Mannose-Decorated Dendritic Polyglycerol Nanocarriers Drive Antiparasitic Drugs To Leishmania infantum-Infected Macrophages

 ,
, 
Abstract
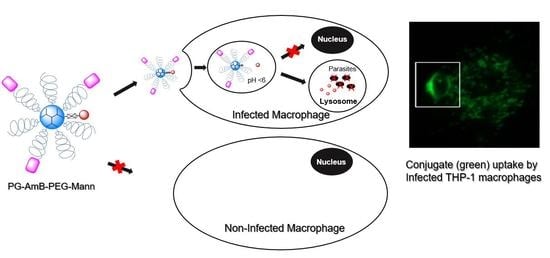
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Conjugates
2.2.1. Synthesis of 2-azidoethyl-α-D-mannopyranoside
Synthesis 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside
Synthesis of 2-bromoethyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside
Synthesis of 2-azidoethyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside
Synthesis of 2-azidoethyl-α-D-mannopyranoside (Mannose-N3)
2.2.2. Synthesis of AmB–EMCH
2.2.3. Synthesis of PG Amine
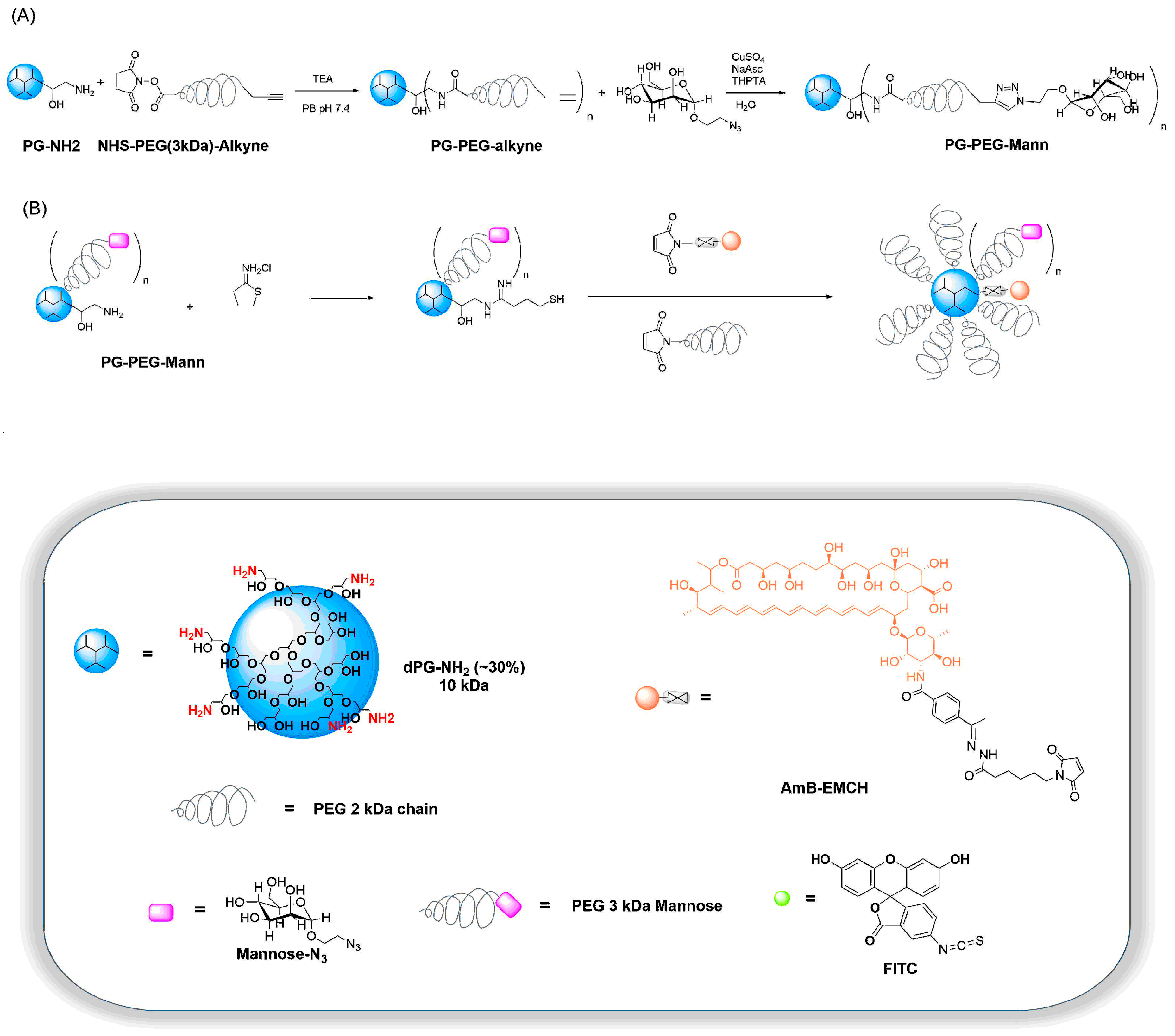
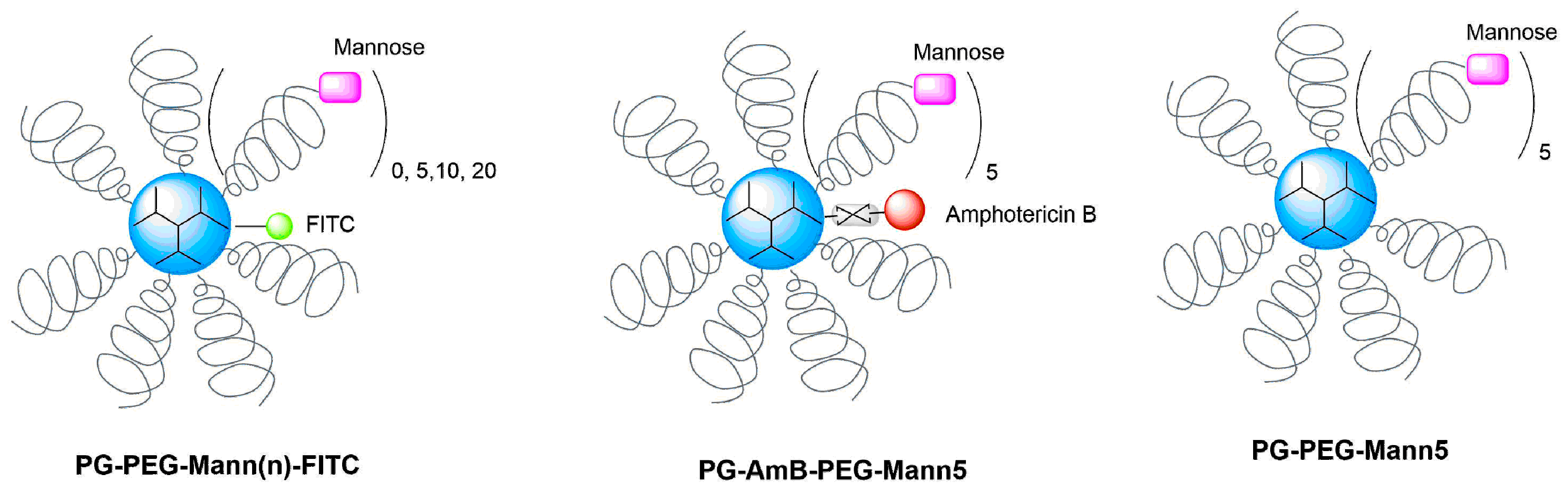
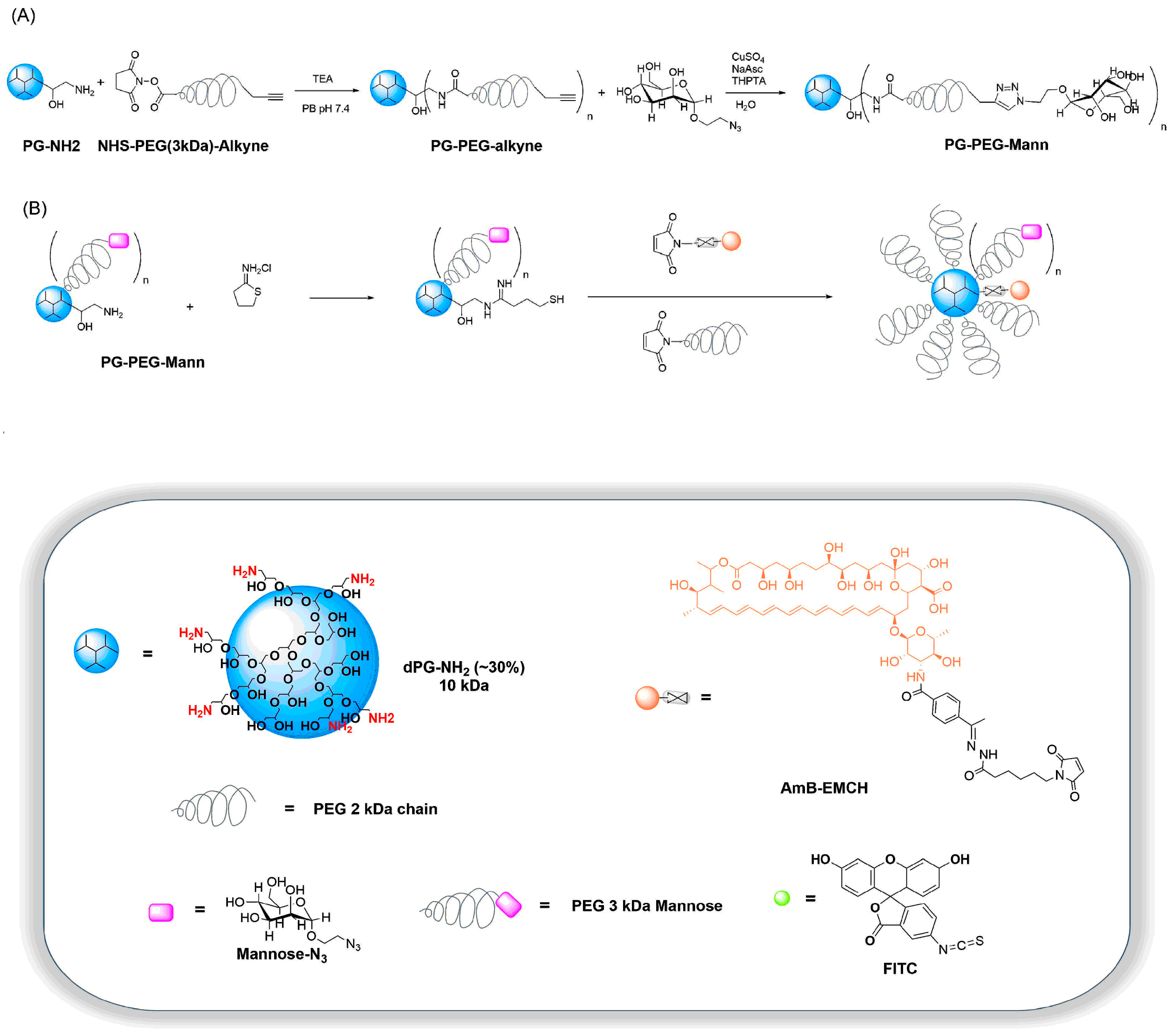
