Development of Amphotericin B Micellar Formulations Based on Copolymers of Poly(ethylene glycol) and Poly(ε-caprolactone) Conjugated with Retinol

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis Procedures
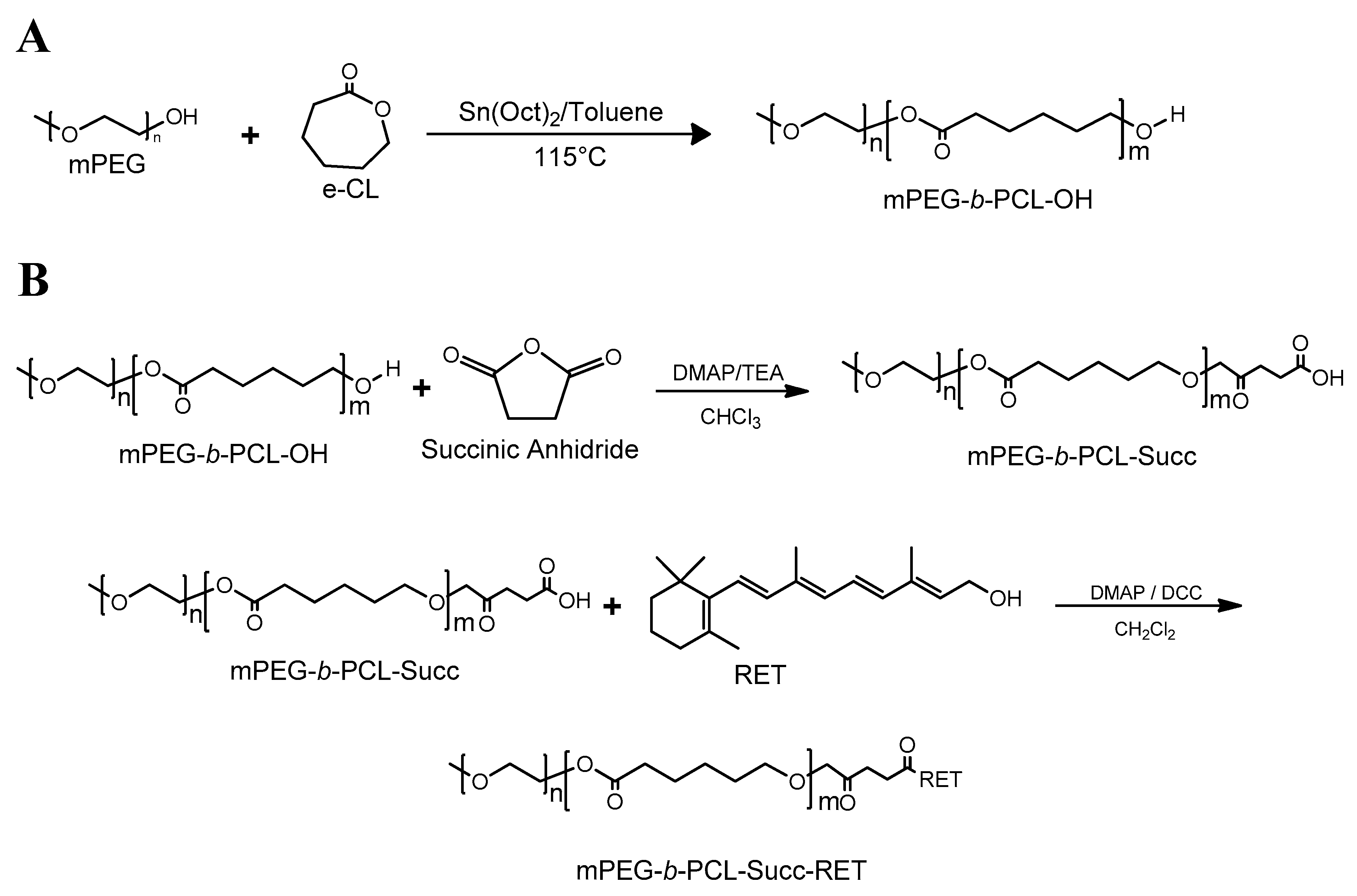
2.2.1. Synthesis of PEG-b-PCL Copolymers
2.2.2. Synthesis of PEG-PCL-COOH
2.2.3. Synthesis of PEG-PCL-RET
2.3. Characterization Techniques
2.4. Critical Micellar Concentration Measurements
2.5. Preparation of AmB Loaded Micelles
2.6. Determination of Encapsulated AmB
2.7. Assessment of Aggregation State
2.8. Release Study
2.9. In Vitro Biocompatibility
2.9.1. Haemolysis
2.9.2. Cytotoxicity against Fibroblasts
2.10. In Vitro Assessment of Antifungal Activity
3. Results
3.1. Synthesis of Polymeric Precursors
3.2. Critical Micellar Concentration
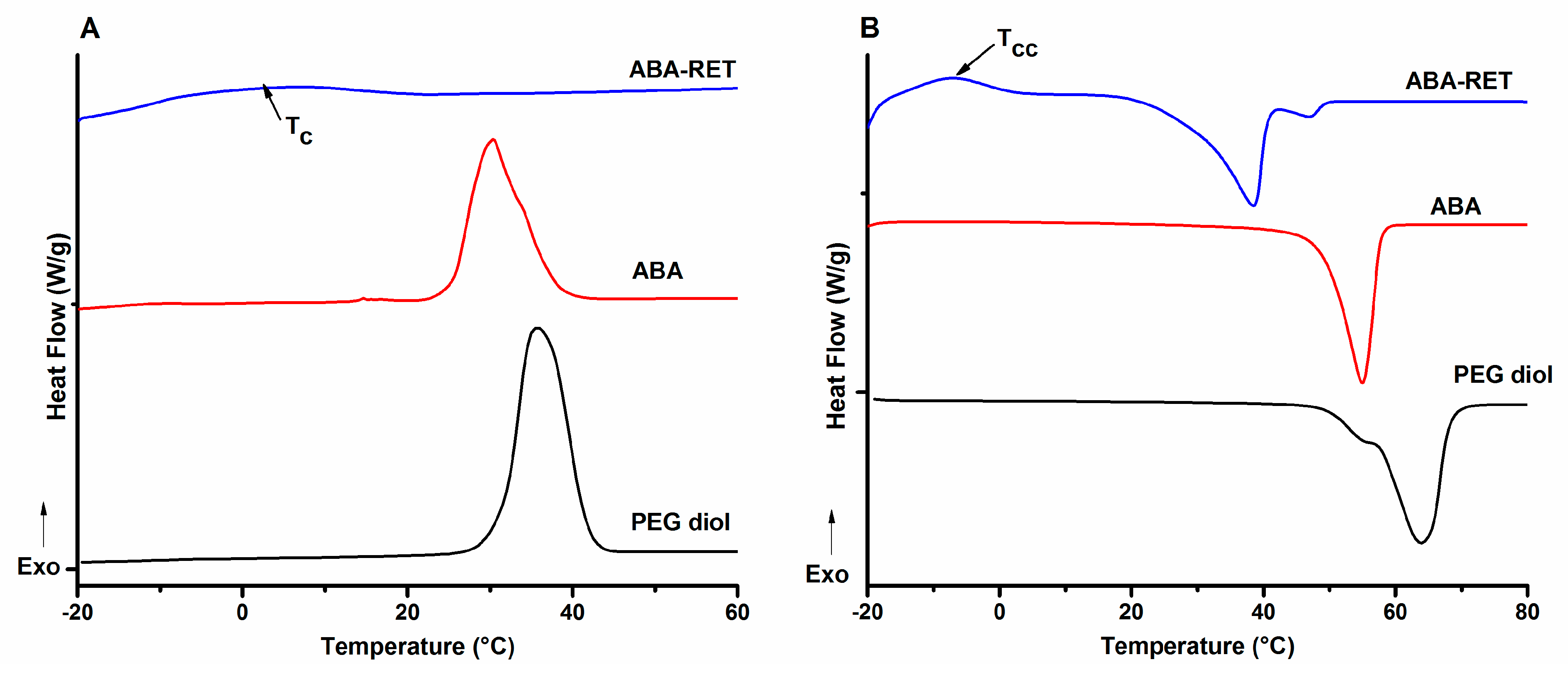
3.3. Differential Scanning Calorimetry
3.4. AmB Encapsulation
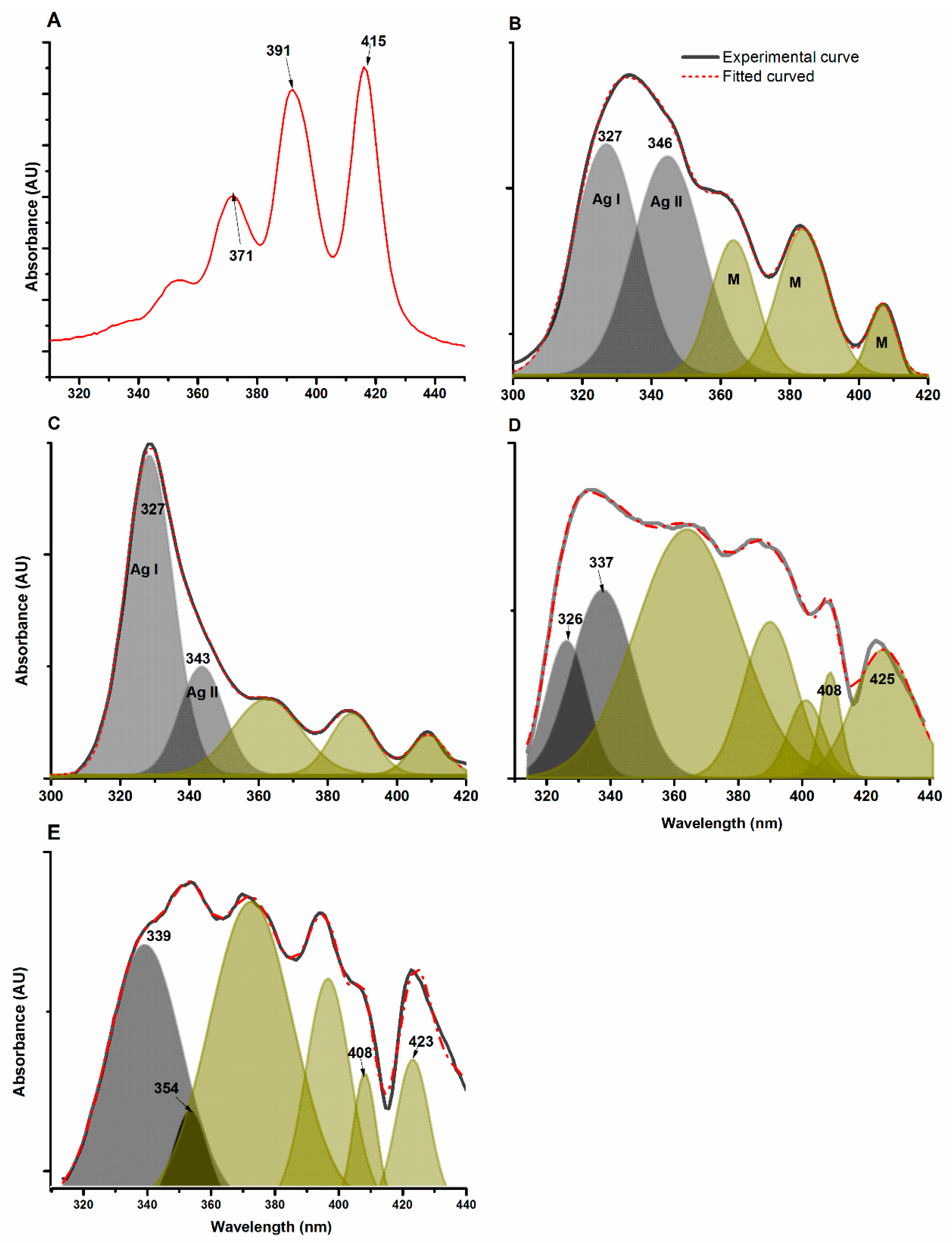
3.5. Assessment of AmB Aggregation
3.6. Release Study
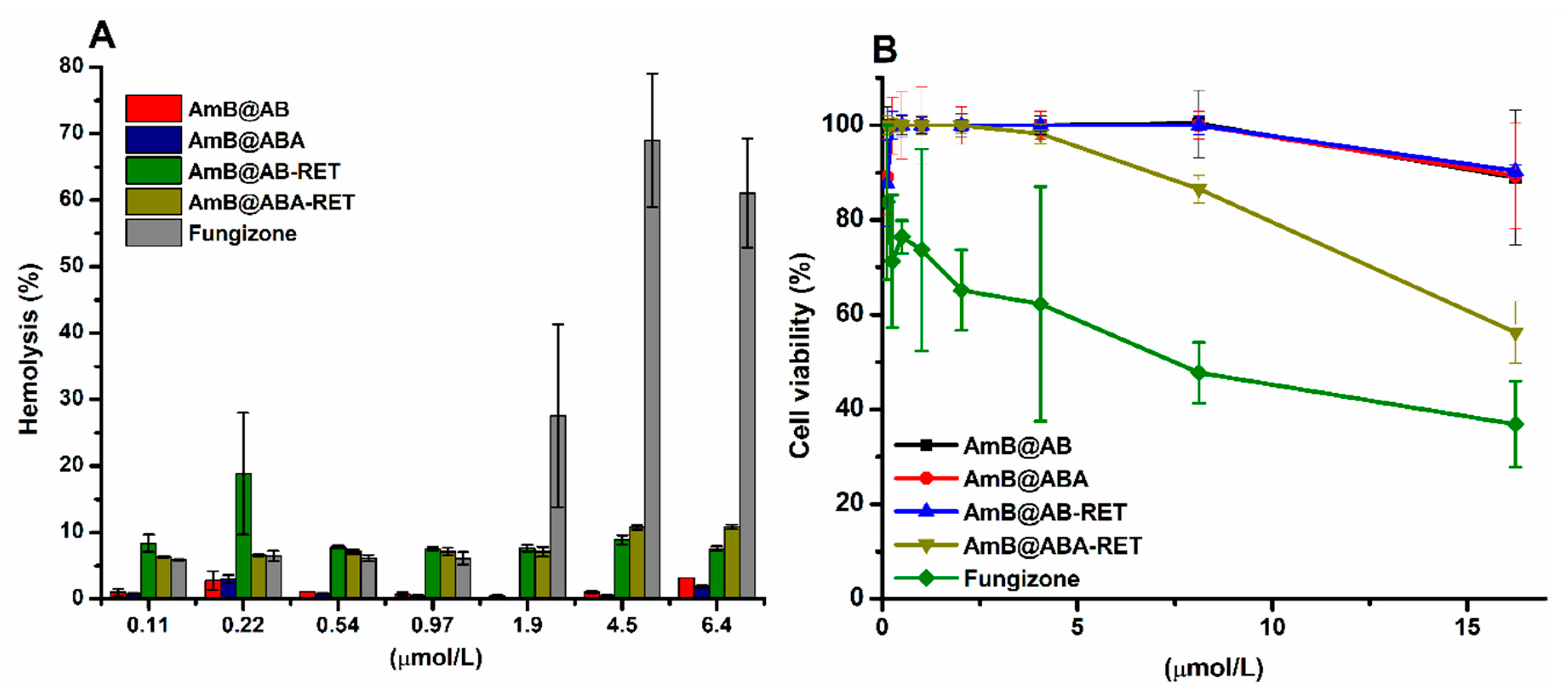
3.7. In Vitro Biocompatibility
3.8. In Vitro Effectiveness
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sifuentes-Osornio, J.; Corzo-León, D.E.; Ponce-De-León, L.A. Epidemiology of invasive fungal infections in Latin America. Curr. Fungal Infec. Rep. 2012, 6, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Ascioglu, S.; Rex, J.; De Pauw, B.; Bennett, J.; Bille, J.; Crokaert, F.; Denning, D.; Donnelly, J.; Edwards, J.; Erjavec, Z. Defining opportunistic invasive fungal infections in immunocompromised patients with cancer and hematopoietic stem cell transplants: An international consensus. Clin. Infect. Dis. 2002, 34, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Mirsaeidi, M.; Motahari, H.; Taghizadeh Khamesi, M.; Sharifi, A.; Campos, M.; Schraufnagel, D.E. Climate Change and Respiratory Infections. Ann. Am. Thorac. Soc. 2016, 13, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wang, Q.; Pleasants, R.A.; Ge, L.; Liu, W.; Peng, K.; Zhai, S. Empiric treatment against invasive fungal diseases in febrile neutropenic patients: A systematic review and network meta-analysis. BMC Infect. Dis. 2017, 17, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saravolatz, L.D.; Ostrosky-Zeichner, L.; Marr, K.A.; Rex, J.H.; Cohen, S.H. Amphotericin B: Time for a New “Gold Standard”. Clin. Infect. Dis. 2003, 37, 415–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarosi, G.A.; Amphotericin, B. Still the ‘gold standard’ for antifungal therapy. Postgrad Med. 1990, 88, 151–152, 155–161, 165–166. [Google Scholar] [CrossRef]
- Calvo, B.; Melo, A.S.A.; Perozo-Mena, A.; Hernandez, M.; Francisco, E.C.; Hagen, F.; Meis, J.F.; Colombo, A.L. First report of Candida auris in America: Clinical and microbiological aspects of 18 episodes of candidemia. J. Infect. 2016, 73, 369–374. [Google Scholar] [CrossRef]
