Bi-Functional Alginate Oligosaccharide–Polymyxin Conjugates for Improved Treatment of Multidrug-Resistant Gram-Negative Bacterial Infections

 , , , and
, , , and
Abstract
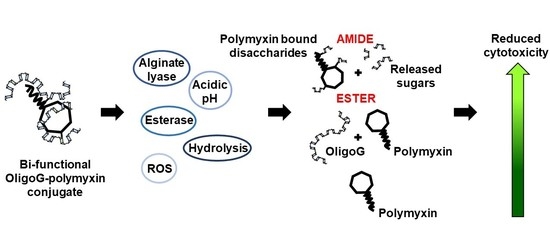
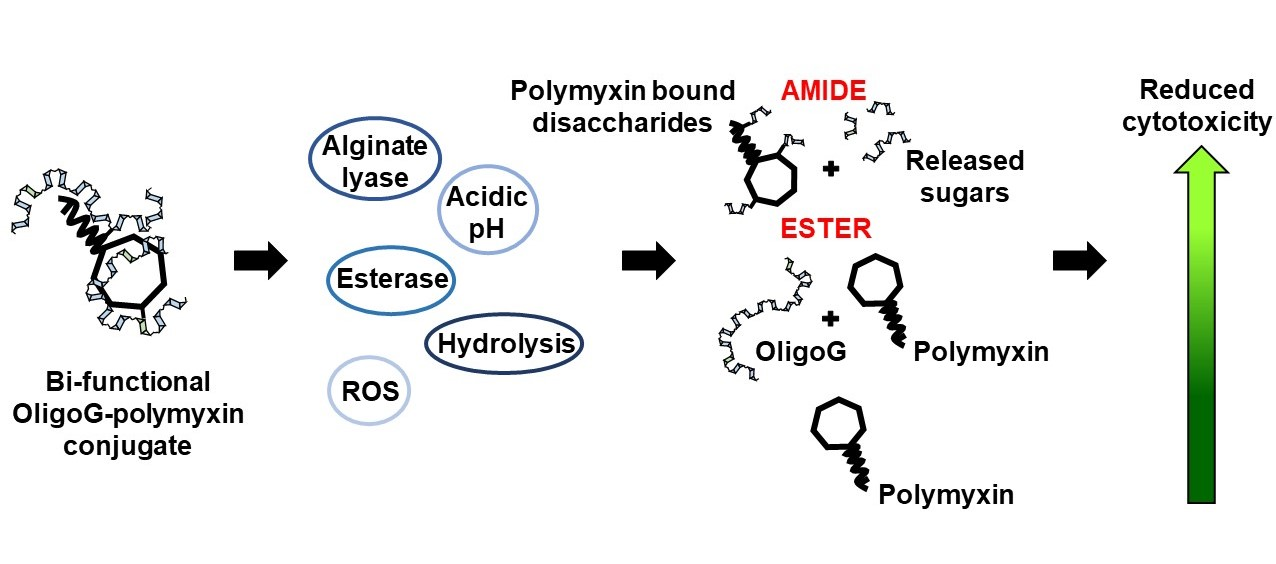
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Lines and Cell Culture
2.3. Bacterial Isolates and Growth Media
2.4. Synthesis of OligoG–Polymyxin Conjugates
2.5. Purification of OligoG–Polymyxin Conjugates
2.6. Characterisation of OligoG–Polymyxin Conjugates
2.7. Drug Release of OligoG–Polymyxin Conjugates
2.8. Characterisation of In Vitro Toxicity
2.9. Antimicrobial Activity of OligoG–Polymyxin Conjugates
2.10. Anti-Biofilm Activity of OligoG–Polymyxin Conjugates
2.11. Pharmacokinetic–Pharmacodynamic (PK–PD) Model
2.12. Statistical Analysis