2.2.4. Synthesis of PG–PEG–Mann (PEG 3 kDa)
2.2.5. Synthesis of PG–PEG–Mann Drug/Dye Conjugates
2.2.6. Anthrone Method for Sugar Loading Determination
2.3. Physicochemical Characterization of PG–AmB–PEG–Mann Conjugates
2.4. Drug Release Profile Determination of PG-AmB–PEG Conjugate
2.5. Mice and Parasites
2.6. Macrophage Cell Cultures and In Vitro Leishmania Infections
2.7. Nanoparticle Uptake Studies
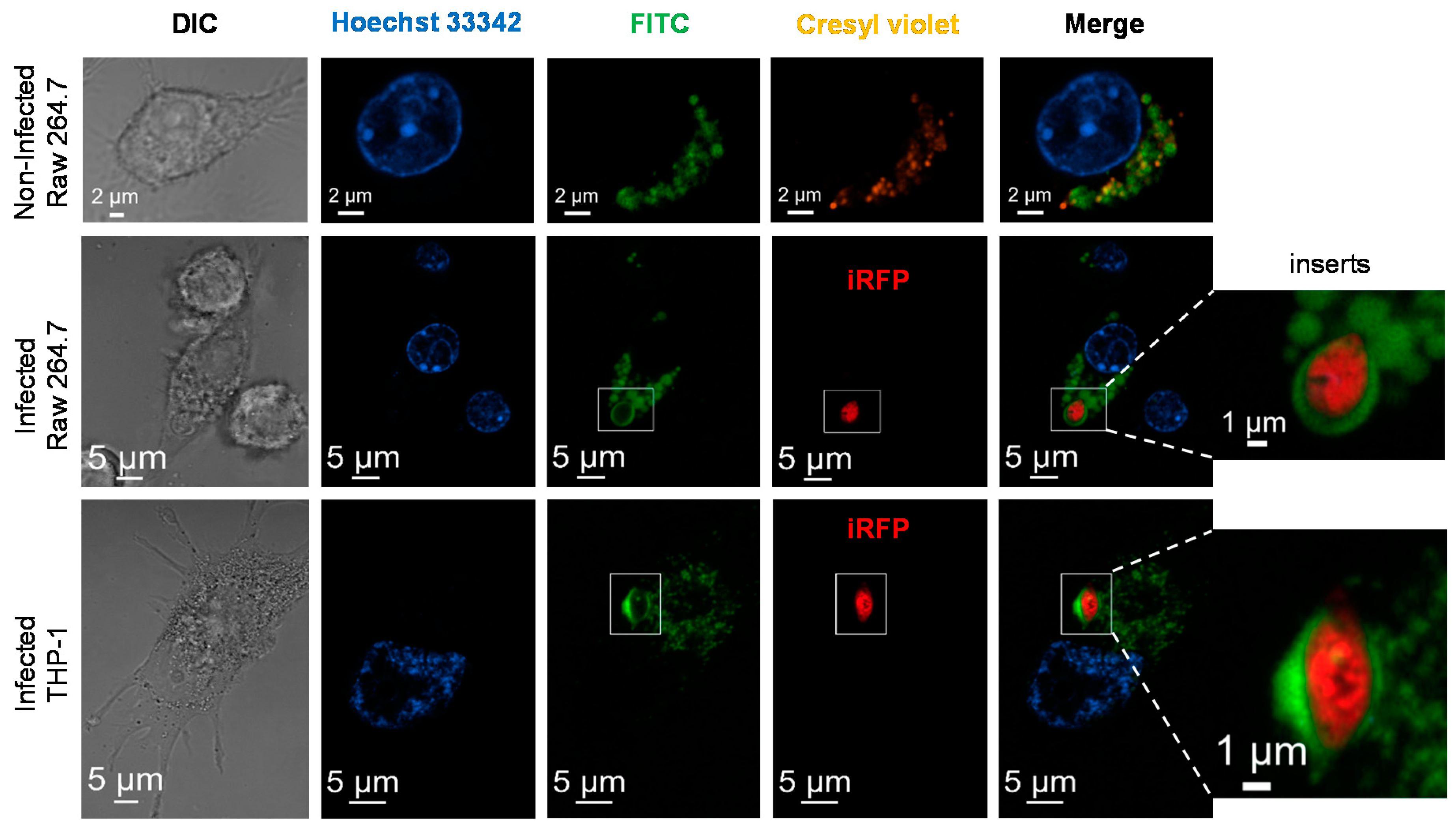
2.8. Subcellular Localization of Nanoparticles
2.9. Assessment of Antileishmanial Effect Using Infected ex vivo Splenic Explant Cultures
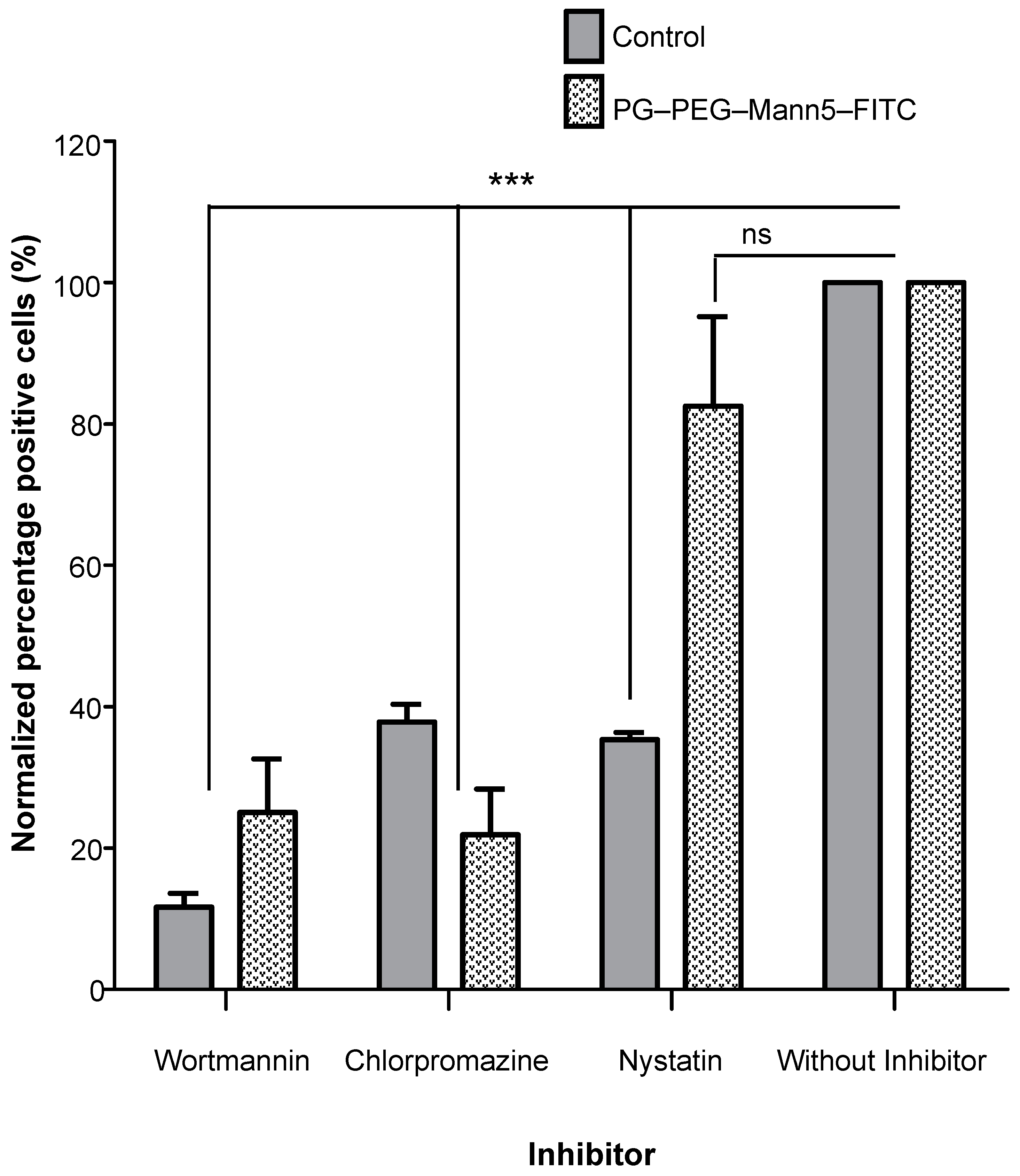
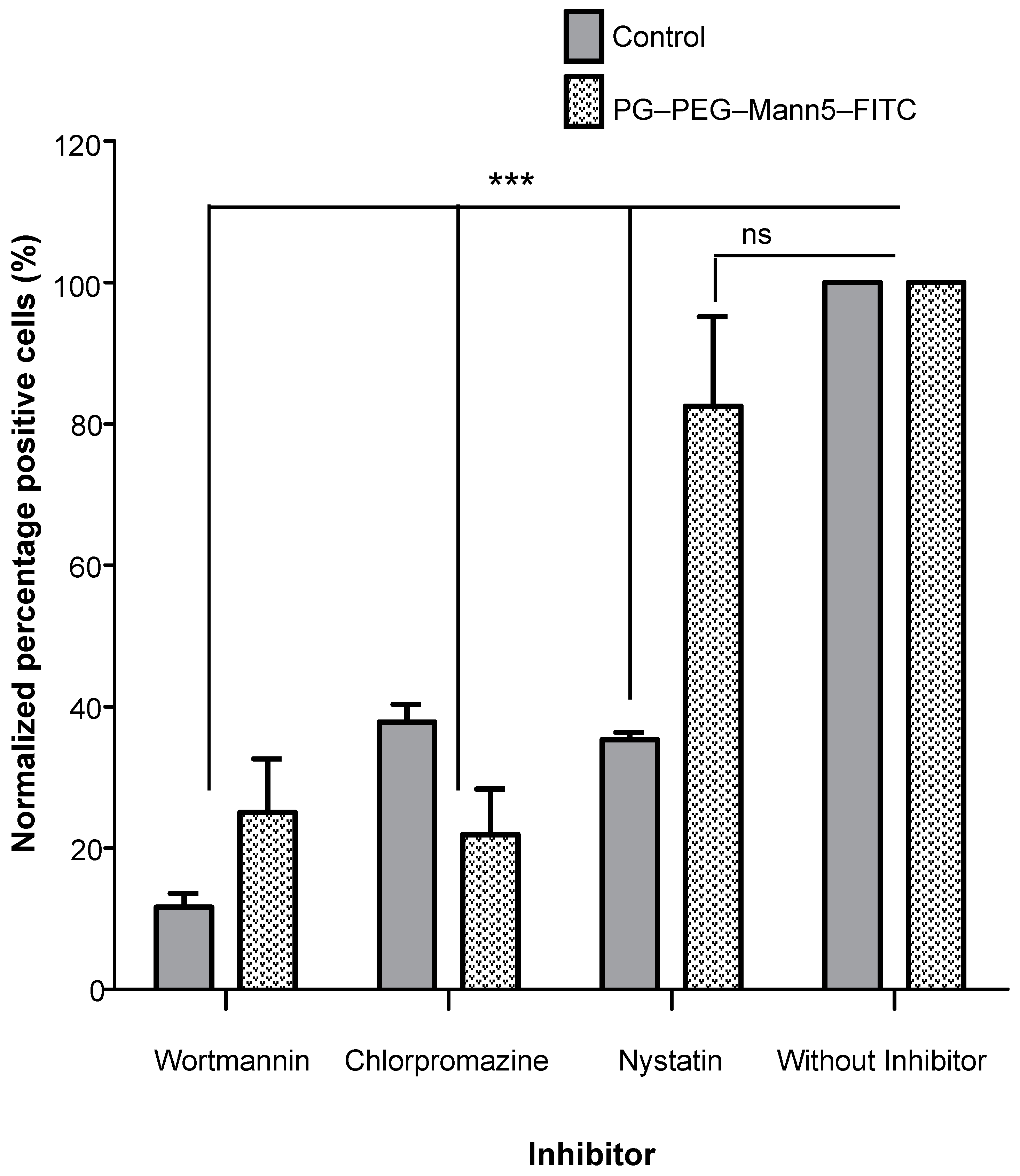
2.10. Inhibition of Endocytosis
3. Results
3.1. Synthesis and Characterization of Targeting Polyglycerol Conjugates
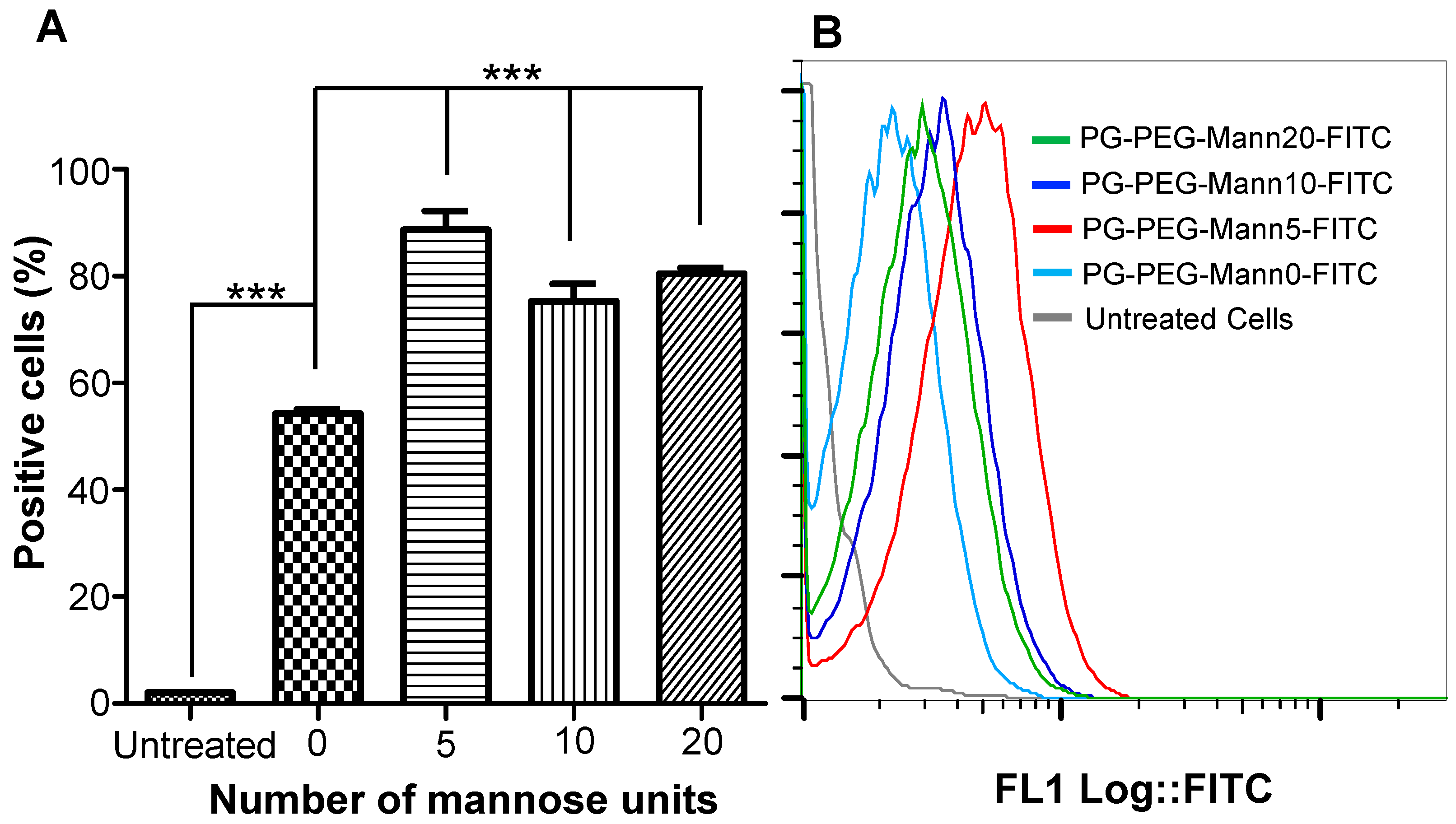