- Vogelsinger, H.; Weiler, S.; Djanani, A.; Kountchev, J.; Bellmann-Weiler, R.; Wiedermann, C.J.; Bellmann, R. Amphotericin B tissue distribution in autopsy material after treatment with liposomal amphotericin B and amphotericin B colloidal dispersion. J. Antimicrob. Chemother. 2006, 57, 1153–1160. [Google Scholar] [CrossRef]
- Kamiński, D.M. Recent progress in the study of the interactions of amphotericin B with cholesterol and ergosterol in lipid environments. Eur. Biophys. J. 2014, 43, 453–467. [Google Scholar] [CrossRef] [Green Version]
- Wingard, J.R.; White, M.H.; Anaissie, E.; Raffalli, J.; Goodman, J.; Arrieta, A. A randomized, double-blind comparative trial evaluating the safety of liposomal amphotericin B versus amphotericin B lipid complex in the empirical treatment of febrile neutropenia. Clin. Infect. Dis. 2000, 31, 1155–1163. [Google Scholar] [CrossRef] [Green Version]
- Bates, D.; Su, L.; Yu, D.; Chertow, G.; Seger, D.; Gomes, D.; Dasbach, E.; Platt, R. Mortality and costs of acute renal failure associated with amphotericin B therapy. Clin. Infect. Dis. 2001, 32, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Adler-Moore, J.P.; Gangneux, J.-P.; Pappas, P.G. Comparison between liposomal formulations of amphotericin B. Med. Mycol. 2016, 54, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Fanos, V.; Cataldi, L. Amphotericin B-induced nephrotoxicity: A review. J. Chemother. 2000, 12, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Ringdén, O.; Meunier, F.; Tollemar, J.; Ricci, P.; Tura, S.; Kuse, E.; Viviani, M.A.; Gorin, N.C.; Klastersky, J.; Fenaux, P.; et al. Efficacy of amphotericin B encapsulated in liposomes (AmBisome) in the treatment of invasive fungal infections in immunocompromised patients. J. Antimicrob. Chemother. 1991, 28 (Suppl. B), 73–82. [Google Scholar]
- Walsh, T.J.; Finberg, R.W.; Arndt, C.; Hiemenz, J.; Schwartz, C.; Bodensteiner, D.; Pappas, P.; Seibel, N.; Greenberg, R.N.; Dummer, S.; et al. Liposomal Amphotericin B for Empirical Therapy in Patients with Persistent Fever and Neutropenia. N. Engl. J. Med. 1999, 340, 764–771. [Google Scholar] [CrossRef]
- Botero Aguirre, J.P.; Restrepo Hamid, A.M. Amphotericin B deoxycholate versus liposomal amphotericin B: Effects on kidney function. Cochrane Database Syst. Rev. 2015, 11. [Google Scholar] [CrossRef] [Green Version]
- Gamboa Garay, O.A.; Fuentes Pachón, J.C.; Cuervo Maldonado, S.I.; Gómez Rincón, J.C.; Castillo Londoño, J.S. Análisis de Costo Efectividad de Estrategias de Tratamiento Antimicótico en Pacientes con Neutropenia Febril Persistente y Tratamiento Antibiótico de Amplio Espectro. Value Health Reg. Issues 2012, 1, 201–210. [Google Scholar] [CrossRef]
