Effects of Carbamazepine and Phenytoin on Pharmacokinetics and Pharmacodynamics of Rivaroxaban

 , and
, and 
Abstract
:1. Introduction
2. Materials and Method
2.1. Materials
2.2. Study Design
2.3. Plasma Concentration and PT Measurement Methods
2.4. Noncompartmental PK and PD Analysis
2.5. Population PK Analysis
2.5.1. Development of a Base Model
2.5.2. Development of a Covariate Model with Investigation of CBZ/PHT Effects
2.5.3. Selection and Evaluation for the Final Model
3. Results
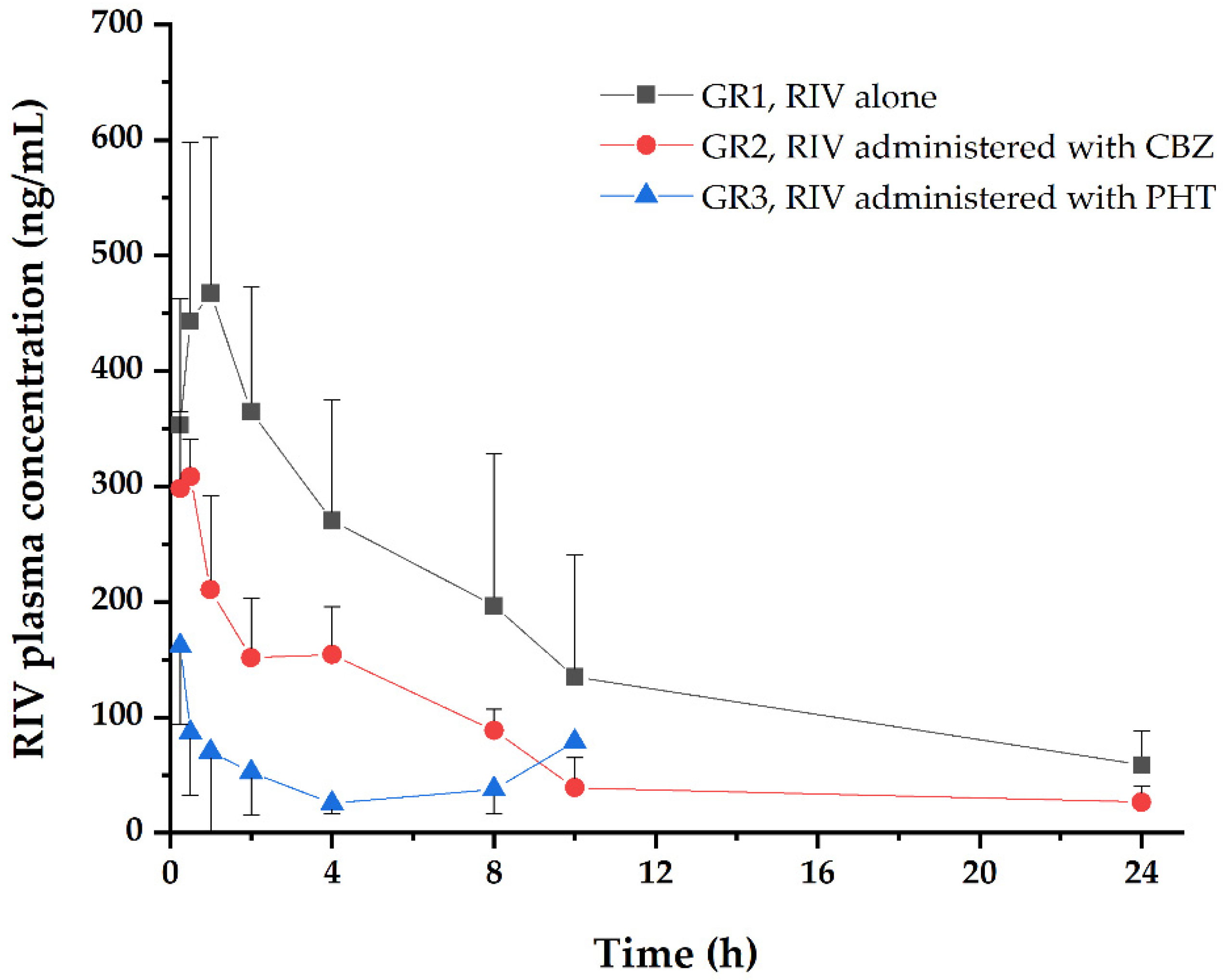
3.1. Noncompartmental PK and PD Analysis
3.2. Population PK Analysis
3.2.1. Development of the Population Pharmacokinetic Model
3.2.2. Development of a Covariate Model with Investigation of CBZ/PHT Effects
3.2.3. Evaluation of Population PK Model for RIV
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Myint, P.K.; Staufenberg, E.F.A.; Sabanathan, K. Post-stroke seizure and post-stroke epilepsy. Postgrad. Med. J. 2006, 82, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Nairooz, R.; Ayoub, K.; Sardar, P.; Payne, J.; Almomani, A.; Pothineni, N.V.; Shailesh, F.; Aronow, W.S.; Mukherjee, D. Uninterrupted New Oral Anticoagulants Compared With Uninterrupted Vitamin K Antagonists in Ablation of Atrial Fibrillation: A Meta-analysis. Can. J. Cardiol. 2016, 32, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Mayer, F.; Kirchmayer, U.; Coletta, P.; Agabiti, N.; Belleudi, V.; Cappai, G.; Di Martino, M.; Schneeweiss, S.; Davoli, M.; Patorno, E. Safety and effectiveness of direct oral anticoagulants versus vitamin K antagonists: Pilot implementation of a near-real-time monitoring program in Italy. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Ruff, C.T.; Giugliano, R.P.; Braunwald, E.; Hoffman, E.B.; Deenadayalu, N.; Ezekowitz, M.D.; Camm, A.J.; Weitz, J.I.; Lewis, B.S.; Parkhomenko, A.; et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: A meta-analysis of randomised trials. Lancet 2014, 383, 955–962. [Google Scholar] [CrossRef]