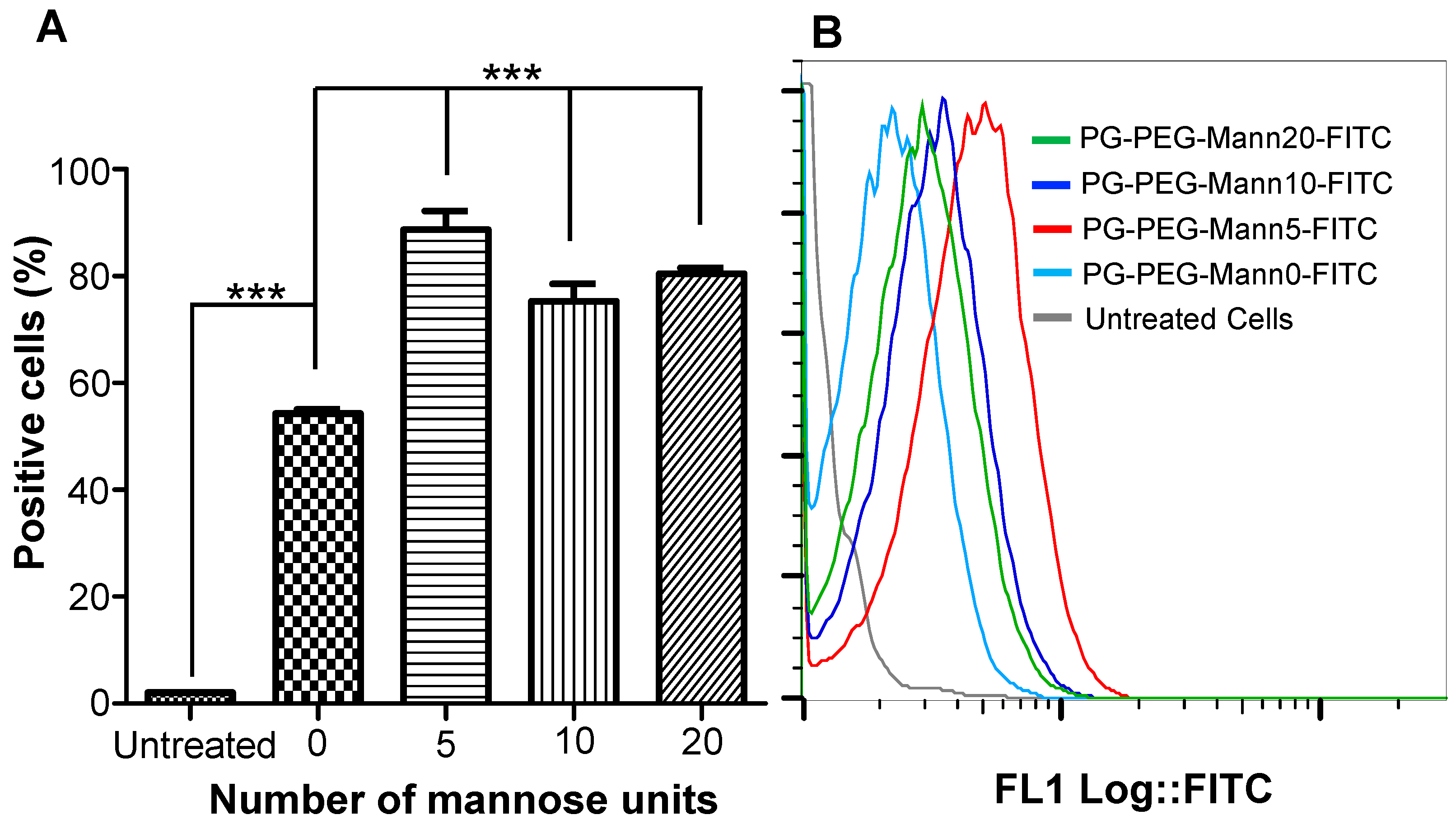
3.2. Internalization of Mannosylated PG–PEG Nanocarrier in Macrophages Depends on CD206 Receptor
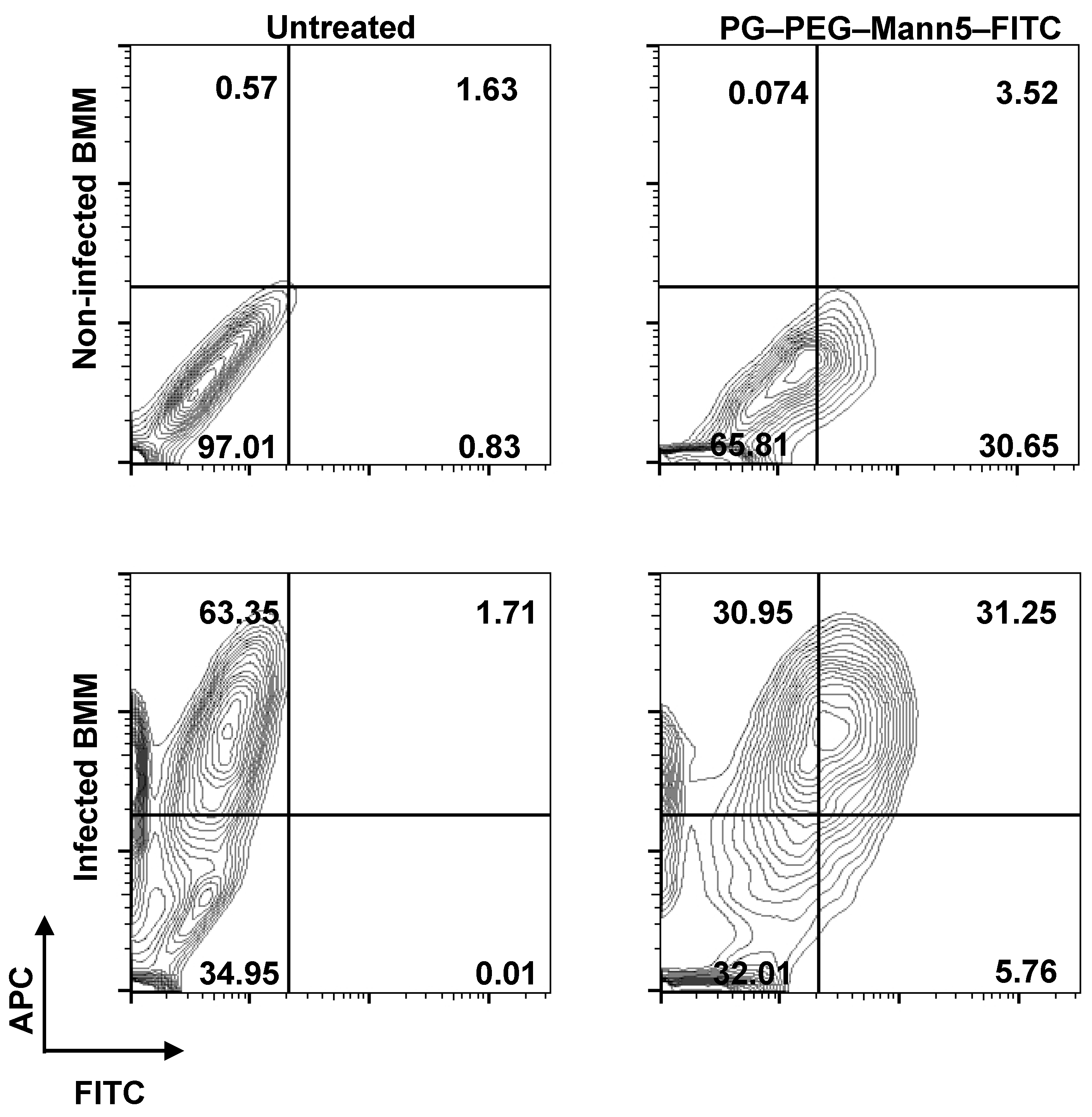
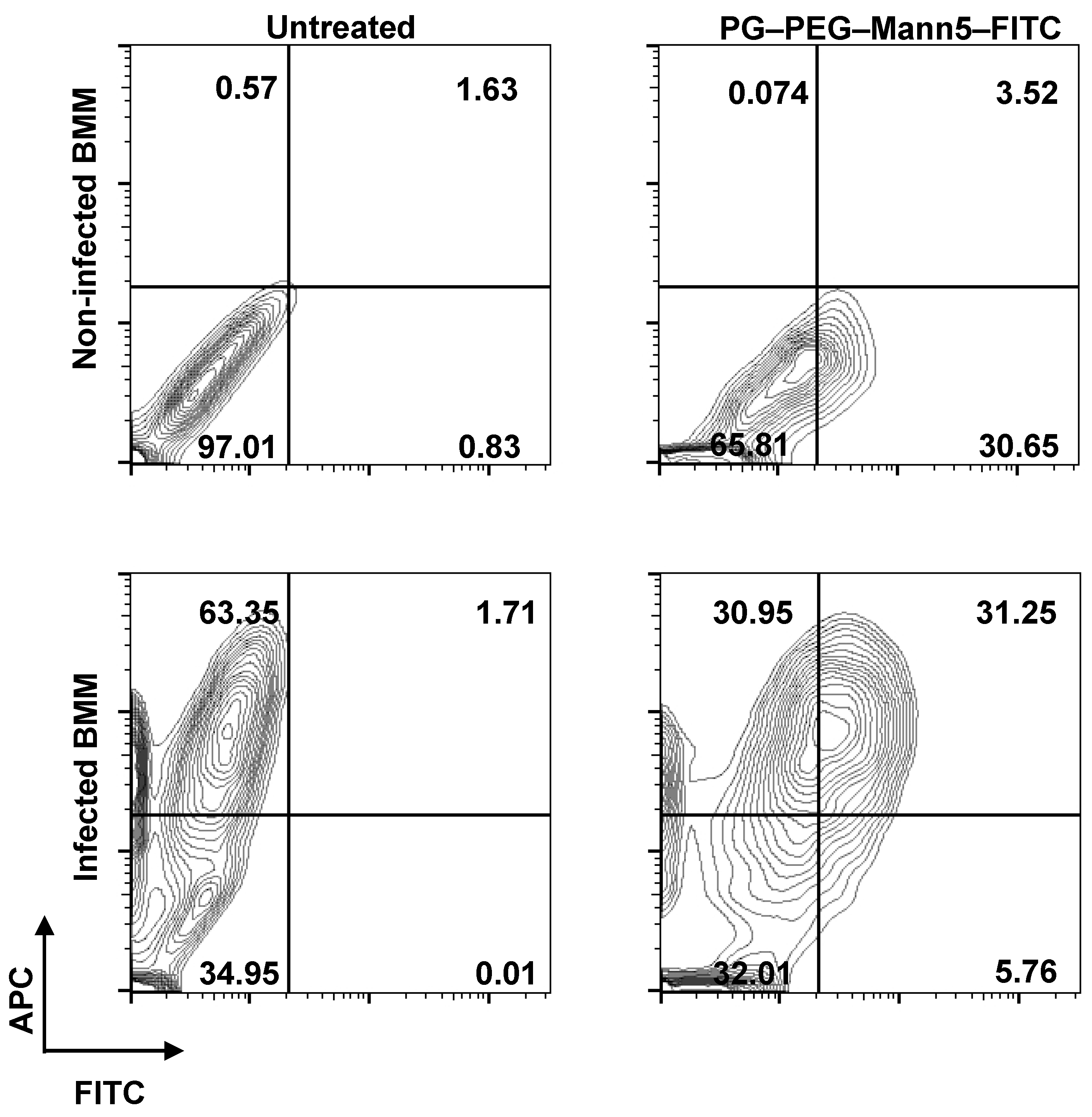
3.3. L. infantum Infected Macrophages Display Higher Avidity for Mannosylated Nanocarriers than Non-Infected Macrophages
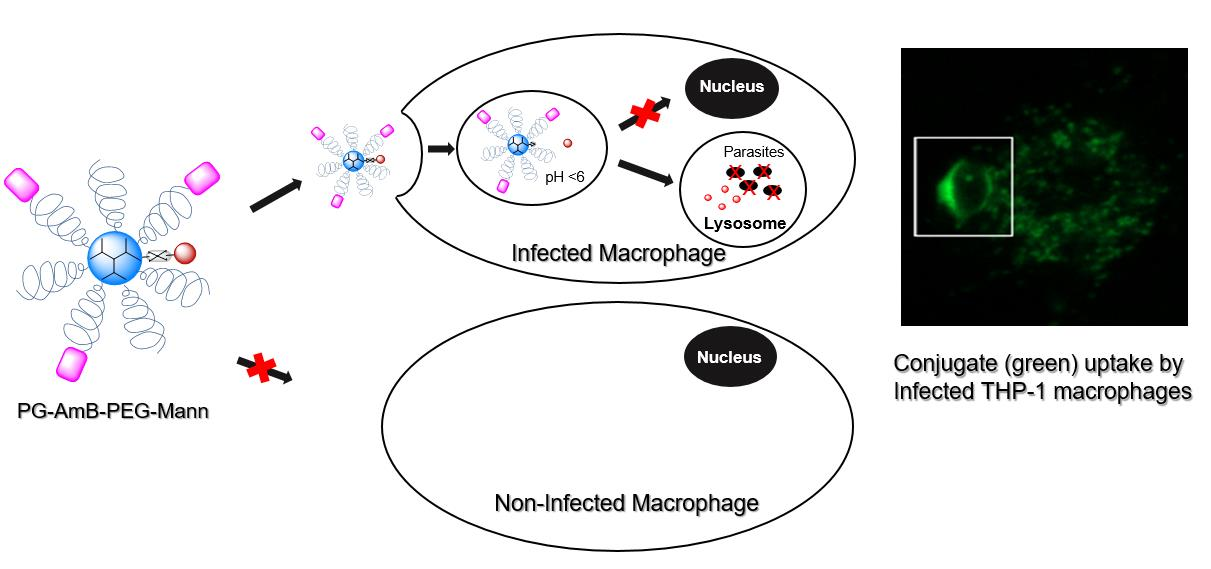
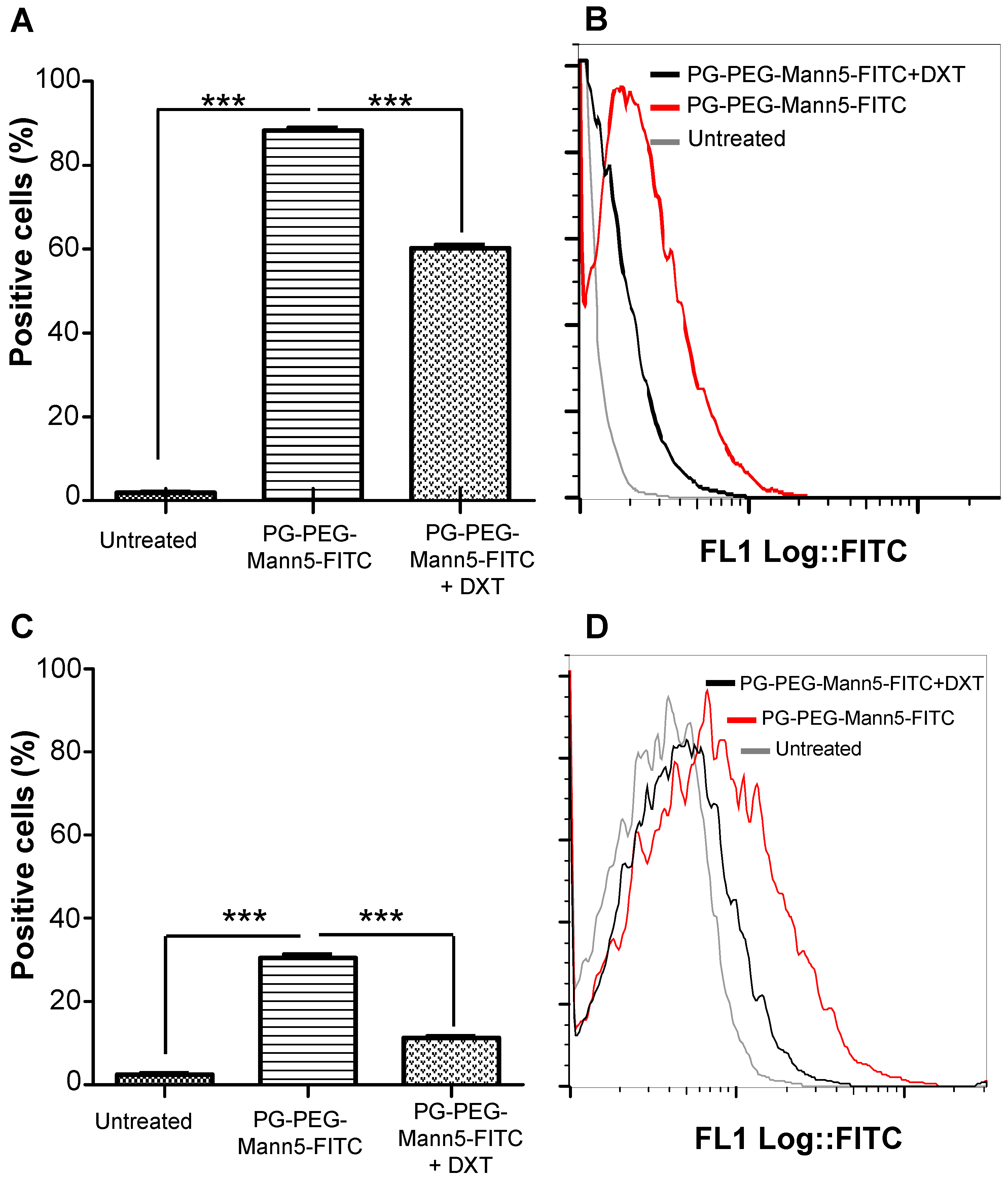
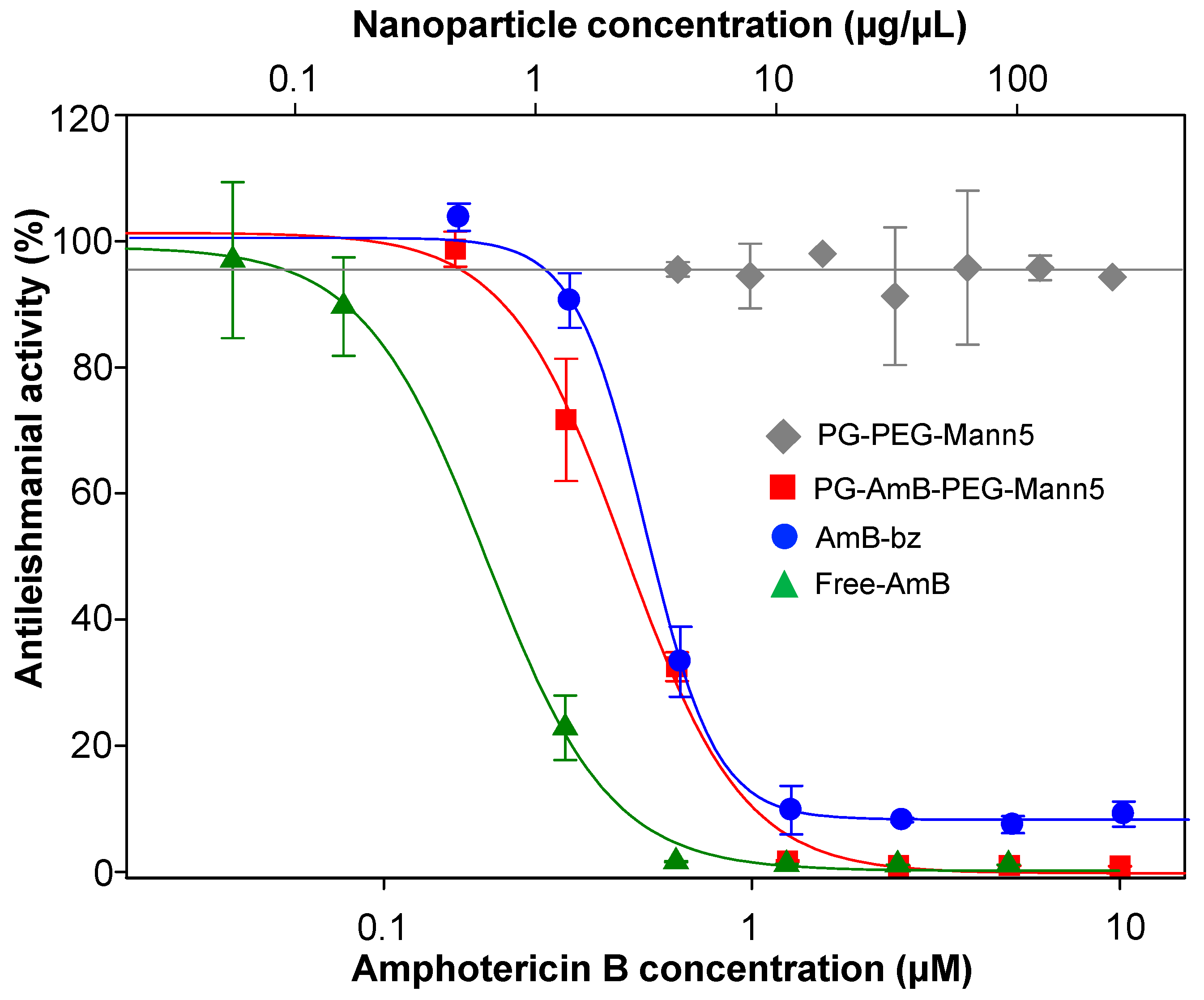
3.4. Mannosylated Nanoparticles as Drug Carriers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- van Griensven, J.; Diro, E. Visceral leishmaniasis. Infect. Dis. Clin. North Am. 2012, 26, 309–322. [Google Scholar]
- Zijlstra, E.E.; Musa, A.M.; Khalil, E.A.; el-Hassan, I.M.; el-Hassan, A.M. Post-kala-azar dermal leishmaniasis. Lancet Infect. Dis. 2003, 3, 87–98. [Google Scholar] [PubMed]