- Jensen, G.; Skenes, C.; Bunch, T.; Weissman, C.; Amirghahari, N.; Satorius, A.; Moynihan, K.; Eley, C. Determination of the relative toxicity of amphotericin B formulations: A red blood cell potassium release assay. Drug Deliv. 1999, 6, 81–88. [Google Scholar] [CrossRef]
- Perlin, D.S.; Rautemaa-Richardson, R.; Alastruey-Izquierdo, A. The global problem of antifungal resistance: Prevalence, mechanisms, and management. Lancet Infect. Dis. 2017, 17, e383–e392. [Google Scholar] [CrossRef]
- Fernández-García, R.; de Pablo, E.; Ballesteros, M.P.; Serrano, D.R. Unmet clinical needs in the treatment of systemic fungal infections: The role of amphotericin B and drug targeting. Int. J. Pharm. 2017, 525, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Kasai, Y.; Nobuaki, M.; Hiroyuki, U.; Kenichi, N.; Shinya, Y.; Murata, M.; Tohru, O. Synthesis of 6-F-Ergosterol and Its Influence on Membrane-Permeabilization of Amphotericin B and Amphidinol 3. Org. Biomol. Chem. 2011, 9, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Mouri, R.; Konoki, K.; Matsumori, N.; Oishi, T.; Murata, M. Complex formation of amphotericin B in sterol-containing membranes as evidenced by surface plasmon resonance. Biochemistry 2008, 47, 7807–7815. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Umegawa, Y.; Nonomura, K.; Matsushita, N.; Takano, T.; Tsuchikawa, H.; Hanashima, S.; Oishi, T.; Matsumori, N.; Murata, M. Axial hydrogen at C7 position and bumpy tetracyclic core markedly reduce sterol’s affinity to amphotericin B in membrane. Biochemistry 2015, 54, 303–312. [Google Scholar] [CrossRef]
- Barwicz, J.; Tancrède, P. The effect of aggregation state of amphotericin-B on its interactions with cholesterol- or ergosterol-containing phosphatidylcholine monolayers. Chem. Phys. Lipids 1997, 85, 145–155. [Google Scholar] [CrossRef]
- Neumann, A.; Baginski, M.; Czub, J. Exploring Amphotericin B-Membrane Interactions: Free Energy Simulations. Biophys. J. 2013, 104 (Suppl. 1), 250. [Google Scholar] [CrossRef] [Green Version]
- Legrand, P.; Romero, E.A.; Cohen, B.E.; Bolard, J. Effects of aggregation and solvent on the toxicity of amphotericin B to human erythrocytes. Antimicrob. Agents Chemother. 1992, 36, 2518–2522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941. [Google Scholar] [CrossRef]
- Cabral, H.; Matsumoto, Y.; Mizuno, K.; Chen, Q.; Murakami, M.; Kimura, M.; Terada, Y.; Kano, M.; Miyazono, K.; Uesaka, M. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat. Nanotechnol. 2011, 6, 815. [Google Scholar] [CrossRef]
- Lavasanifar, A.; Samuel, J.; Sattari, S.; Kwon, G.S. Block copolymer micelles for the encapsulation and delivery of amphotericin B. Pharm. Res. 2002, 19, 418–422. [Google Scholar] [CrossRef]
- Kataoka, K.; Harada, A.; Nagasaki, Y. Block copolymer micelles for drug delivery: Design, characterization and biological significance. Adv. Drug Deliv. Rev. 2012, 64, 37–48. [Google Scholar] [CrossRef]
