.9898 | 35.91 | 0.9841 | 0.33 | 0.9952 | 0.04 | 0.9964 | 0.38 | 0.53 |
| SP Liposomes | 0.9728 | 9.05 | −0.9935 | 0.16 | 0.9967 | 27.87 | 0.9881 | 0.21 | 0.9987 | 0.02 | 0.9963 | 0.27 | 0.53 |
| SP Liposome gel | 0.9786 | 8.43 | −0.9942 | 0.14 | 0.9974 | 25.83 | 0.9901 | 0.18 | 0.9985 | 0.02 | 0.9948 | 0.25 | 0.50 |
| SP LeciPlex F1 | 0.9935 | 7.50 | −0.9968 | 0.12 | 0.9932 | 22.56 | 0.9968 | 0.16 | 0.9848 | 0.01 | 0.9921 | 0.21 | 0.50 |
| SP LeciPlex F1 gel | 0.9937 | 6.97 | −0.9965 | 0.10 | 0.9943 | 20.99 | 0.9965 | 0.14 | 0.9821 | 0.01 | 0.9973 | 0.19 | 0.53 |
| Parameter | Qa | Jsb | Papp × 10−3 c | Q (SC) d | Q (Rest of Skin) e |
|---|---|---|---|---|---|
| SP LeciPlex | 273.0 ± 8.2 | 3.9 ± 0.1 | 14.6 ± 3.7 | 20.8 ± 2.2 | 4.1 ± 0.3 |
| Control SP | 139.6 ± 7.2 | 2.0 ± 0.1 | 4.1 ± 0.4 | 11.7 ± 1.5 | 1.6 ± 0.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salama, A.; Badran, M.; Elmowafy, M.; Soliman, G.M. Spironolactone-Loaded LeciPlexes as Potential Topical Delivery Systems for Female Acne: In Vitro Appraisal and Ex Vivo Skin Permeability Studies. Pharmaceutics 2020, 12, 25. https://doi.org/10.3390/pharmaceutics12010025
Salama A, Badran M, Elmowafy M, Soliman GM. Spironolactone-Loaded LeciPlexes as Potential Topical Delivery Systems for Female Acne: In Vitro Appraisal and Ex Vivo Skin Permeability Studies. Pharmaceutics. 2020; 12(1):25. https://doi.org/10.3390/pharmaceutics12010025
Chicago/Turabian StyleSalama, Ayman, Mohamed Badran, Mohammed Elmowafy, and Ghareb M. Soliman. 2020. "Spironolactone-Loaded LeciPlexes as Potential Topical Delivery Systems for Female Acne: In Vitro Appraisal and Ex Vivo Skin Permeability Studies" Pharmaceutics 12, no. 1: 25. https://doi.org/10.3390/pharmaceutics12010025