- Granger, C.B.; Alexander, J.H.; McMurray, J.J.V.; Lopes, R.D.; Hylek, E.M.; Hanna, M.; Al-Khalidi, H.R.; Ansell, J.; Atar, D.; Avezum, A.; et al. Apixaban versus warfarin in patients with atrial fibrillation. N. Engl. J. Med. 2011, 365, 981–992. [Google Scholar] [CrossRef]
- Liakishev, A.A. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. Results of the RE-COVER study. Kardiologiia 2010, 50, 80–81. [Google Scholar] [PubMed]
- Jeong, H.K.; Lee, K.H.; Park, H.W.; Yoon, N.S.; Kim, M.C.; Lee, N.; Kim, J.S.; Ahn, Y.; Jeong, M.H.; Park, J.C.; et al. Real World Comparison of Rivaroxaban and Warfarin in Korean Patients with Atrial Fibrillation: Propensity Matching Cohort Analysis. Chonnam Med. J. 2019, 55, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Romero, J.; Cerrud-Rodriguez, R.C.; Alviz, I.; Diaz, J.C.; Rodriguez, D.; Arshad, S.; Cerna, L.; Taveras, J.; Grupposo, V.; Natale, A.; et al. Significant Benefit of Uninterrupted DOACs Versus VKA During Catheter Ablation of Atrial Fibrillation. JACC Clin. Electrophysiol. 2019, 5, 1396–1405. [Google Scholar] [CrossRef] [PubMed]
- Harskamp, R.E.; Teichert, M.; Lucassen, W.A.M.; van Weert, H.C.P.M.; Lopes, R.D. Impact of Polypharmacy and P-Glycoprotein- and CYP3A4-Modulating Drugs on Safety and Efficacy of Oral Anticoagulation Therapy in Patients with Atrial Fibrillation. Cardiovasc. Drugs Ther. 2019, 33, 615–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Direct-Acting Oral Anticoagulants and Vitamin K Antagonists Executive Summary Background and Prior P & T Committee Actions. 2019. Available online: https://health.mil/Reference-Center/Meeting-References/2017/05/11/DoD-PT-Committee-Executive-Summary-Acting-Anticoagulants (accessed on 20 February 2020).