- Sun, X.; Wang, G.; Zhang, H.; Hu, S.; Liu, X.; Tang, J.; Shen, Y. The Blood Clearance Kinetics and Pathway of Polymeric Micelles in Cancer Drug Delivery. ACS Nano 2018, 12, 6179–6192. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Miah, J.; Shanmugam, S.; Yong, C.S.; Choi, H.-G.; Kim, J.A.; Yoo, B.K. Mixed micellar nanoparticle of amphotericin B and poly styrene-block-poly ethylene oxide reduces nephrotoxicity but retains antifungal activity. Arch. Pharmacal Res. 2007, 30, 1344–1349. [Google Scholar]
- Xu, H.; Teng, F.; Zhou, F.; Zhu, L.; Wen, Y.; Feng, R.; Song, Z. Linolenic acid-modified MPEG-PEI micelles for encapsulation of amphotericin B. Future Med. Chem. 2019, 11, 2647–2662. [Google Scholar] [CrossRef] [PubMed]
- Diezi, T.A.; Takemoto, J.K.; Davies, N.M.; Kwon, G.S. Pharmacokinetics and nephrotoxicity of amphotericin B-incorporated poly(ethylene glycol)-block-poly(N-hexyl stearate l-aspartamide) micelles. J. Pharm. Sci. 2011, 100, 2064–2070. [Google Scholar] [CrossRef]
- Yoo, B.K.; Jalil Miah, M.A.; Lee, E.S.; Han, K. Reduced renal toxicity of nanoparticular amphotericin B micelles prepared with partially benzylated poly-L-aspartic acid. Biol. Pharm. Bull. 2006, 29, 1700–1705. [Google Scholar] [CrossRef] [Green Version]
- Alconcel, S.N.; Baas, A.S.; Maynard, H.D. FDA-approved poly (ethylene glycol)–protein conjugate drugs. Polym. Chem. 2011, 2, 1442–1448. [Google Scholar] [CrossRef]
- Williford, J.-M.; Archang, M.M.; Minn, I.; Ren, Y.; Wo, M.; Vandermark, J.; Fisher, P.B.; Pomper, M.G.; Mao, H.-Q. Critical length of PEG grafts on lPEI/DNA nanoparticles for efficient in vivo delivery. ACS Biomater. Sci. Eng. 2016, 2, 567–578. [Google Scholar] [CrossRef]
- Pozzi, D.; Colapicchioni, V.; Caracciolo, G.; Piovesana, S.; Capriotti, A.L.; Palchetti, S.; De Grossi, S.; Riccioli, A.; Amenitsch, H.; Laganà, A. Effect of polyethyleneglycol (PEG) chain length on the bio–nano-interactions between PEGylated lipid nanoparticles and biological fluids: From nanostructure to uptake in cancer cells. Nanoscale 2014, 6, 2782–2792. [Google Scholar] [CrossRef]
- Yoon, K.; Kang, H.C.; Li, L.; Cho, H.; Park, M.-K.; Lee, E.; Bae, Y.H.; Huh, K.M. Amphiphilic poly (ethylene glycol)-poly (ε-caprolactone) AB 2 miktoarm copolymers for self-assembled nanocarrier systems: Synthesis, characterization, and effects of morphology on antitumor activity. Polym. Chem. 2015, 6, 531–542. [Google Scholar] [CrossRef]
- Castleberry, S.A.; Quadir, M.A.; Sharkh, M.A.; Shopsowitz, K.E.; Hammond, P.T. Polymer conjugated retinoids for controlled transdermal delivery. J. Control. Release 2017, 262, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Maiti, S.; Chatterji, P.R.; Nisha, C.; Manorama, S.; Aswal, V.K.; Goyal, P.S. Aggregation and polymerization of PEG-based macromonomers with methacryloyl group as the only hydrophobic segment. J. Colloid Interface Sci. 2001, 240, 630–635. [Google Scholar] [CrossRef]