- Alvar, J.; Velez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M. and WHO Leishmaniasis Control Team. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar]
- World Health Organization. Leishmaniasis; World Health Organization: Geneva, Switzerland; Available online: http://www.who.int/mediacentre/factsheets/fs375/en/ (accessed on 9 January 2019).
- Reguera, R.M.; Gutiérrez-Corbo, C.; Ordóñez, C.; Pérez-Pertejo, M.Y.; Balaña-Fouce, R. Current and promising novel drug candidates against visceral leishmaniasis. Pure Appl. Chem. 2019, 91, 1385–1404. [Google Scholar]
- Alves, F.; Bilbe, G.; Blesson, S.; Goyal, V.; Monnerat, S.; Mowbray, C.; Muthoni Ouattara, G.; Pécoul, B.; Rijal, S.; Rode, J.; et al. Recent development of visceral leishmaniasis treatments: Successes, pitfalls, and perspectives. Clin. Microbiol. Rev. 2018, 31, e00048-18. [Google Scholar] [PubMed] [Green Version]
- Caridha, D.; Vesely, B.; van Bocxlaer, K.; Arana, B.; Mowbray, C.E.; Rafati, S.; Uliana, S.; Reguera, R.; Kreishman-Deitrick, M.; Sciotti, R.; et al. Route map for the discovery and pre-clinical development of new drugs and treatments for cutaneous leishmaniasis. Int. J. Parasitol. Drugs Drug. Resist. 2019, 11, 106–117. [Google Scholar]
- Sundar, S.; Chakravarty, J. Leishmaniasis: An update of current pharmacotherapy. Expert Opin. Pharmacother. 2012, 14, 53–63. [Google Scholar]
- Hendrickx, S.; Guerin, P.J.; Caljon, G.; Croft, S.L.; Maes, L. Evaluating drug resistance in visceral leishmaniasis: The challenges. Parasitology 2018, 145, 453–463. [Google Scholar]
- Sundar, S.; Singh, A. Chemotherapeutics of visceral leishmaniasis: Present and future developments. Parasitology 2018, 145, 481–489. [Google Scholar]
- Stone, N.R.H.; Bicanic, T.; Salim, R.; Hope, W. Liposomal amphotericin B (AmBisome®): A review of the pharmacokinetics, pharmacodynamics, clinical experience and future directions. Drugs 2016, 76, 485–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorlo, T.P.; Balasegaram, M.; Beijnen, J.H.; de Vries, P.J. Miltefosine: A review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J. Antimicrob. Chemother. 2012, 67, 2576–2597. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://dndi.org/diseases/visceral-leishmaniasis/ (accessed on 23 September 2020).