- Lühdorf, K.; Jensen, L.K.; Plesner, A.M. Etiology of Seizures in the Elderly. Epilepsia 1986, 27, 458–463. [Google Scholar] [CrossRef]
- Xu, M.Y. Poststroke seizure: Optimising its management. Stroke Vasc. Neurol. 2019, 4, 48–56. [Google Scholar] [CrossRef]
- Johannessen Landmark, C.; Patsalos, P.N. Drug interactions involving the new second- and third-generation antiepileptic drugs. Expert Rev. Neurother. 2010, 10, 119–140. [Google Scholar] [CrossRef]
- De Reuck, J.L. Management of stoke-related seizures. Acta Neurol. Belg. 2009, 109, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Ferro, J.M.; Pinto, F. Poststroke epilepsy: Epidemiology, pathophysiology and management. Drugs and Aging 2004, 21, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, S.I.; Landmark, C.J. Antiepileptic drug interactions - principles and clinical implications. Curr. Neuropharmacol. 2010, 8, 254–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, A.; Goldring, C.; Morgan, P.; Park, B.K.; Pirmohamed, M. Induction of P-glycoprotein in lymphocytes by carbamazepine and rifampicin: The role of nuclear hormone response elements. Br. J. Clin. Pharmacol. 2006, 62, 237–242. [Google Scholar] [CrossRef] [Green Version]
- Alvariza, S.; Fagiolino, P.; Vázquez, M.; Feria-Romero, I.; Orozco-Suárez, S. Chronic administration of phenytoin induces efflux transporter overexpression in rats. Pharmacol. Rep. 2014, 66, 946–951. [Google Scholar] [CrossRef]
- Jing, X.; Liu, X.; Wen, T.; Xie, S.; Yao, D.; Liu, X.; Wang, G.; Xie, L. Combined effects of epileptic seizure and phenobarbital induced overexpression of P-glycoprotein in brain of chemically kindled rats. Br. J. Pharmacol. 2010, 159, 1511–1522. [Google Scholar] [CrossRef]
- Eyal, S.; Lamb, J.G.; Smith-Yockman, M.; Yagen, B.; Fibach, E.; Altschuler, Y.; White, H.S.; Bialer, M. The antiepileptic and anticancer agent, valproic acid, induces P-glycoprotein in human tumour cell lines and in rat liver. Br. J. Pharmacol. 2006, 149, 250–260. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Stearns, R. Heterotropic Cooperativity of Cytochrome P450 3A4 and Potential Drug-Drug Interactions. Curr. Drug Metab. 2001, 2, 185–198. [Google Scholar] [CrossRef]
- Giessmann, T.; May, K.; Modess, C.; Wegner, D.; Hecker, U.; Zschiesche, M.; Dazert, P.; Grube, M.; Schroeder, E.; Warzok, R. Carbamazepine regulates intestinal P-glycoprotein and multidrug resistance protein MRP2 and influences disposition of talinolol in humans. Clin. Pharmacol. Ther. 2004, 76, 192–200. [Google Scholar] [CrossRef]
- Vlase, L.; Popa, A.; Neag, M.; Muntean, D.; Bĝldea, I.; Leucuţa, S.E. Pharmacokinetic interaction between zolpidem and carbamazepine in healthy volunteers. J. Clin. Pharmacol. 2011, 51, 1233–1236. [Google Scholar] [CrossRef]
- Yamada, S.; Yasui-Furukori, N.; Akamine, Y.; Kaneko, S.; Uno, T. Effects of the P-glycoprotein inducer carbamazepine on fexofenadine pharmacokinetics. Ther. Drug Monit. 2009, 31, 764–768. [Google Scholar] [CrossRef] [PubMed]