- Shim, W.S.; Kim, S.W.; Choi, E.K.; Park, H.J.; Kim, J.S.; Lee, D.S. Novel pH sensitive block copolymer micelles for solvent free drug loading. Macromol. Biosci. 2006, 6, 179–186. [Google Scholar] [CrossRef]
- Diaz, I.L.; Perez, L.D. Synthesis and micellization properties of triblock copolymers PDMAEMA-b-PCL-b-PDMAEMA and their applications in the fabrication of amphotericin B-loaded nanocontainers. Colloid Polym. Sci. 2015, 293, 913–923. [Google Scholar] [CrossRef]
- Villamil, J.C.; Parra-Giraldo, C.M.; Pérez, L.D. Enhancing the performance of PEG-b-PCL copolymers as precursors of micellar vehicles for amphotericin B through its conjugation with cholesterol. Colloids Surf. A Physicochem. Eng. Asp. 2019, 572, 79–87. [Google Scholar] [CrossRef]
- Ramage, G.; Walle, K.V.; Wickes, B.L.; López-Ribot, J.L. Standardized method for in vitro antifungal susceptibility testing of Candida albicansbiofilms. Antimicrob. Agents Chemother. 2001, 45, 2475–2479. [Google Scholar] [CrossRef] [Green Version]
- Lockhart, S.R.; Bolden, C.B.; Iqbal, N.; Kuykendall, R.J. Validation of 24-hour flucytosine MIC determination by comparison with 48-hour determination by the Clinical and Laboratory Standards Institute M27-A3 broth microdilution reference method. J. Clin. Microbiol. 2011, 49, 4322–4325. [Google Scholar] [CrossRef] [Green Version]
- Fai, P.B.; Grant, A. A rapid resazurin bioassay for assessing the toxicity of fungicides. Chemosphere 2009, 74, 1165–1170. [Google Scholar] [CrossRef]
- O’brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef]
- Chen, Y.J.; Fang, H.J.; Hsu, S.C.N.; Jheng, N.Y.; Chang, H.C.; Ou, S.W.; Peng, W.T.; Lai, Y.C.; Chen, J.Y.; Chen, P.L.; et al. Improving the ring-opening polymerization of ε-caprolactone and l-lactide using stannous octanoate. Polym. Bull. 2013, 70, 993–1001. [Google Scholar] [CrossRef]
- Erothu, H.; Sohdi, A.A.; Kumar, A.C.; Sutherland, A.J.; Dagron-Lartigau, C.; Allal, A.; Hiorns, R.C.; Topham, P.D. Facile synthesis of poly (3-hexylthiophene)-block-poly (ethylene oxide) copolymers via Steglich esterification. Polym. Chem. 2013, 4, 3652–3655. [Google Scholar] [CrossRef]
- Liu, J.; Zeng, F.; Allen, C. In vivo fate of unimers and micelles of a poly(ethylene glycol)-block-poly(caprolactone) copolymer in mice following intravenous administration. Eur. J. Pharm. Biopharm. 2007, 65, 309–319. [Google Scholar] [CrossRef]
- Kim, S.; Shi, Y.; Kim, J.Y.; Park, K.; Cheng, J.-X. Overcoming the barriers in micellar drug delivery: Loading efficiency, in vivo stability, and micelle—cell interaction. Expert Opin. Drug Deliv. 2010, 7, 49–62. [Google Scholar] [CrossRef]