3. Results
3.1. Synthesis and Characterisation of OligoG–Polymyxin Conjugates
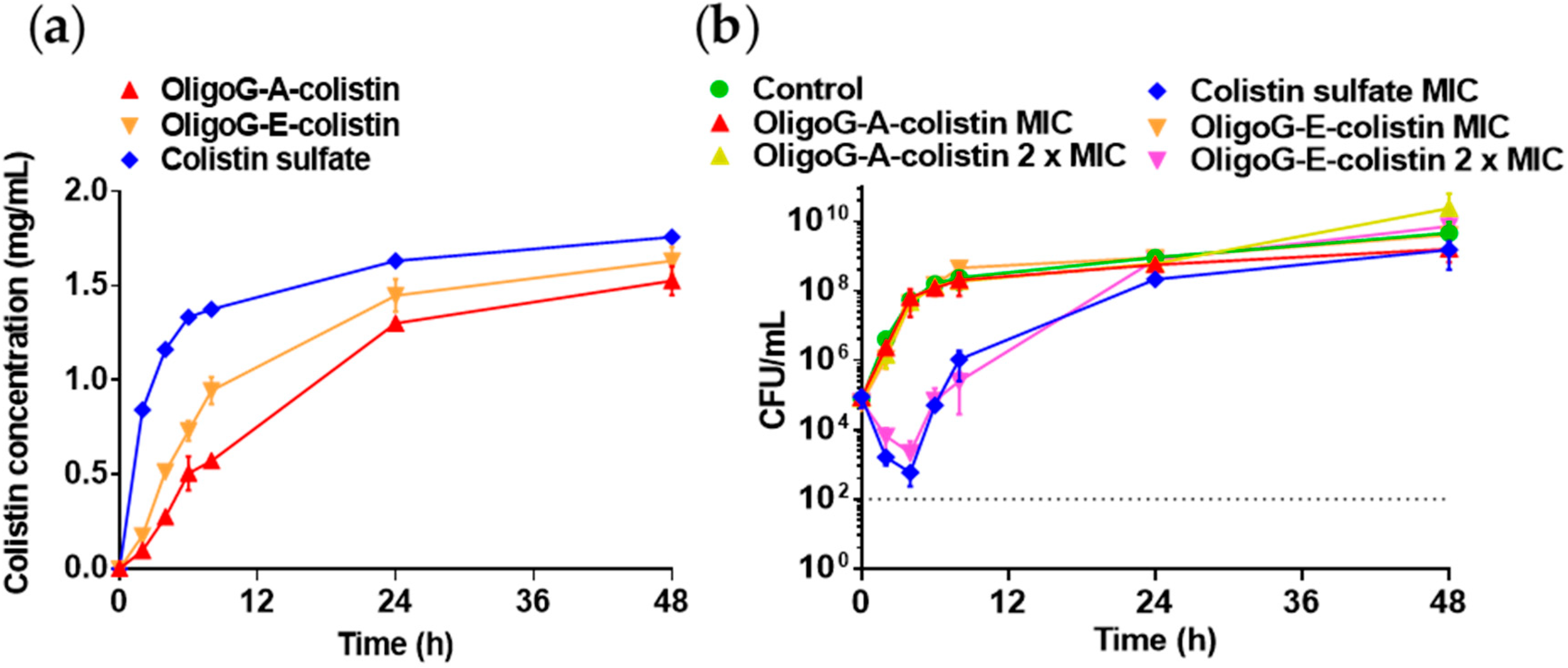
3.2. Stability of OligoG–Polymyxin Conjugates
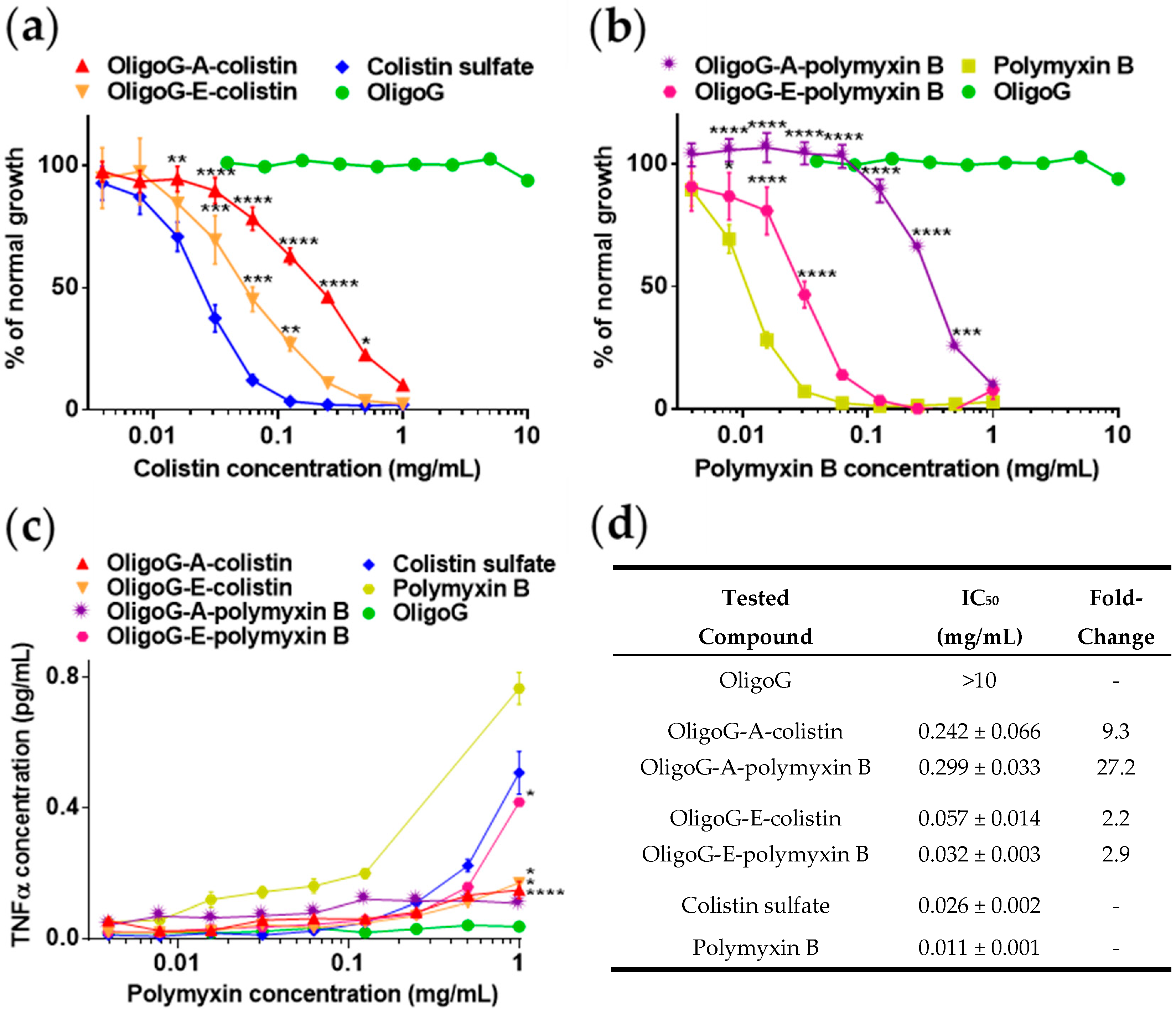
3.3. Cytotoxicity of OligoG–Polymyxin Conjugates
3.4. Antimicrobial Activity of OligoG–Polymyxin Conjugates
3.5. Anti-Biofilm Activity of OligoG–Polymyxin Conjugates
3.6. Pharmacokinetic–Pharmacodynamic (PK–PD) Model
4. Discussion
4.1. Rationale for Development of OligoG–Polymyxin Conjugates
4.2. Physicochemical Characterisation of OligoG–Polymyxin Conjugates
4.3. Biological Characterisation of OligoG–Polymyxin Conjugates
4.4. PK–PD Modelling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations. 2016. Available online: https://amr-review.org/Publications.html (accessed on 6 November 2020).
