Nanocrystals of Fusidic Acid for Dual Enhancement of Dermal Delivery and Antibacterial Activity: In Vitro, Ex Vivo and In Vivo Evaluation


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of FA-NC
2.3. Response Surface Planning of FA-NC Formulations
2.4. Selection of the Optimized Formulation
2.5. Preparation of Freeze-Dried FA-NC
2.6. Micromeritics of FA-NC
2.6.1. Particle Size and Span Value Measurement
2.6.2. Determination of Zeta Potential
2.7. Physico-Chemical Characterization of Optimized FA-NC
2.7.1. Determination of Saturated Solubility
2.7.2. In-Vitro Dissolution Studies
2.7.3. Differential Scanning Calorimetry (DSC) Studies
2.7.4. Powder X-ray Diffraction (XRD)
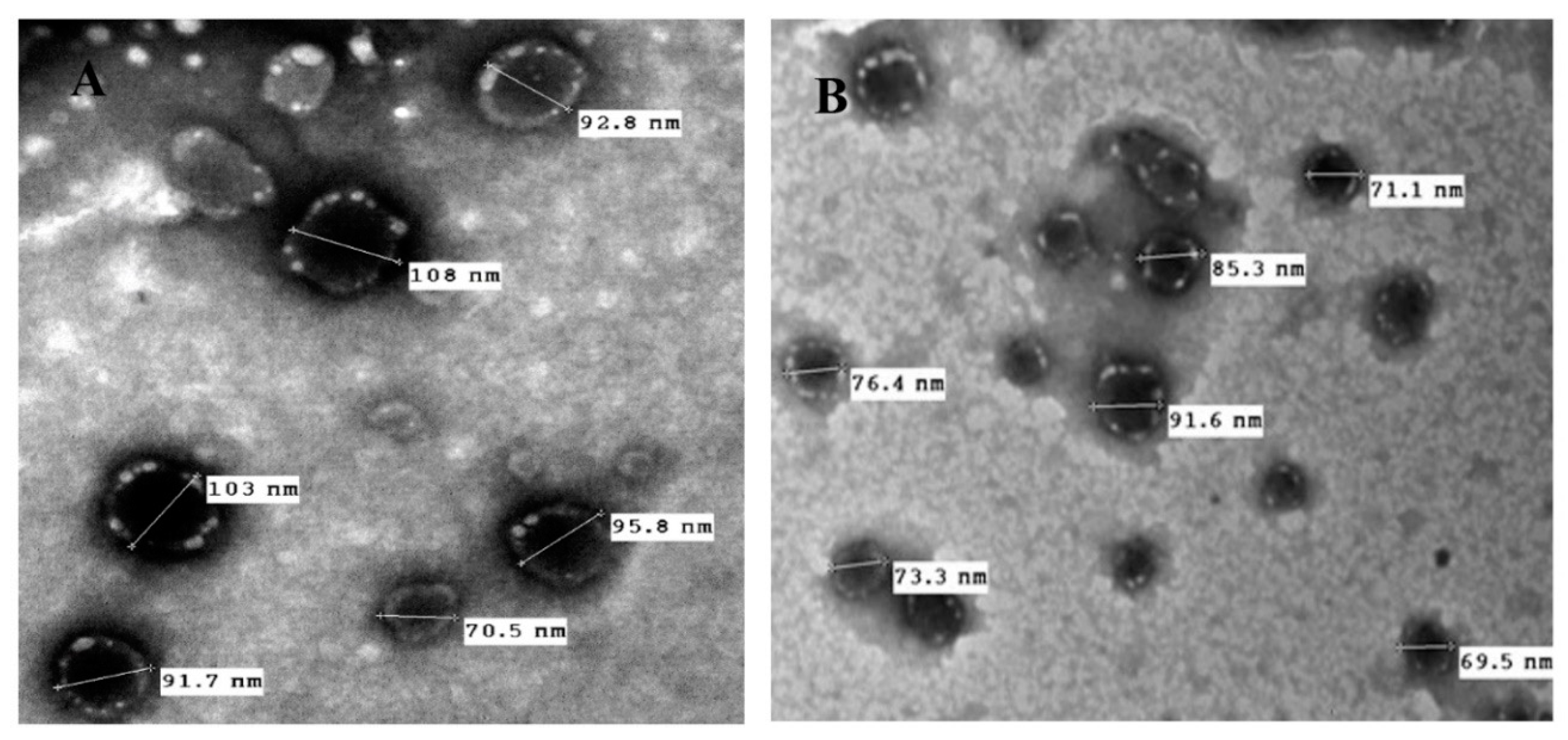
2.8. Morphological Characterization
2.9. Short-Term Stability Studies
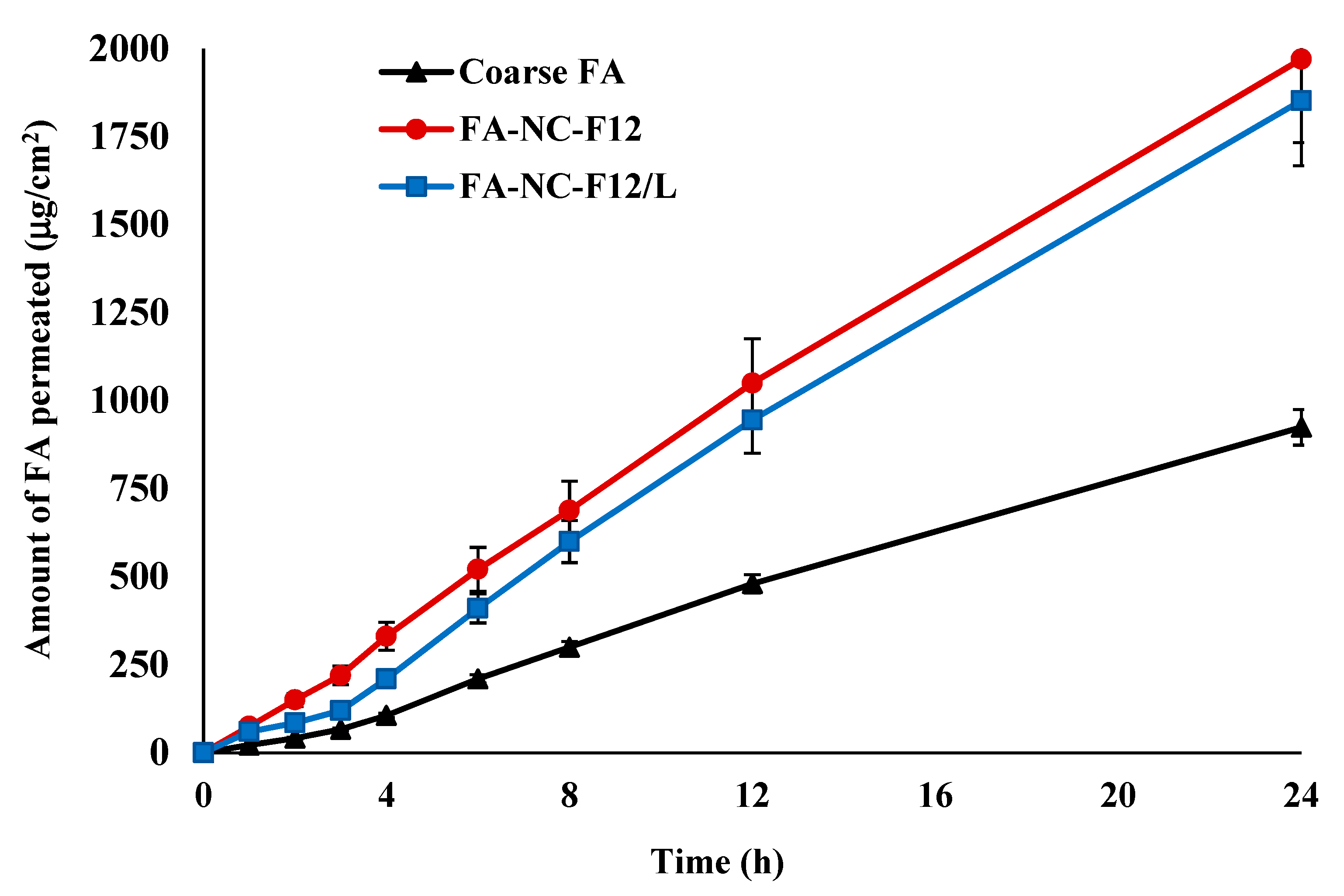