- Ohyashiki, T.; Mohri, T. Fluorometric Analysis of the Micelle Formation Process of Surfactants in Aqueous Solution. I. Utility of Pyrene in Determination of the Critical Micelle Concentration. Chem. Pharm. Bull. 1983, 31, 1296–1300. [Google Scholar] [CrossRef] [Green Version]
- Scholz, N.; Behnke, T.; Resch-Genger, U. Determination of the Critical Micelle Concentration of Neutral and Ionic Surfactants with Fluorometry, Conductometry, and Surface Tension—A Method Comparison. J. Fluoresc. 2018, 28, 465–476. [Google Scholar] [CrossRef]
- Bogdanov, B.; Vidts, A.; Van Den Buicke, A.; Verbeeck, R.; Schacht, E. Synthesis and thermal properties of poly(ethylene glycol)-poly(ϵ-caprolactone) copolymers. Polymer 1998, 39, 1631–1636. [Google Scholar] [CrossRef]
- Huang, Y.; Li, L.; Li, G. An enzyme-catalysed access to amphiphilic triblock copolymer of PCL-b-PEG-b-PCL: Synthesis, characterization and self-assembly properties. Des. Monomers Polym. 2015, 18, 799–806. [Google Scholar] [CrossRef]
- Verheyen, S.; Augustijns, P.; Kinget, R.; Van den Mooter, G. Melting behavior of pure polyethylene glycol 6000 and polyethylene glycol 6000 in solid dispersions containing diazepam or temazepam: A DSC study. Thermochim. Acta 2001, 380, 153–164. [Google Scholar] [CrossRef]
- Liu, X.-Q.; Bao, R.-Y.; Wu, X.-J.; Yang, W.; Xie, B.-H.; Yang, M.-B. Temperature induced gelation transition of a fumed silica/PEG shear thickening fluid. RSC Adv. 2015, 5, 18367–18374. [Google Scholar] [CrossRef]
- Raman, A.S.; Vishnyakov, A.; Chiew, Y.C. A coarse-grained model for PCL: Conformation, self-assembly of MePEG-b-PCL amphiphilic diblock copolymers. Mol. Simul. 2017, 43, 92–101. [Google Scholar] [CrossRef]
- Linse, P.; Malmsten, M. Temperature-dependent micellization in aqueous block copolymer solutions. Macromolecules 1992, 25, 5434–5439. [Google Scholar] [CrossRef]
- Zielińska, J.; Wieczór, M.; Bączek, T.; Gruszecki, M.; Czub, J. Thermodynamics and kinetics of amphotericin B self-association in aqueous solution characterized in molecular detail. Sci. Rep. 2016, 6, 19109. [Google Scholar] [CrossRef] [Green Version]
- Espada, R.; Valdespina, S.; Alfonso, C.; Rivas, G.; Ballesteros, M.P.; Torrado, J.J. Effect of aggregation state on the toxicity of different amphotericin B preparations. Int. J. Pharm. 2008, 361, 64–69. [Google Scholar] [CrossRef] [Green Version]
- Torrado, J.J.; Espada, R.; Ballesteros, M.P.; Torrado-Santiago, S. Amphotericin B formulations and drug targeting. J. Pharm. Sci. 2008, 97, 2405–2425. [Google Scholar] [CrossRef]
- Fujii, G.; Chang, J.-E.; Coley, T.; Steere, B. The formation of amphotericin B ion channels in lipid bilayers. Biochemistry 1997, 36, 4959–4968. [Google Scholar] [CrossRef]
- Barwicz, J.; Gruszecki, W.I.; Gruda, I. Spontaneous Organization of Amphotericin B in Aqueous Medium. J. Colloid Interface Sci. 1993, 158, 71–76. [Google Scholar] [CrossRef]