- Vranckx, P.; Valgimigli, M.; Heidbuchel, H. The Significance of Drug-Drug and Drug-Food Interactions of Oral Anticoagulation. Arrhythmia Electrophysiol. Rev. 2018, 7, 55–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galgani, A.; Palleria, C.; Iannone, L.F.; De Sarro, G.; Giorgi, F.S.; Maschio, M.; Russo, E. Pharmacokinetic Interactions of Clinical Interest Between Direct Oral Anticoagulants and Antiepileptic Drugs. Front. Neurol. 2018, 9, 1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.-H.; Chou, I.-J.; Yeh, Y.-H.; Chiou, M.-J.; Wen, M.-S.; Kuo, C.-T.; See, L.-C.; Kuo, C.-F. Association Between Use of Non-Vitamin K Oral Anticoagulants With and Without Concurrent Medications and Risk of Major Bleeding in Nonvalvular Atrial Fibrillation. JAMA 2017, 318, 1250–1259. [Google Scholar] [CrossRef]
- Risselada, A.J.; Visser, M.J.; van Roon, E.N. Pulmonary embolism due to interaction between rivaroxaban and carbamazepine]. Ned. Tijdschr. Geneeskd. 2013, 157, A6568. [Google Scholar]
- Stöllberger, C.; Finsterer, J. Recurrent venous thrombosis under rivaroxaban and carbamazepine for symptomatic epilepsy. Neurol. Neurochir. Pol. 2017, 51, 194–196. [Google Scholar] [CrossRef]
- Becerra, A.F.; Amuchastegui, T.; Tabares, A.H. Decreased Rivaroxaban Levels in a Patient with Cerebral Vein Thrombosis Receiving Phenytoin. Case Rep. Hematol. 2017, 2017, 4760612. [Google Scholar] [CrossRef] [Green Version]
- Reddy, Y.K.; Reddy, T.R. Pharmacokinetic Interactions between Concomitantly Administered Phenytoin with Rivaroxaban. Int. J. Pharm. Sci. Drug Res. 2018, 10. [Google Scholar] [CrossRef]
- Deeb, S.; McKeown, D.A.; Torrance, H.J.; Wylie, F.M.; Logan, B.K.; Scott, K.S. Simultaneous analysis of 22 antiepileptic drugs in postmortem blood, serum and plasma using LC-MS-MS with a focus on their role in forensic cases. J. Anal. Toxicol. 2014, 38, 485–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.M.N.; Yetal, S.M.; Vaidya, V.V.; Joshi, S.S. Determination and quantification of phenytoin in human plasma by liquid chromatography with electrospray ionization tandem mass spectrometry. E-J. Chem. 2008, 5, 169–176. [Google Scholar] [CrossRef]
- Derogis, P.B.M.; Sanches, L.R.; De Aranda, V.F.; Colombini, M.P.; Mangueira, C.L.P.; Katz, M.; Faulhaber, A.C.L.; Mendes, C.E.A.; Ferreira, C.E.D.S.; França, C.N.; et al. Determination of rivaroxaban in patient’s plasma samples by anti-Xa chromogenic test associated to high performance liquid chromatography tandem mass spectrometry (HPLC-MS/MS). PLoS ONE 2017, 12, e0171272. [Google Scholar] [CrossRef]
- Johnson, J.R. Methods for Handling Concentration Values Below the Limit of Quantification in PK Studies. PhUSE US Connect 2018, 2018, 1–9. [Google Scholar]
- Keizer, R.J.; Karlsson, M.O.; Hooker, A. Modeling and Simulation Workbench for NONMEM: Tutorial on Pirana, PsN, and Xpose. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, e50. [Google Scholar] [CrossRef] [PubMed]
- Boeckmann, A.J.; Beal, S.L.; Sheiner, L.B. NONMEM User’s Guide, Part I; NONMEM Project Group, University of California at San Francisco: San Francisco, CA, USA, 1998. [Google Scholar]
- Burden, T.; Thompson, C.; Bonanos, E.; Medford, A.R. Lesson of the month 2: Pulmonary embolism in a patient on rivaroxaban and concurrent carbamazepine. Clin. Med. 2018, 18, 103–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, H.L.; Polasek, T.M. Potential drug–drug interactions with direct oral anticoagulants in elderly hospitalized patients. Ther. Adv. Drug Saf. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- U.S. Food and Drug Administration. XARELTO® (Rivaroxaban): Prescribing Information. 2011. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/202439s001lbl.pdf (accessed on 14 February 2020).