- Zaman, S.B.; Hussain, M.A.; Nye, R.; Mehta, V.; Mamun, K.T.; Hossain, N. A review on antibiotic resistance: Alarm bells are ringing. Cureus 2017, 9, e1403. [Google Scholar] [CrossRef] [Green Version]
- Schäberle, T.F.; Hack, I.M. Overcoming the current deadlock in antibiotic research. Trends Microbiol. 2014, 22, 165–167. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, J. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. 2014. Available online: https://amr-review.org/Publications.html (accessed on 6 November 2020).
- World Health Organization (WHO). Antibacterial Agents in Clinical Development. 2017. Available online: https://www.who.int/medicines/areas/rational_use/antibacterial_agents_clinical_development/en/ (accessed on 6 November 2020).
- Nouvellet, P.; Robotham, J.V.; Naylor, N.R.; Woodford, N.; Ferguson, N.M. Potential impact of novel diagnostics and treatments on the burden of antibiotic resistant in Escherichia coli. BioRxiv 2016, 052944. [Google Scholar]
- Falagas, M.E.; Kasiakou, S.K.; Saravolatz, L.D. Colistin: The revival of polymyxins for the management of multidrug-resistant Gram-negative bacterial infections. Clin. Infect. Dis. 2005, 40, 1333–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, P.; Abbott, E.; Abdulle, O.; Boakes, S.; Coleman, S.; Divall, N.; Duperchy, E.; Moss, S.; Rivers, D.; Simonovic, M.; et al. Design of next generation polymyxins with lower toxicity: The discovery of SPR206. ACS Infect. Dis. 2019, 5, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Wang, M.; Hong, Y.; Nimmagadda, A.; Shen, N.; Shi, Y.; Gao, R.; Zhang, E.; Cao, C.; Cai, J. Polymyxin derivatives as broad-spectrum antibiotic agents. Chem. Commun. 2019, 55, 13104–13107. [Google Scholar] [CrossRef]
- Vaara, M. Polymyxin derivatives that sensitize Gram-negative bacteria to other antibiotics. Molecules 2019, 24, 249. [Google Scholar] [CrossRef] [Green Version]
- Vaara, M. New polymyxin derivatives that display improved efficacy in animal infection models as compared to polymyxin B and colistin. Med. Res. Rev. 2018, 38, 1661–1673. [Google Scholar] [CrossRef]
- Kanazawa, K.; Sato, Y.; Ohki, K.; Okimura, K.; Uchida, Y.; Shindo, M.; Sakura, N. Contribution of each amino acid residue in polymyxin B3 to antimicrobial and lipopolysaccharide binding activity. Chem. Pharm. Bull. 2009, 57, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Velkov, T.; Roberts, K.D.; Thompson, P.E.; Li, J. Polymyxins: A new hope in combating Gram-negative superbugs? Future Med. Chem. 2016, 8, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Cal, P.M.; Matos, M.J.; Bernardes, G.J. Trends in therapeutic drug conjugates for bacterial diseases: A patent review. Expert. Opin. Ther. Pat. 2017, 27, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Azzopardi, E.A.; Ferguson, E.L.; Thomas, D.W. The enhanced permeability retention effect: A new paradigm for drug targeting in infection. J. Antimicrob. Chemother. 