- Stoodley, R.; Wasan, K.M.; Bizzotto, D. Fluorescence of Amphotericin B-Deoxycholate (Fungizone) Monomers and Aggregates and the Effect of Heat-Treatment. Langmuir 2007, 23, 8718–8725. [Google Scholar] [CrossRef]
- Son, G.-H.; Lee, B.-J.; Cho, C.-W. Mechanisms of drug release from advanced drug formulations such as polymeric-based drug-delivery systems and lipid nanoparticles. J. Pharm. Investig. 2017, 47, 287–296. [Google Scholar] [CrossRef]
- De Oliveira, E.F.; Paula, H.C.; de Paula, R.C. Alginate/cashew gum nanoparticles for essential oil encapsulation. Colloids Surfaces B Biointerfaces 2014, 113, 146–151. [Google Scholar] [CrossRef]
- Italia, J.L.; Yahya, M.M.; Singh, D.; Ravi Kumar, M.N.V. Biodegradable nanoparticles improve oral bioavailability of amphotericin B and show reduced nephrotoxicity compared to intravenous fungizone. Pharm. Res. 2009, 26, 1324–1331. [Google Scholar] [CrossRef]
- Jung, O.; Smeets, R.; Porchetta, D.; Kopp, A.; Ptock, C.; Müller, U.; Heiland, M.; Schwade, M.; Behr, B.; Kröger, N.; et al. Optimized in vitro procedure for assessing the cytocompatibility of magnesium-based biomaterials. Acta Biomater. 2015, 23, 354–363. [Google Scholar] [CrossRef]









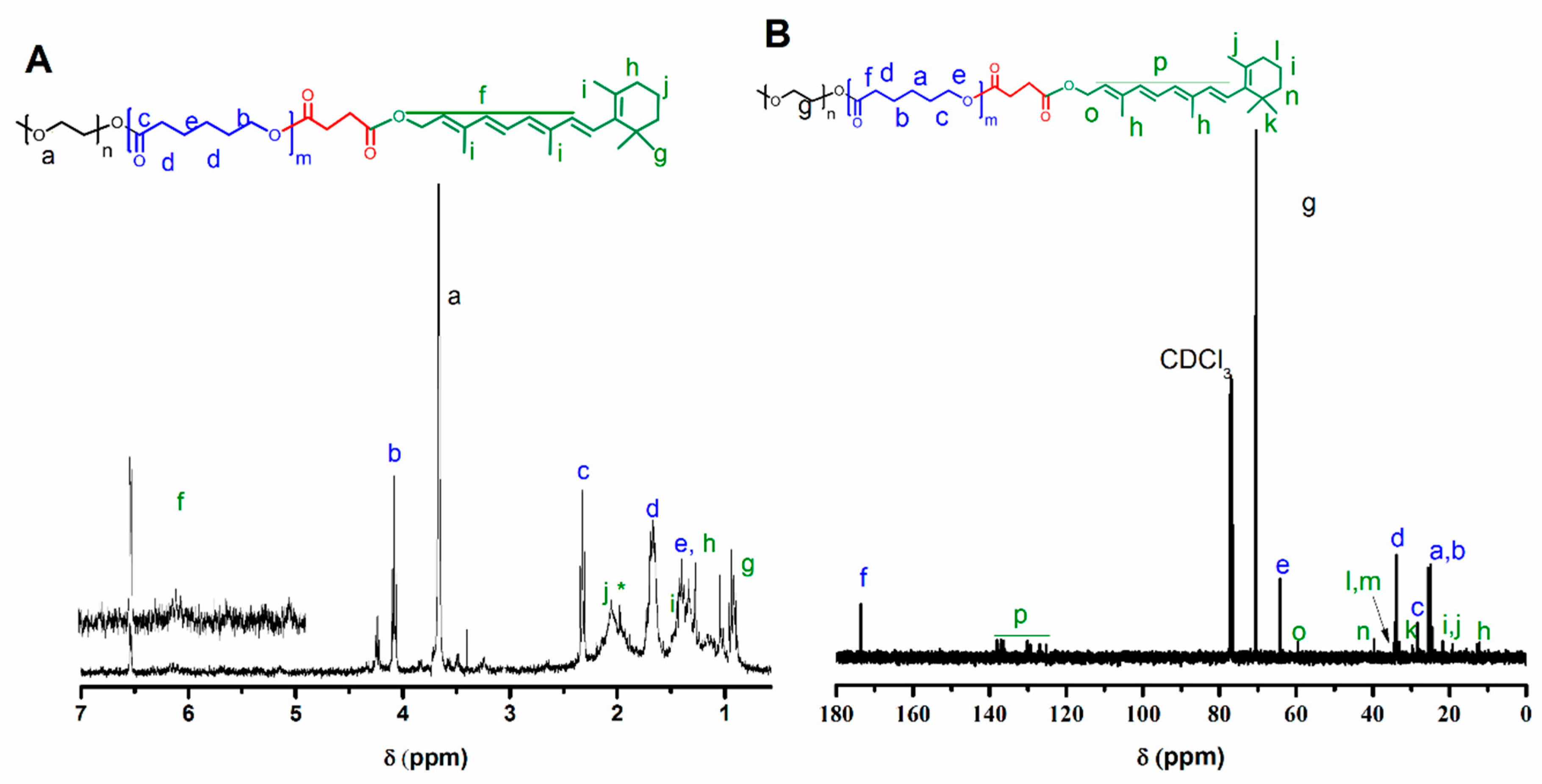{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Composition a | Mn (kDa) a | Mw/Mn b | CMC | |
|---|---|---|---|---|---|
| mg/L | μM | ||||
| AB | (mPEG)114-(PCL)11-OH | 6.27 | 1.20 | 4.5 ± 0.26 | 4.9 |
| AB-RET | (mPEG)114-b-(PCL)11-RET | 6.56 | 1.24 | 7.4 ± 0.78 | 8.0 |
| ABA | HO-(PCL)9-(PEG)136-(PCL)9-OH | 8.04 | 1.13 | 2.7 ± 0.86 | 2.9 |
| ABA-RET | RET-(PCL)9-(PEG)136-(PCL)9-RET | 8.61 | 1.29 | 5.5 ± 0.72 | 5.9 |
| Sample | Thermal Properties | ||
|---|---|---|---|
| Tc (°C) | Tm (°C) | ΔHm (J/g) | |
| mPEG | 40.6 | 60.2 | 171.9 |
| AB | 28.2 18.6 | 48.0 | 86.2 |
| AB-RET | 23.3 −7.15 | 39.4 | 61.9 |
| PEG-diol | 35.4 | 63.9 | 107.9 |
| ABA | 30.8 | 54.4 | 110.7 |
| ABA-RET | 2.7 | 38.41 49.1 | 41.8 |
| Formulation | EE (%) | DL (%) |
|---|---|---|
| AmB@AB | 13.7 ± 0.71 | 2.74 ± 0.17 |
| AmB@AB-RET | 51.05 ± 0.79 | 10.21 ± 0.21 |
| AmB@ABA | 10.19 ± 0.12 | 2.38 ± 0.03 |
| AmB@ABA-RET | 33.65 ± 1.23 | 6.73 ± 0.28 |
| Formulation | Empty Micelles | AmB@PMs | ||||||
|---|---|---|---|---|---|---|---|---|
| 25 °C | 37 °C | 25 °C | 37 °C | |||||
| Dh (nm) | PDI | Dh (nm) | PDI | Dh (nm) | PDI | Dh (nm) | PDI | |
| AmB@AB | 31.1 ± 1.7 | 0.172 ± 0.032 | 35.5 ± 2.8 | 0.189 ± 0.081 | 57.7 ± 4.3 | 0.324 ± 0.072 | 69.0 ± 6.0 | 0.234 ± 0.032 |
| AmB@AB-RET | 57.5 ± 4.2 | 0.213 ± 0.023 | 80.5 ± 7.5 | 0.204 ± 0.035 | 91.0 ± 3.5 | 0.391 ± 0.058 | 115.3 ± 7.2 | 0.359 ± 0.043 |
| AmB@ABA | 22.0 ± 2.3 | 0.198 ± 0.035 | 32.4 ± 3.3 | 0.212 ± 0.014 | 45.9 ± 4.7 | 0.429 ± 0.063 | 67.8 ± 3.5 | 0.354 ± 0.065 |
| AmB@ABA-RET | 53.4 ± 7.3 | 0.321 ± 0.041 | 77.6 ± 9.1 | 0.271 ± 0.037 | 80.2 ± 5.9 | 0.407 ± 0.024 | 81.7 ± 5.2 | 0.428 ± 0.027 |