- U.S. Food and Drug Administration. Tegretol® (Carbamazepine USP): Prescribing Information. 2007. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2007/016608s096,018281s044,018927s035,020234s025REVISED_LABEL.pdf (accessed on 14 February 2020).
- U.S. Food and Drug Administration. Dilantin® (Phenytoin Sodium) 100 mg Extended Oral Capsule: Prescribing Information. 2009. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/084349s060lbl.pdf (accessed on 23 February 2020).
- U.S. Food and Drug Administration. INFATABS® DILANTIN® (Phenytoin Chewable Tablets) for oral use: Prescribing Information. 1953. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/084427s017s022s025s030lbl.pdf (accessed on 20 February 2020).
- U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research. Guidance for Industry Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. 2005. Available online: https://www.federalregister.gov/documents/2005/07/22/05-14456/guidance-for-industry-on-estimating-the-maximum-safe-starting-dose-in-initial-clinical-trials-for (accessed on 15 February 2020).
- Zhou, H. Population-Based Assessments of Clinical Drug-Drug Interactions: Qualitative Indices or Quantitative Measures? J. Clin. Pharmacol. 2006, 46, 1268–1289. [Google Scholar] [CrossRef] [PubMed]
- Kubitza, D.; Roth, A.; Becka, M.; Alatrach, A.; Halabi, A.; Hinrichsen, H.; Mueck, W. Effect of hepatic impairment on the pharmacokinetics and pharmacodynamics of a single dose of rivaroxaban, an oral, direct Factor Xa inhibitor. Br. J. Clin. Pharmacol. 2013, 76, 89–98. [Google Scholar] [CrossRef]
- Kubitza, D.; Becka, M.; Voith, B.; Zuehlsdorf, M.; Wensing, G. Safety, pharmacodynamics, and pharmacokinetics of single doses of BAY 59-7939, an oral, direct factor Xa inhibitor. Clin. Pharmacol. Ther. 2005, 78, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Noda, K.; Kohno, K.; Samejima, M. Pharmacokinetic analysis of concentration data of drugs with irregular absorption profiles using multi-fraction absorption models. J. Pharm. Sci. 1987, 76, 109–113. [Google Scholar] [CrossRef]
- Jin, S.J.; Bae, K.S.; Cho, S.H.; Jung, J.A.; Kim, U.; Choe, S.; Ghim, J.L.; Noh, Y.H.; Park, H.J.; Kim, H.S.; et al. Population pharmacokinetic analysis of simvastatin and its active metabolite with the characterization of atypical complex absorption Kinetics. Pharm. Res. 2014, 31, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H. Pharmacokinetic strategies in deciphering atypical drug absorption profiles. J. Clin. Pharmacol. 2003, 43, 211–227. [Google Scholar] [CrossRef]
- Ngo, L.; Cho, H.-Y.; Lee, Y. Effects of hydrochlorothiazide and amlodipine on single oral dose pharmacokinetics of valsartan in healthy Korean subjects: Population model-based approach. Eur. J. Pharm. Sci. 2018, 118, 154–164. [Google Scholar] [CrossRef]
- Center for Drug Evaluation and Research. Application Number: 202439Orig1s000 for XARELTO® (Rivaroxaban). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202439Orig1s000PharmR.pdf (accessed on 15 February 2020).