2013, 68, 257–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, E.L.; Azzopardi, E.; Roberts, J.L.; Walsh, T.R.; Thomas, D.W. Dextrin-colistin conjugates as a model bioresponsive treatment for multidrug resistant bacterial infections. Mol. Pharm. 2014, 11, 4437–4447. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Schneider, E.K.; Wang, J.; Kempe, K.; Wilson, P.; Velkov, T.; Li, J.; Davis, T.P.; Whittaker, M.R.; Haddleton, D.M. A traceless reversible polymeric colistin prodrug to combat multidrug-resistant (MDR) Gram-negative bacteria. J. Control. Release 2017, 259, 83–91. [Google Scholar] [CrossRef]
- Varache, M.; Powell, L.C.; Aarstad, O.A.; Williams, T.L.; Wenzel, M.N.; Thomas, D.W.; Ferguson, E.L. Polymer masked-unmasked protein therapy: Identification of the active species after amylase activation of dextrin-colistin conjugates. Mol. Pharm. 2019, 16, 3199–3207. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.S.; Xie, Y.J.; He, W. Research progress on chemical modification of alginate: A review. Carbohydr. Polym. 2011, 84, 33–39. [Google Scholar] [CrossRef]
- Khan, S.; Tøndervik, A.; Sletta, H.; Klinkenberg, G.; Emanuel, C.; Onsøyen, E.; Myrvold, R.; Howe, R.A.; Walsh, T.R.; Hill, K.E.; et al. Overcoming drug resistance with alginate oligosaccharides able to potentiate the action of selected antibiotics. Antimicrob. Agents Chemother. 2012, 56, 5134–5141. [Google Scholar] [CrossRef] [Green Version]
- Powell, L.C.; Sowedan, A.; Khan, S.; Wright, C.J.; Hawkins, K.; Onsøyen, E.; Myrvold, R.; Hill, K.E.; Thomas, D.W. The effect of alginate oligosaccharides on the mechanical properties of Gram-negative biofilms. Biofouling 2013, 29, 413–421. [Google Scholar] [CrossRef]
- Powell, L.C.; Pritchard, M.F.; Emanuel, C.; Onsøyen, E.; Rye, P.D.; Wright, C.J.; Hill, K.E.; Thomas, D.W. A nanoscale characterization of the interaction of a novel alginate oligomer with the cell surface and motility of Pseudomonas aeruginosa. Am. J. Respir. Cell Mol. Biol. 2014, 50, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Powell, L.C.; Pritchard, M.F.; Ferguson, E.L.; Powell, K.A.; Patel, S.U.; Rye, P.D.; Sakellakou, S.M.; Buurma, N.J.; Brilliant, C.D.; Copping, J.M.; et al. Targeted disruption of the extracellular polymeric network of Pseudomonas aeruginosa biofilms by alginate oligosaccharides. NPJ Biofilms Microbiomes 2018, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, M.F.; Powell, L.C.; Jack, A.A.; Powell, K.; Beck, K.; Florance, H.; Forton, J.; Rye, P.D.; Dessen, A.; Hill, K.E.; et al. A low-molecular-weight alginate oligosaccharide disrupts pseudomonal microcolony formation and enhances antibiotic effectiveness. Antimicrob. Agents Chemother. 2017, 61, e00762-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, R. The dawning era of polymer therapeutics. Nat. Rev. Drug. Discov. 2003, 2, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Hengzhuang, W.; Song, Z.; Ciofu, O.; Onsøyen, E.; Rye, P.D.; Høiby, N. OligoG CF-5/20 disruption of mucoid Pseudomonas aeruginosa biofilm in a murine lung infection model. Antimicrob. Agents Chemother. 