| Sample | Aggregation Ratio * | AgI/Ag ** |
|---|---|---|
| AmB in PBS | 0.66 | 0.49 |
| Fungizone® | 0.70 | 0.74 |
| AmB@AB | 0.21 | 0.17 |
| AmB@AB-RET | 0.32 | 0.75 |
| AmB@ABA | 0.23 | 0.14 |
| AmB@ABA-RET | 0.37 | 0.81 |
| Model | Parameter | ABA | ABA-RET | ABA | ABA-RET |
|---|---|---|---|---|---|
| Zero order | R2 | 0.975 | 0.903 | 0.973 | 0.969 |
| K (%·h−1) | 2.426 | 2.061 | 0.138 | 0.173 | |
| First order | R2 | 0.917 | 0.878 | 0.950 | 0.950 |
| k (h−1) | 0.207 | 0.095 | 0.006 | 0.005 | |
| Higuchi | R2 | 0.992 | 0.955 | 0.996 | 0.994 |
| k (%·h−0.5) | 0.085 | 0.074 | 0.021 | 0.026 | |
| Korsmeyer–Peppas | R2 | 0.994 | 0.975 | 0.999 | 0.999 |
| n | 0.608 | 0.283 | 0.613 | 0.496 | |
| k (h−n) | 0.063 | 0.166 | 0.072 | 0.141 |
| Yeast | Reference | CLSI | Formulations | |||||
|---|---|---|---|---|---|---|---|---|
| FLC * | CAS * | Fungizone® | AmB@AB | AmB@ABA | AmB@AB-RET | AmB@ABA-RET | ||
| MIC (µg/mL) | ||||||||
| C. albicans | SC5314 | 1 | 0.06 | 0.46 | <0.11 | <0.11 | <0.11 | <0.11 |
| C. glabrata | ATCC 2001 | 0.25 | 0.06 | 0.46 | <0.11 | <0.11 | <0.11 | <0.11 |
| C. krusei | ATCC 6258 | 32 | 0.25 | 1.875 | 0.23 | 0.23 | 0.43 | 0.23 |
| C. parapsilosis | ATCC 22019 | 1 | 0.25 | 0.23 | <0.11 | <0.11 | <0.11 | <0.11 |
| C. auris | 435-PUJ-HUSI | 8 | 0.25 | 0.93 | 0.23 | 0.23 | 0.46 | 0.23 |
| C. auris * | 537-PUJ-HUSI | 128 ** | <0.05 | 3.75 | 1.865 | 0.93 | 3.75 | 3.75 |
| C. parapsilosis | 75-PUJ-FVL | 0.125 | <0.05 | 0.11 | <0.11 | <0.11 | 0.11 | <0.11 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez, Y.J.; Quejada, L.F.; Villamil, J.C.; Baena, Y.; Parra-Giraldo, C.M.; Perez, L.D. Development of Amphotericin B Micellar Formulations Based on Copolymers of Poly(ethylene glycol) and Poly(ε-caprolactone) Conjugated with Retinol. Pharmaceutics 2020, 12, 196. https://doi.org/10.3390/pharmaceutics12030196
Rodriguez YJ, Quejada LF, Villamil JC, Baena Y, Parra-Giraldo CM, Perez LD. Development of Amphotericin B Micellar Formulations Based on Copolymers of Poly(ethylene glycol) and Poly(ε-caprolactone) Conjugated with Retinol. Pharmaceutics. 2020; 12(3):196. https://doi.org/10.3390/pharmaceutics12030196
Chicago/Turabian StyleRodriguez, Yeimy J., Luis F. Quejada, Jean C. Villamil, Yolima Baena, Claudia M. Parra-Giraldo, and Leon D. Perez. 2020. "Development of Amphotericin B Micellar Formulations Based on Copolymers of Poly(ethylene glycol) and Poly(ε-caprolactone) Conjugated with Retinol" Pharmaceutics 12, no. 3: 196. https://doi.org/10.3390/pharmaceutics12030196
APA StyleRodriguez, Y. J., Quejada, L. F., Villamil, J. C., Baena, Y., Parra-Giraldo, C. M., & Perez, L. D. (2020). Development of Amphotericin B Micellar Formulations Based on Copolymers of Poly(ethylene glycol) and Poly(ε-caprolactone) Conjugated with Retinol. Pharmaceutics, 12(3), 196. https://doi.org/10.3390/pharmaceutics12030196