- Xue, X.; Cao, M.; Ren, L.; Qian, Y.; Chen, G. Preparation and Optimization of Rivaroxaban by Self-Nanoemulsifying Drug Delivery System (SNEDDS) for Enhanced Oral Bioavailability and No Food Effect. AAPS PharmSciTech 2018, 19, 1847–1859. [Google Scholar] [CrossRef]
- Cifarelli, V.; Eichmann, A. The Intestinal Lymphatic System: Functions and Metabolic Implications. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 503–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuber, R.; Anzenbacherová, E.; Anzenbacher, P. Cytochromes P450 and experimental models of drug metabolism. J. Cell. Mol. Med. 2002, 6, 189–198. [Google Scholar] [CrossRef]
- Martignoni, M.; Groothuis, G.M.M.; de Kanter, R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin. Drug Metab. Toxicol. 2006, 2, 875–894. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PK Parameters | GR1 | GR2 | GR3 |
|---|---|---|---|
| ‡Tmax (h) | 1.00 (0.250–8.00) | 0.250 (0.250–0.500) * | 0.250 (0.250–0.250) * |
| Cmax (ng/mL) | 540 ± 106 | 306 ± 62.4 * | 162 ± 67.8 * |
| AUCt (ng/mL*h) | 3088 ± 1103 | 1299 ± 417 * | 317 ± 188 * |
| AUCinf (ng*h/mL) | 3565 ± 1648 | 1595 ± 645 * | 524 ± 234* |
| V/F (L/kg) | 7.21 ± 3.58 | 14.9 ± 5.93 * | 49.7 ± 27.6 * |
| CL/F (L/h/kg) | 1.03 ± 0.495 | 2.19 ± 0.933 * | 7.46 ± 5.27 * |
| Parameter | GR1 | GR2 | GR3 |
|---|---|---|---|
| Emax (%) | 51.7 ± 29.1 | 36.8 ± 27.7 | 25.6 ± 24.7 |
| AUCPT (%*h) | 212 ± 128 | 158 ± 150 | 132 ± 158 |
| Model Building Step | Model No. | Description | Df | OFV | ΔOFV | Compared to Model No. | |
|---|---|---|---|---|---|---|---|
| Step 1. Development of a base model. Dataset: PKs of RIV administered alone (GR1) | 1.1 | 1-compartment model | First-order kinetics | - | 860.38 | - | - |
| 1.2 | Zero-order kinetics | 0 | 861.42 | 1.034 | 1.1 | ||
| 1.3 | Combined first- and zero-order kinetics | 4 | 859.71 | 0.669 | 1.1 | ||
| 1.4 | 2-compartment model | First-order kinetics | - | 858.71 | - | - | |
| 1.5 | Zero-order kinetics | 0 | 859.92 | 1.783 | 1.4 | ||
| 1.6 ¥ | Combined first- and zero-order kinetics | 4 | 839.38 | 18.75 | 1.4 | ||
| 1.6 ¥ | Combined first- and zero-order kinetics | 1 | 839.38 | 21.00 | 1.1 | ||
| Step 2. Development of covariate models. Dataset: Full PKs of RIV (GR1, GR2, GR3) | 1.6 ¥ | Base model | - | 1470.68 | - | - | |
| 2.1 | Base model with addition of IIV for D2 | 1 | 1460.11 | 10.56 | 1.6 | ||
| 2.2 | Model 2.1 with addition of a covariate for D2 | 2 | 1435.24 | 24.88 | 1.6 | ||