2016, 60, 2620–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Li, M.; Spiller, O.B.; Andrey, D.O.; Hinchliffe, P.; Li, H.; Maclean, C.; Niumsup, P.; Powell, L.C.; Pritchard, M.F.; et al. Balancing mcr-1 expression and bacterial survival is a delicate equilibrium between essential cellular defence mechanisms. Nat. Commun. 2017, 8, 2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, J.H.; Turnidge, J.D. Chapter 71—Susceptibility test methods: Dilution and disk diffusion methods. In Manual of Clinical Microbiology, 11th ed.; Jorgensen, J.H., Pfaller, M., Carroll, K., Funke, G., Landry, M., Richter, S., Warnock, D., Eds.; ASM Press: Washington, DC, USA, 2015; pp. 1253–1273. [Google Scholar]
- Hsieh, M.H.; Yu, C.M.; Yu, V.L.; Chow, J.W. Synergy assessed by checkerboard: A critical analysis. Diagn. Microbiol. Infect. Dis. 1993, 16, 343–349. [Google Scholar] [CrossRef]
- Bonapace, C.R.; Bosso, J.A.; Friedrich, L.V.; White, R.L. Comparison of methods of interpretation of checkerboard synergy testing. Diagn. Microbiol. Infect. Dis. 2002, 44, 363–366. [Google Scholar] [CrossRef]
- Heydorn, A.; Nielsen, A.T.; Hentzer, M.; Sternberg, C.; Givskov, M.; Ersbøll, B.K.; Molin, S. Quantification of biofilm structures by the novel computer program comstat. Microbiology 2000, 146, 2395–2407. [Google Scholar] [CrossRef] [Green Version]
- Azzopardi, E.A.; Ferguson, E.L.; Thomas, D.W. Development and validation of an in vitro pharmacokinetic/pharmacodynamic model to test the antibacterial efficacy of antibiotic polymer conjugates. Antimicrob. Agents. Chemother. 2015, 59, 1837–1843. [Google Scholar] [CrossRef] [Green Version]
- Clinical and Laboratory Standards Institute (CLSI). Methods for Determining Bactericidal Activity of Antimicrobial Agents; Approved Guideline (M26-A). 1999. Available online: https://clsi.org/standards/products/microbiology/documents/m26/ (accessed on 6 November 2020).
- Levison, M.E.; Levison, J.H. Pharmacokinetics and pharmacodynamics of antibacterial agents. Infect. Dis. Clin. N. Am. 2009, 23, 791–815. [Google Scholar] [CrossRef] [Green Version]
- Vaara, M. Polymyxins and their potential next generation as therapeutic antibiotics. Front. Microbiol. 2019, 10, 1689. [Google Scholar] [CrossRef]
- Rabanal, F.; Cajal, Y. Recent advances and perspectives in the design and development of polymyxins. Nat. Prod. Rep. 2017, 34, 886–908. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.; Dawson, M.J. Development of new polymyxin derivatives for multi-drug resistant Gram-negative infections. J. Antibiot. 2017, 70, 386–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermanson, G.T. Zero-length crosslinkers. In Bioconjugate Techniques, 3rd ed.; Academic Press: Boston, MA, USA, 2013; Chapter 4; pp. 259–273. [Google Scholar]
- Simpson, J.A.; Smith, S.E.; Dean, R.T. Alginate may accumulate in cystic fibrosis lung because the enzymatic and free radical capacities of phagocytic cells are inadequate for its degradation. Biochem. Mol. Biol. Int. 1993, 30, 1021–1034. [Google Scholar] [PubMed]