- Saha, A.; Basu, M.; Ukil, A. Recent advances in understanding Leishmania donovani infection: The importance of diverse host regulatory pathways. IUBMB Life 2018, 70, 593–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueno, N.; Wilson, M.E. Receptor-mediated phagocytosis of Leishmania: Implications for intracellular survival. Trends Parasitol. 2012, 28, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinet, A.F.; Fukuda, M.; Turco, S.J.; Descoteaux, A. The Leishmania donovani lipophosphoglycan excludes the vesicular proton-ATPase from phagosomes by impairing the recruitment of synaptotagmin V. PLoS Pathog. 2009, 5, e1000628. [Google Scholar] [CrossRef] [Green Version]
- Polando, R.E.; Jones, B.C.; Ricardo, C.; Whitcomb, J.; Ballhorn, W.; McDowell, M.A. Mannose receptor (MR) and Toll-like receptor 2 (TLR2) influence phagosome maturation during Leishmania infection. Parasite Immunol. 2018, 40, e12521. [Google Scholar] [CrossRef]
- Tomiotto-Pellissier, F.; Bortoleti, B.T.D.S.; Assolini, J.P.; Gonçalves, M.D.; Carloto, A.C.M.; Miranda-Sapla, M.M.; Conchon-Costa, I.; Bordignon, J.; Pavanelli, W.R. Macrophage Polarization in Leishmaniasis: Broadening Horizons. Front. Immunol. 2018, 9, 2529. [Google Scholar] [CrossRef] [Green Version]
- Lefèvre, L.; Lugo-Villarino, G.; Meunier, E.; Valentin, A.; Olagnier, D.; Authier, H.; Duval, C.; Dardenne, C.; Bernad, J.; Lemesre, J.L.; et al. The C-type lectin receptors dectin-1, MR, and SIGNR3 contribute both positively and negatively to the macrophage response to Leishmania infantum. Immunity 2013, 38, 1038–1049. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, D.; Mukherjee, S.; Roy, S.; Dalton, J.E.; Kundu, S.; Sarkar, A.; Das, N.K.; Kaye, P.M.; Chatterjee, M. M2 polarization of monocytes-macrophages is a hallmark of Indian post kala-azar dermal leishmaniasis. PLoS Negl. Trop. Dis. 2015, 9, e0004145. [Google Scholar] [CrossRef]
- Martinez-Pomares, L. The mannose receptor. J. Leukoc Biol. 2012, 92, 1177–1186. [Google Scholar] [CrossRef]
- Azad, A.K.; Rajaram, M.V.; Metz, W.L.; Cope, F.O.; Blue, M.S.; Vera, D.R.; Schlesinger, L.S. γ-Tilmanocept, a New Radiopharmaceutical Tracer for Cancer Sentinel Lymph Nodes, Binds to the Mannose Receptor (CD206). J. Immunol. 2015, 195, 2019–2029. [Google Scholar]
- Shaw, C.D.; Carter, K.C. Drug delivery: Lessons to be learnt from Leishmania studies. Nanomedicine (Lond) 2014, 9, 1531–1544. [Google Scholar]
- Gutiérrez, V.; Seabra, A.B.; Reguera, R.M.; Khandare, J.; Calderón, M. New approaches from nanomedicine for treating leishmaniasis. Chem. Soc. Rev. 2016, 45, 152–168. [Google Scholar]
- Gutierrez-Corbo, C.; Dominguez-Asenjo, B.; Vossen, L.I.; Pérez-Pertejo, Y.; Muñoz-Fenández, M.A.; Balaña-Fouce, R.; Calderón, M.; Reguera, R.M. PEGylated dendritic polyglycerol conjugate delivers doxorubicin to the parasitophorous vacuole in Leishmania infantum infections. Macromol. Biosci. 2017, 17. [Google Scholar] [CrossRef] [Green Version]
- Willner, D.; Trail, P.A.; Hofstead, S.J.; King, H.D.; Lasch, S.J.; Braslawsky, G.R.; Greenfield, R.S.; Kaneko, T.; Firestone, R.A. (6-Maleimidocaproyl)hydrazone of doxorubicin--a new derivative for the preparation of immunoconjugates of doxorubicin. Bioconjugate Chem. 1993, 4, 521–527. [Google Scholar]
- Kratz, K. DOXO-EMCH (INNO-206): The first albumin-binding prodrug of doxorubicin to enter clinical trials. Expert Opin. Inv. Drug. 2007, 16, 855–866. [Google Scholar]
- Charville, H.; Jin, J.; Evans, C.W.; Brimble, M.A.; Williams, D.E. The synthesis and lectin-binding properties of novel mannose-functionalised polymers. RSC Adv. 2013, 3, 15435–15441. [Google Scholar]
- Rodrigues, P.C.A.; Scheuermann, K.; Stockmar, C.; Maier, G.; Fiebig, H.H.; Unger, C.; Mülhaupt, R.; Kratz, F. Synthesis and In vitro efficacy of acid-Sensitive poly(ethylene glycol) paclitaxel conjugates. Bioorg. Med. Chem. Lett. 2003, 13, 355–360. [Google Scholar]
- Roller, S.; Zhou, H.; Haag, R. High-loading polyglycerol supported reagents for Mitsunobu- and acylation-reactions and other useful polyglycerol derivatives. Mol. Divers. 2005, 9, 305–316. [Google Scholar]
- Sunder, A.; Mülhaupt, R.; Haag, R.; Frey, H. Hyperbranched Polyether Polyols: A Modular Approach to Complex Polymer Architectures. Adv. Mater. 2000, 12, 235–239. [Google Scholar]
- Calvo-Alvarez, E.; Stamatakis, K.; Punzón, C.; Álvarez-Velilla, R.; Tejeria, A.; Escudero-Martinez, J.M.; Perez-Pertejo, Y.; Fresno, M.; Balana-Fouce, R.; Reguera, R.M. Infrared fluorescent imaging as a potent tool for in vitro, ex vivo and in vivo models of visceral leishmaniasis. PLoS Negl. Trop. Dis. 2015, 9, e0003666. [Google Scholar]
- Daigneault, M.; Preston, J.A.; Marriott, H.M.; Whyte, M.K.; Dockrell, D.H. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS ONE 2010, 5, e8668. [Google Scholar]
- Weischenfeldt, J.; Porse, B. Bone marrow-derived macrophages (BMM): Isolation and applications. CSH Protoc. 2008. [Google Scholar] [CrossRef] [Green Version]
- Ostrowski, P.P.; Fairn, G.D.; Grinstein, S.; Johnson, D.E. Cresyl violet: A superior fluorescent lysosomal marker. Traffic 2016, 17, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, M.R.; Reis, R.L.; Oliveira, J.M. Dendrimer nanoparticles for colorectal cancer applications. J. Mater. Chem. B 2020, 8, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Wilms, D.; Stiriba, S.E.; Frey, H. Hyperbranched Polyglycerols: From the Controlled Synthesis of Biocompatible Polyether Polyols to Multipurpose Applications. Acc. Chem. Res. 2010, 43, 129–141. [Google Scholar] [CrossRef]
- Calderón, M.; Quadir, M.A.; Sharma, S.K.; Haag, R. Dendritic Polyglycerols for Biomedical Applications. Adv. Mater. 2010, 22, 190–218. [Google Scholar] [CrossRef]
- Calderón, M.; Graeser, R.; Kratz, F.; Haag, R. Development of enzymatically cleavable prodrugs derived from dendritic polyglycerol. Bioorg. Med. Chem. Lett. 2009, 19, 3725–3728. [Google Scholar] [CrossRef]
- Calderón, M.; Welker, P.; Licha, K.; Fichtner, I.; Graeser, R.; Haag, R.; Kratz, F. Development of efficient acid cleavable multifunctional prodrugs derived from dendritic polyglycerol with a poly(ethylene glycol) shell. J. Controll. Release 2011, 151, 295–301. [Google Scholar] [CrossRef]
- Vossen, L.I.; Wedepohl, S.; Calderón, M. A Facile, One-Pot, Surfactant-Free Nanoprecipitation Method for the Preparation of Nanogels from Polyglycerol–Drug Conjugates that Can Be Freely Assembled for Combination Therapy Applications. Polymers 2018, 10, 398. [Google Scholar] [CrossRef] [Green Version]
- Baabur-Cohen, H.; Vossen, L.I.; Krüger, H.R.; Eldar-Boock, A.; Yeini, E.; Landa-Rouben, N.; Tiram, G.; Wedepohl, S.; Markovsky, E.; Leor, J.; et al. In vivo comparative study of distinct polymeric architectures bearing a combination of paclitaxel and doxorubicin at a synergistic ratio. J. Controll. Release 2017, 257, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Nagel, G.; Sousa-Herves, A.; Wedepohl, S.; Calderón, M. Matrix Metalloproteinase-sensitive Multistage Nanogels Promote Drug Transport in 3D Tumor Model. Theranostics 2020, 10, 91–108. [Google Scholar] [CrossRef]