| 2.3 | Model 2.1 addition of a covariate for CL/F | 2 | 1419.07 | 41.05 | 1.6 | ||
| 2.4 | Model 2.1 with addition of a covariate for both CL/F and D2 | 2 | 1397.64 | 21.42 | 2.3 | ||
| 2.4 | Final covariate model | 4 | 1397.4 | 73.04 | 1.6 | ||
| Parameters | Unit | Estimates | RSE (%) | Shrinkage (%) | Bootstrap Replicates (n = 1000) | |||
|---|---|---|---|---|---|---|---|---|
| Median | 95% CI | |||||||
| TVCL/F | L/h/kg | 0.61 | 36.9 | 0.609 | 0.456 | − | 0.817 | |
| 2.11 | 69.7 | 2.11 | 1.15 | − | 3.48 | |||
| 10.30 | 71.8 | 10.9 | 6.28 | − | 17.4 | |||
| TV | h | 6.62 (FIXED) | N.E. | 6.62 | 6.62 | − | 6.62 | |
| 0.339 | 17.2 | 0.335 | 0.255 | − | 0.380 | |||
| 0.434 (FIXED) | N.E. | 0.434 | 0.434 | − | 0.434 | |||
| /F | L/kg | 0.693 | 44.2 | 0.701 | 0.514 | − | 1.06 | |
| Ka | 1/h | 2.27 | 33.6 | 2.31 | 1.46 | − | 3.11 | |
| Q/F | L/h/kg | 0.660 | 21.4 | 0.665 | 0.520 | − | 0.875 | |
| /F | L/kg | 5.57 | 38.1 | 5.60 | 3.83 | − | 8.93 | |
| F1 | 0.250 | 9.10 | 0.260 | 0.211 | − | 0.351 | ||
| Alag2 | h | 0.501 (FIXED) | N.E. | 0.501 | 0.501 | − | 0.501 | |
| Inter-individual variability | ||||||||
| IIV for /F | % | 47.0 | 33.0 | 4.0 | 44.5 | 30.9 | − | 59.7 |
| IIV for CL/F | % | 49.0 | 35.0 | 23.0 | 46.7 | 19.2 | − | 63.3 |
| Residual random variability | ||||||||
| Additive error | ng/mL | 13.6 | 35.8 | 13.5 | 6.33 | − | 23.7 | |
| Proportional error | % | 23.2 | 17.2 | 22.0 | 16.0 | − | 26.1 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ngo, L.T.; Yang, S.-y.; Tran, Q.T.; Kim, S.K.; Yun, H.-y.; Chae, J.-w. Effects of Carbamazepine and Phenytoin on Pharmacokinetics and Pharmacodynamics of Rivaroxaban. Pharmaceutics 2020, 12, 1040. https://doi.org/10.3390/pharmaceutics12111040
Ngo LT, Yang S-y, Tran QT, Kim SK, Yun H-y, Chae J-w. Effects of Carbamazepine and Phenytoin on Pharmacokinetics and Pharmacodynamics of Rivaroxaban. Pharmaceutics. 2020; 12(11):1040. https://doi.org/10.3390/pharmaceutics12111040
Chicago/Turabian StyleNgo, Lien Thi, Sung-yoon Yang, Quyen Thi Tran, Sang Kyum Kim, Hwi-yeol Yun, and Jung-woo Chae. 2020. "Effects of Carbamazepine and Phenytoin on Pharmacokinetics and Pharmacodynamics of Rivaroxaban" Pharmaceutics 12, no. 11: 1040. https://doi.org/10.3390/pharmaceutics12111040
APA StyleNgo, L. T., Yang, S.-y., Tran, Q. T., Kim, S. K., Yun, H.-y., & Chae, J.-w. (2020). Effects of Carbamazepine and Phenytoin on Pharmacokinetics and Pharmacodynamics of Rivaroxaban. Pharmaceutics, 12(11), 1040. https://doi.org/10.3390/pharmaceutics12111040