- Wong, T.Y.; Preston, L.A.; Schiller, N.L. Alginate lyase: Review of major sources and enzyme characteristics, structure-function analysis, biological roles, and applications. Annu. Rev. Microbiol. 2000, 54, 289–340. [Google Scholar] [CrossRef]
- Greco, F.; Vicent, M. Polymer-drug conjugates: Current status and future trends. Front. Biosci. 2008, 13, 2744–2756. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Yu, D.F.; Newman, R.A.; Cabral, F.; Stephens, L.C.; Hunter, N.; Milas, L.; Wallace, S. Complete regression of well-established tumors using a novel water-soluble poly(l-glutamic acid)-paclitaxel conjugate. Cancer Res. 1998, 58, 2404–2409. [Google Scholar]
- Tsakos, M.; Schaffert, E.S.; Clement, L.L.; Villadsen, N.L.; Poulsen, T.B. Ester coupling reactions-an enduring challenge in the chemical synthesis of bioactive natural products. Nat. Prod. Rep. 2015, 32, 605–632. [Google Scholar] [CrossRef]
- Wong, P.T.; Choi, S.K. Mechanisms of drug release in nanotherapeutic delivery systems. Chem. Rev. 2015, 115, 3388–3432. [Google Scholar] [CrossRef]
- Dalheim, M.Ø.; Vanacker, J.; Najmi, M.A.; Aachmann, F.L.; Strand, B.L.; Christensen, B.E. Efficient functionalization of alginate biomaterials. Biomaterials 2016, 80, 146–156. [Google Scholar] [CrossRef]
- Spapen, H.; Jacobs, R.; Gorp, V.V.; Troubleyn, J.; Honoré, P.M. Renal and neurological side effects of colistin in critically ill patients. Ann. Intensive Care 2011, 1, 14. [Google Scholar] [CrossRef] [Green Version]
- Sandri, A.M.; Landersdorfer, C.B.; Jacob, J.; Boniatti, M.M.; Dalarosa, M.G.; Falci, D.R.; Behle, T.F.; Bordinhão, R.C.; Wang, J.; Forrest, A.; et al. Population pharmacokinetics of intravenous polymyxin B in critically ill patients: Implications for selection of dosage regimens. Clin. Infect. Dis. 2013, 57, 524–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinical and Laboratory Standards Institute (CLSI). Performance Standards for Antimicrobial Susceptibility Testing (M100). 2020. Available online: https://clsi.org/standards/products/microbiology/documents/m100/ (accessed on 6 November 2020).
- European Committee on Antimicrobial Susceptibility Testing (ECAST). Breakpoint Tables for Interpretation of MICs and Zone Diameters. 2019. Available online: http://www.eucast.org/clinical_breakpoints/ (accessed on 6 November 2020).
- Huang, J.X.; Blaskovich, M.A.T.; Pelingon, R.; Ramu, S.; Kavanagh, A.; Elliott, A.G.; Butler, M.S.; Montgomery, A.B.; Cooper, M.A. Mucin binding reduces colistin antimicrobial activity. Antimicrob. Agents Chemother. 2015, 59, 5925–5931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchard, M.F.; Oakley, J.L.; Brilliant, C.D.; Rye, P.D.; Forton, J.; Doull, I.J.M.; Ketchell, I.; Hill, K.E.; Thomas, D.W.; Lewis, P.D. Mucin structural interactions with an alginate oligomer mucolytic in cystic fibrosis sputum. Vib. Spectrosc. 2019, 103, 102932. [Google Scholar] [CrossRef]