- Hussain, A.F.; Krüger, H.R.; Kampmeier, F.; Weissbach, T.; Licha, K.; Kratz, F.; Haag, R.; Calderón, M.; Barth, S. Targeted Delivery of Dendritic Polyglycerol–Doxorubicin Conjugates by scFv-SNAP Fusion Protein Suppresses EGFR+ Cancer Cell Growth. Biomacromolecules 2013, 14, 2510–2520. [Google Scholar] [CrossRef] [PubMed]
- Ferber, S.; Tiram, G.; Sousa-Herves, A.; Eldar-Boock, A.; Krivitsky, A.; Scomparin, A.; Yeini, E.; Ofek, P.; Ben-Shushan, D.; Vossen, L.I.; et al. Co-targeting the tumor endothelium and P-selectin-expressing glioblastoma cells leads to a remarkable therapeutic outcome. eLife 2017, 6, e25281. [Google Scholar] [CrossRef]
- Vossen, L.I.; Markovsky, E.; Eldar-Boock, A.; Tschiche, H.R.; Wedepohl, S.; Pisarevsky, E.; Satchi-Fainaro, R.; Calderón, M. PEGylated dendritic polyglycerol conjugate targeting NCAM-expressing neuroblastoma: Limitations and challenges. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Nagel, G.; Tschiche, H.R.; Wedepohl, S.; Calderón, M. Modular approach for theranostic polymer conjugates with activatable fluorescence: Impact of linker design on the stimuli-induced release of doxorubicin. J. Controll. Release 2018, 285, 200–211. [Google Scholar] [CrossRef]
- Krüger, H.R.; Schütz, I.; Justies, A.; Licha, K.; Welker, P.; Haucke, V.; Calderón, M. Imaging of doxorubicin release from theranostic macromolecular prodrugs via fluorescence resonance energy transfer. J. Controll. Release 2014, 194, 189–196. [Google Scholar] [CrossRef]
- Antoine, J.C.; Prina, E.; Jouanne, C.; Bongrand, P. Parasitophorous vacuoles of Leishmania amazonensis-infected macrophages maintain an acidic pH. Infect. Immun. 1990, 58, 779–787. [Google Scholar] [CrossRef] [Green Version]
- Yeeprae, W.; Kawakami, S.; Yamashita, F.; Hashida, M. Effect of mannose density on mannose receptor-mediated cellular uptake of mannosylated O/W emulsions by macrophages. J. Controll. Release 2006, 114, 193–201. [Google Scholar] [CrossRef]
- Chen, P.; Zhang, X.; Jia, L.; Prud’homme, R.K.; Szekely, Z.; Sinko, P.J. Optimal structural design of mannosylated nanocarriers for macrophage targeting. J. Controll. Release 2014, 194, 341–349. [Google Scholar] [CrossRef] [Green Version]
- Patil, T.S.; Deshpande, A.S. Mannosylated nanocarriers mediated site-specific drug delivery for the treatment of cancer and other infectious diseases: A state of the art review. J. Controll. Release 2020, 320, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Pustylnikov, S.; Sagar, D.; Jain, P.; Khan, Z.K. Targeting the C-type lectins-mediated host-pathogen interactions with dextran. J. Pharm. Pharm. Sci. 2014, 17, 371–392. [Google Scholar] [PubMed] [Green Version]
- Hopkins, C.R.; Trowbridge, I.S. Internalization and processing of transferrin and the transferrin receptor in human carcinoma A431 cells. J. Cell Biol. 1983, 97, 508–521. [Google Scholar]
- Eidels, L.; Proia, R.L.; Hart, D.A. Membrane receptors for bacterial toxins. Microbiol. Rev. 1983, 47, 596–620. [Google Scholar] [PubMed]
- Falcone, S.; Cocucci, E.; Podini, P.; Kirchhausen, T.; Clementi, E.; Meldolesi, J. Macropinocytosis: Regulated coordination of endocytic and exocytic membrane traffic events. J. Cell. Sci. 2006, 119, 4758–4769. [Google Scholar] [PubMed] [Green Version]
- Kumar, G.A.; Karmakar, J.; Mandal, C.; Chattopadhyay, A. Leishmania donovani internalizes into host cells via caveolin-mediated endocytosis. Sci. Rep. 2019, 9, 12636. [Google Scholar]
- Johnson, D.E.; Ostrowski, P.; Jaumoille, V.; Grinstein, S. The position of lysosomes within the cell determines their luminal pH. J. Cell Biol. 2016, 212, 677–692. [Google Scholar]
- Chen, P.; Zhang, X.; Venosa, A.; Lee, I.H.; Myers, D.; Holloway, J.A.; Prud’homme, R.K.; Gao, D.; Szekely, Z.; Laskin, J.D.; et al. A Novel Bivalent Mannosylated Targeting Ligand Displayed on Nanoparticles Selectively Targets Anti-Inflammatory M2 Macrophages. Pharmaceutics 2020, 12, 243. [Google Scholar]
- Huang, Z.; Luo, Q.; Guo, Y.; Chen, J.; Xiong, G.; Peng, Y.; Ye, J.; Li, J. Mycobacterium tuberculosis-Induced Polarization of Human Macrophage Orchestrates the Formation and Development of Tuberculous Granulomas In Vitro. PLoS ONE 2015, 10, e0129744. [Google Scholar]
- Wagener, J.; MacCallum, D.M.; Brown, G.D.; Gow, N.A. Candida albicans Chitin Increases Arginase-1 Activity in Human Macrophages, with an Impact on Macrophage Antimicrobial Functions. mBio 2017, 8, e01820-16. [Google Scholar]
- Saliba, A.E.; Li, L.; Westermann, A.J.; Appenzeller, S.; Stapels, D.A.; Schulte, L.N.; Helaine, S.; Vogel, J. Single-cell RNA-seq ties macrophage polarization to growth rate of intracellular Salmonella. Nat. Microbiol. 2016, 2, 16206. [Google Scholar] [CrossRef] [PubMed]
- Balaña-Fouce, R.; Pérez-Pertejo, M.Y.; Domínguez-Asenjo, B.; Gutierrez-Corbo, C.; Reguera, R.M. Walking a tightrope: Drug Discovery in visceral leishmaniasis. Drug Discov. Today 2019, 24, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Velilla, R.; Gutiérrez-Corbo, M.; Punzón, C.; Pérez-Pertejo, M.Y.; Balaña-Fouce, R.; Fresno, M.; Reguera, R.M. A chronic bioluminescent model of experimental visceral leishmaniasis for accelerating drug discovery. PLoS Negl. Trop. Dis. 2019, 13, e0007133. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MW of PG Core a | Reactive Groups per PG b | AmB Loading in weight% c | FITC Loading in Weight% d | Mannose Loading in Weight% e |
|---|---|---|---|---|---|
| PG–PEG–FITC | 10 kDa | 40 NH2 | – | 0.23 | – |
| PG–PEG–Mann5–FITC | 10 kDa | 40 NH2 | – | 0.29 | 0.77 |
| PG–PEG–Mann10–FITC | 10 kDa | 40 NH2 | – | 0.29 | 1.66 |
| PG–PEG–Mann20–FITC | 10 kDa | 40 NH2 | – | 0.29 | 4.71 |
| PG–PEG–Mann5 | 10 kDa | 40 NH2 | – | – | 0.83 |
| PG–AmB–PEG–Mann5 | 10 kDa | 40 NH2 | 0.43 | – | 0.41 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vossen, L.I.; Domínguez-Asenjo, B.; Gutiérrez-Corbo, C.; Pérez-Pertejo, M.Y.; Balaña-Fouce, R.; Reguera, R.M.; Calderón, M. Mannose-Decorated Dendritic Polyglycerol Nanocarriers Drive Antiparasitic Drugs To Leishmania infantum-Infected Macrophages. Pharmaceutics 2020, 12, 915. https://doi.org/10.3390/pharmaceutics12100915
Vossen LI, Domínguez-Asenjo B, Gutiérrez-Corbo C, Pérez-Pertejo MY, Balaña-Fouce R, Reguera RM, Calderón M. Mannose-Decorated Dendritic Polyglycerol Nanocarriers Drive Antiparasitic Drugs To Leishmania infantum-Infected Macrophages. Pharmaceutics. 2020; 12(10):915. https://doi.org/10.3390/pharmaceutics12100915
Chicago/Turabian StyleVossen, Laura I., Bárbara Domínguez-Asenjo, Camino Gutiérrez-Corbo, M. Yolanda Pérez-Pertejo, Rafael Balaña-Fouce, Rosa María Reguera, and Marcelo Calderón. 2020. "Mannose-Decorated Dendritic Polyglycerol Nanocarriers Drive Antiparasitic Drugs To Leishmania infantum-Infected Macrophages" Pharmaceutics 12, no. 10: 915. https://doi.org/10.3390/pharmaceutics12100915
APA StyleVossen, L. I., Domínguez-Asenjo, B., Gutiérrez-Corbo, C., Pérez-Pertejo, M. Y., Balaña-Fouce, R., Reguera, R. M., & Calderón, M. (2020). Mannose-Decorated Dendritic Polyglycerol Nanocarriers Drive Antiparasitic Drugs To Leishmania infantum-Infected Macrophages. Pharmaceutics, 12(10), 915. https://doi.org/10.3390/pharmaceutics12100915