- He, X.; Hwang, H.M.; Aker, W.G.; Wang, P.; Lin, Y.; Jiang, X.; He, X. Synergistic combination of marine oligosaccharides and azithromycin against Pseudomonas aeruginosa. Microbiol. Res. 2014, 169, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, M.C.; Couet, W.; Olivier, J.C.; Pais, A.A.C.C.; Sousa, J.J.S. Pseudomonas aeruginosa infection in cystic fibrosis lung disease and new perspectives of treatment: A review. Eur. J. Clin. Microbiol. Infect. Dis. 2013, 32, 1231–1252. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Rafailidis, P.I. Attributable mortality of Acinetobacter baumannii: No longer a controversial issue. Crit. Care 2007, 11, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Rayner, C.R.; Nation, R.L.; Owen, R.J.; Spelman, D.; Tan, K.E.; Liolios, L. Heteroresistance to colistin in multidrug-resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 2006, 50, 2946–2950. [Google Scholar] [CrossRef] [Green Version]
- Owen, R.J.; Li, J.; Nation, R.L.; Spelman, D. In vitro pharmacodynamics of colistin against Acinetobacter baumannii clinical isolates. J. Antimicrob. Chemother. 2007, 59, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Tan, T.Y.; Ng, L.S.; Tan, E.; Huang, G. In vitro effect of minocycline and colistin combinations on imipenem-resistant Acinetobacter baumannii clinical isolates. J. Antimicrob. Chemother. 2007, 60, 421–423. [Google Scholar] [CrossRef]
- Song, J.Y.; Kee, S.Y.; Hwang, I.S.; Seo, Y.B.; Jeong, H.W.; Kim, W.J.; Cheong, H.J. In vitro activities of carbapenem/sulbactam combination, colistin, colistin/rifampicin combination and tigecycline against carbapenem-resistant Acinetobacter baumannii. J. Antimicrob. Chemother. 2007, 60, 317–322. [Google Scholar] [CrossRef]
- Nation, R.L.; Garonzik, S.M.; Li, J.; Thamlikitkul, V.; Giamarellos-Bourboulis, E.J.; Paterson, D.L.; Turnidge, J.D.; Forrest, A.; Silveira, F.P. Updated US and European dose recommendations for intravenous colistin: How do they perform? Clin. Infect. Dis. 2016, 62, 552–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landersdorfer, C.B.; Nation, R.L. Colistin: How should it be dosed for the critically ill? Semin. Respir. Crit. Care Med. 2015, 36, 126–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorlí, L.; Luque, S.; Grau, S.; Berenguer, N.; Segura, C.; Montero, M.M.; Álvarez-Lerma, F.; Knobel, H.; Benito, N.; Horcajada, J.P. Trough colistin plasma level is an independent risk factor for nephrotoxicity: A prospective observational cohort study. BMC Infect. Dis. 2013, 13, 380. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
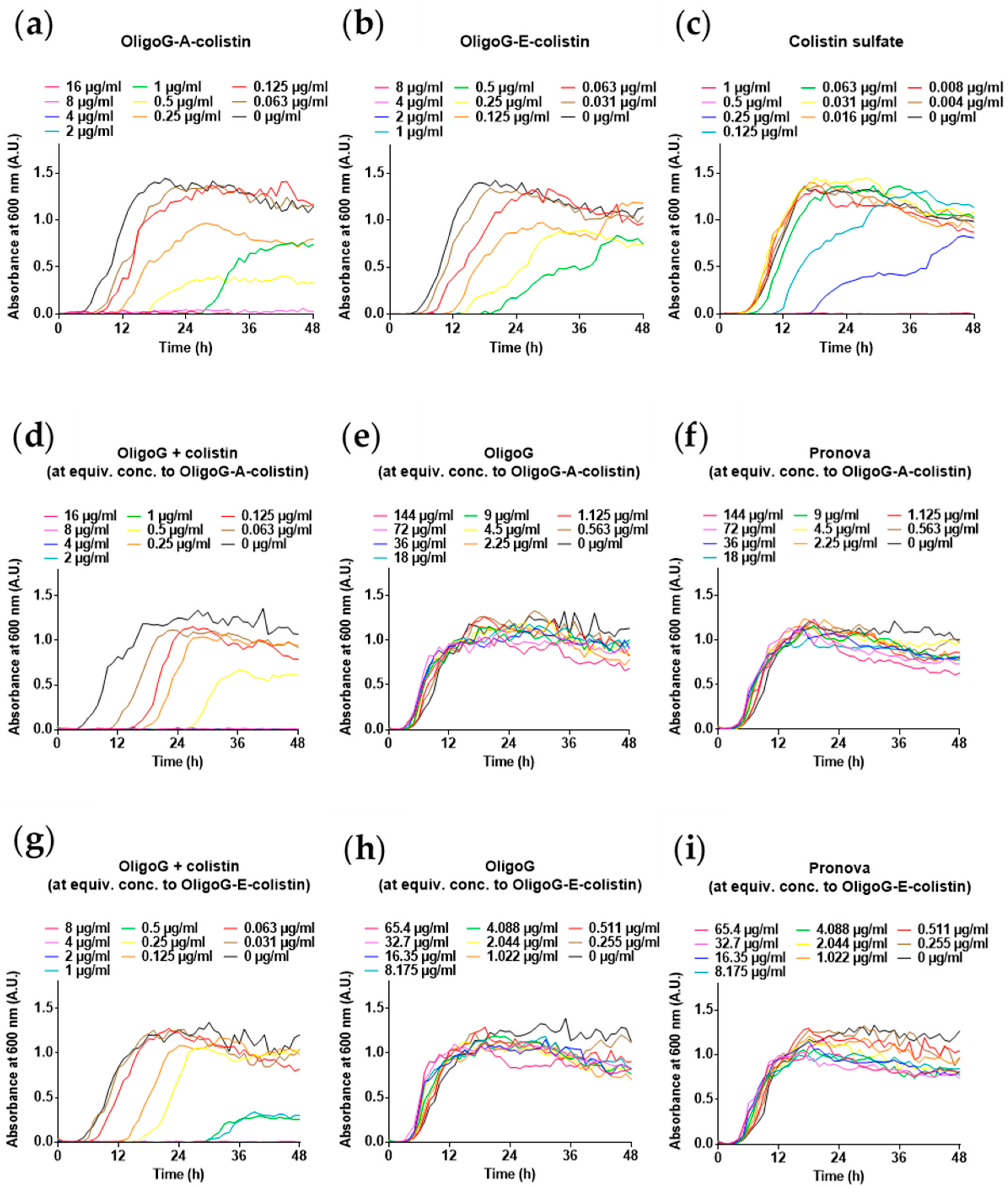{kind=link}
{kind=link}
{kind=link}
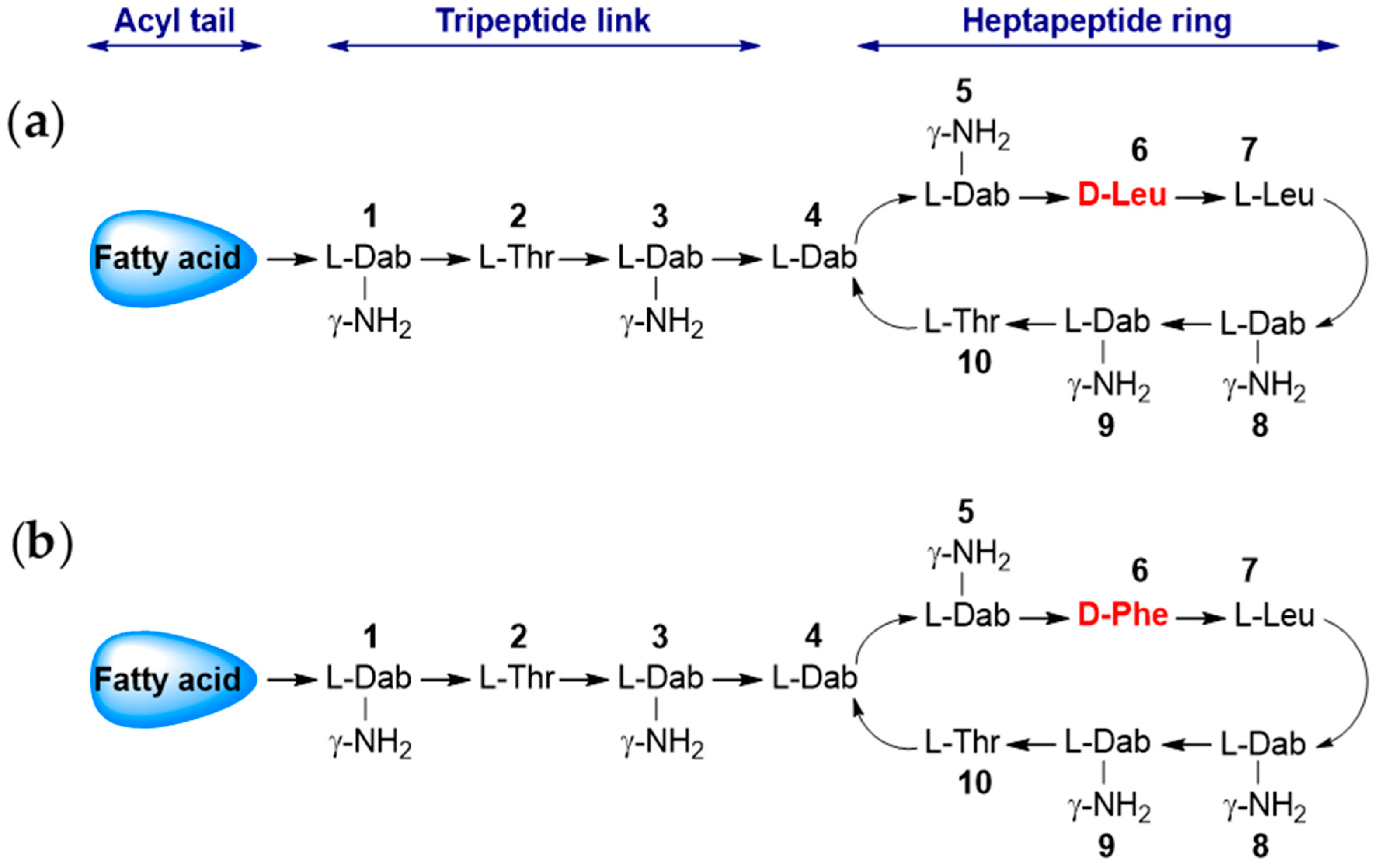{kind=link}
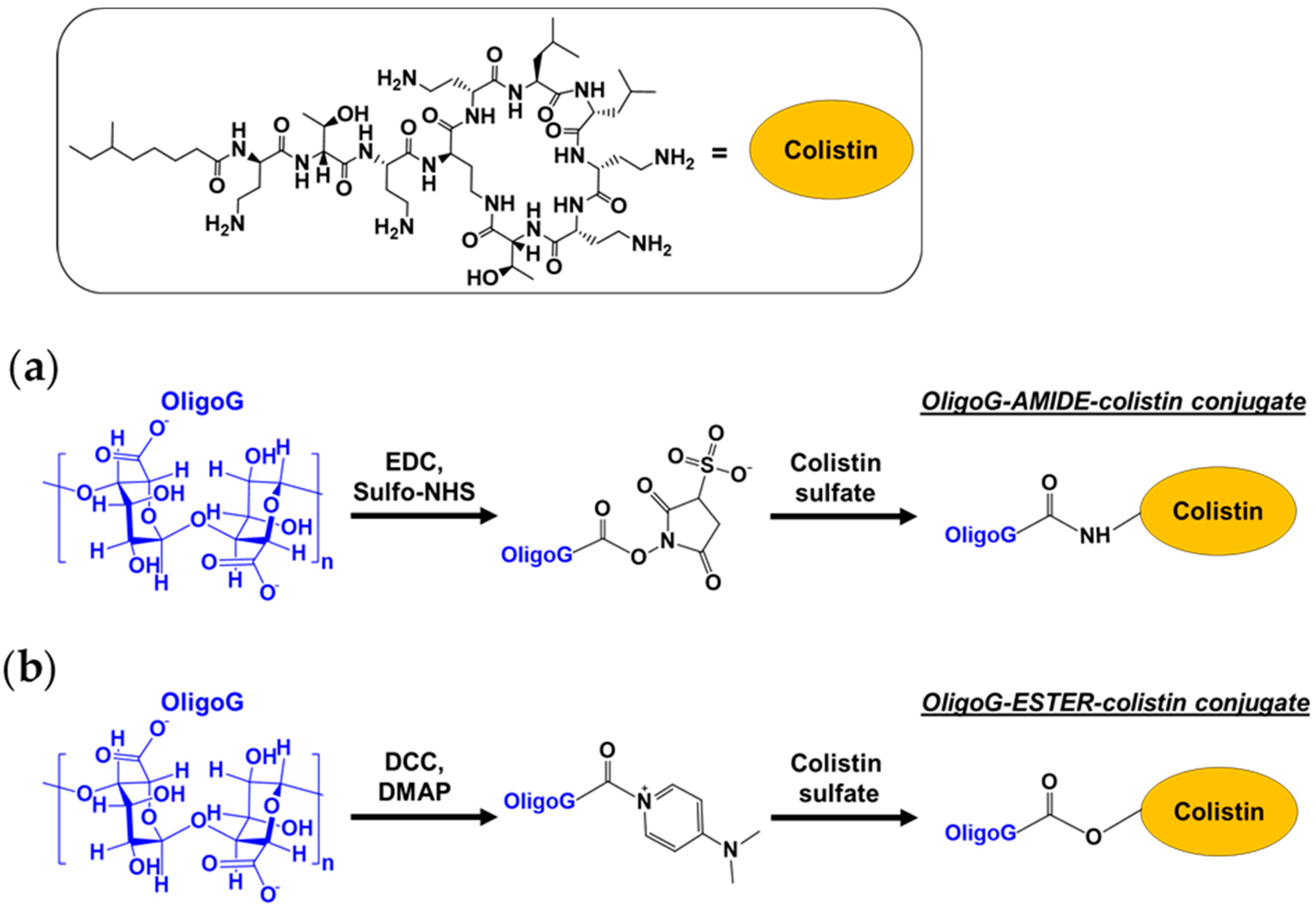{kind=link}
| Tested | Mw (g/mol) (PDI) by SEC-MALS | Polymyxin Content (% w/w) | Molar Ratio (per Colistin) | Conjugated NH2 per Molecule | Free Polymyxin (%) |
|---|---|---|---|---|---|
| OligoG–A–colistin | |||||
| Mean | 9220 (1.3) | 9.4 | 4.3 | 3.4 | 3.2 |
| Range | 8200–12,300 (1.2–1.4) | 8.1–12.5 | 3.1–5.0 | 2.7–4.6 | 1.5–5.7 |
| OligoG–E–colistin | |||||
| Mean | 5550 (1.3) | 10.9 | 3.8 | N/A | 2.7 |
| Range | 5200–5900 (1.2–1.3) | 8.3–12.9 | 3.0–4.9 | N/A | 2.0–3.5 |
| OligoG–A–polymyxin B | |||||
| Mean | 10,950 (1.4) | 7.1 | 6.0 | 2.0 | 1.6 |
| Range | 9100–12,800 (1.3–1.5) | 6.1–8.0 | 5.1–6.8 | 1.9–2.0 | 1.6 |
| OligoG–E–polymyxin B | 6200 (1.2) | 7.0 | 5.9 | N/A | 2.7 |
| Isolate | Tested Compound MIC (μg/mL Drug Base) | |||||
|---|---|---|---|---|---|---|
| Colistin Sulphate | Polymyxin B | OligoG–E–Colistin | OligoG–E–Polymyxin B | OligoG–A–Colistin | OligoG–A–Polymyxin B | |
| P. aeruginosa R22 | 0.5 | 0.25 | 1 | 0.25 | 2 | 4 |
| P. aeruginosa MDR 301 | 0.5 | 0.5 | 0.5 | 0.5 | 1 | 2 |
| P. aeruginosa NH57388A | 0.25 | 0.25 | 0.25 | 0.25 | 0.5 | 1 |
| P. aeruginosa NCTC 10662 | 0.125 | 0.063 | 0.25 | 0.25 | 1 | 4 |
| K. pneumoniae KP05 506 | 0.125 | 0.125 | 0.125 | 0.25 | 0.125 | 0.5 |
| K. pneumoniae IR25 | 0.063 | 0.125 | 0.125 | 0.125 | 1 | 4 |
| A. baumannii MDR ACB | 0.5 | 0.125 | 0.25 | 0.063 | 1 | 2 |
| A. baumannii 7789 | 0.25 | 0.125 | 0.125 | 0.5 | 0.125 | 0.5 |
| E. coli AIM-1 | <0.008 | <0.004 | 0.008 | 0.016 | 0.008 | 0.063 |
| E. coli IR57 | 0.125 | 0.5 | 0.25 | 0.5 | 0.125 | 2 |
| E. coli 5702 | 0.031 | 0.063 | 0.031 | 0.063 | 0.063 | 0.25 |
| E. coli NCTC 10418 | 0.125 | 0.25 | 0.5 | 0.25 | 0.25 | 4 |
| E. coli PN21 | 8 | 8 | 16 | 8 | 32 | 32 |
| E. coli PN25 | 8 | 4 | 8 | 4 | 32 | 32 |
| E. coli PN26 | 0.125 | 0.125 | 0.25 | 0.25 | 0.5 | 0.5 |
| E. coli ATCC 25922 | 0.25 | 0.5 | 1 | 0.5 | 1 | 16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stokniene, J.; Powell, L.C.; Aarstad, O.A.; Aachmann, F.L.; Rye, P.D.; Hill, K.E.; Thomas, D.W.; Ferguson, E.L. Bi-Functional Alginate Oligosaccharide–Polymyxin Conjugates for Improved Treatment of Multidrug-Resistant Gram-Negative Bacterial Infections. Pharmaceutics 2020, 12, 1080. https://doi.org/10.3390/pharmaceutics12111080
Stokniene J, Powell LC, Aarstad OA, Aachmann FL, Rye PD, Hill KE, Thomas DW, Ferguson EL. Bi-Functional Alginate Oligosaccharide–Polymyxin Conjugates for Improved Treatment of Multidrug-Resistant Gram-Negative Bacterial Infections. Pharmaceutics. 2020; 12(11):1080. https://doi.org/10.3390/pharmaceutics12111080
Chicago/Turabian StyleStokniene, Joana, Lydia C. Powell, Olav A. Aarstad, Finn L. Aachmann, Philip D. Rye, Katja E. Hill, David W. Thomas, and Elaine L. Ferguson. 2020. "Bi-Functional Alginate Oligosaccharide–Polymyxin Conjugates for Improved Treatment of Multidrug-Resistant Gram-Negative Bacterial Infections" Pharmaceutics 12, no. 11: 1080. https://doi.org/10.3390/pharmaceutics12111080
APA StyleStokniene, J., Powell, L. C., Aarstad, O. A., Aachmann, F. L., Rye, P. D., Hill, K. E., Thomas, D. W., & Ferguson, E. L. (2020). Bi-Functional Alginate Oligosaccharide–Polymyxin Conjugates for Improved Treatment of Multidrug-Resistant Gram-Negative Bacterial Infections. Pharmaceutics, 12(11), 1080. https://doi.org/10.3390/pharmaceutics12111080