2.10. Skin Permeation Studies
2.10.1. Ex-Vivo Permeation
2.10.2. Determination of Dermal Penetration (Retention Studies)
2.11. Incorporation of FA-NC in Topical Formulation
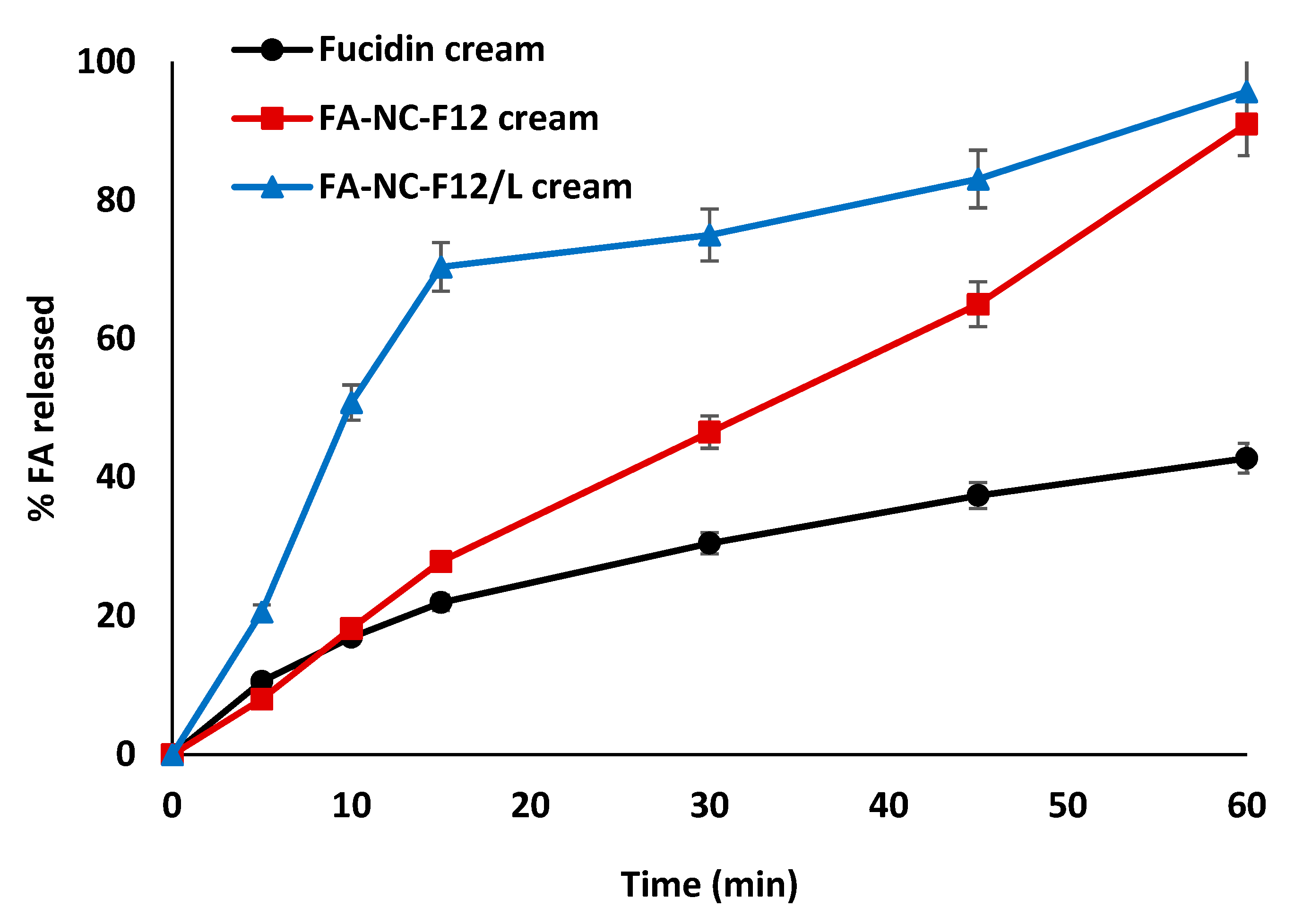
2.11.1. In-Vitro Release Studies
2.11.2. Microbiological Studies
2.12. In-Vivo Antibacterial Activity
2.12.1. Animals
2.12.2. Induction of Infected Wounds
2.12.3. Drug Administration and Dosing
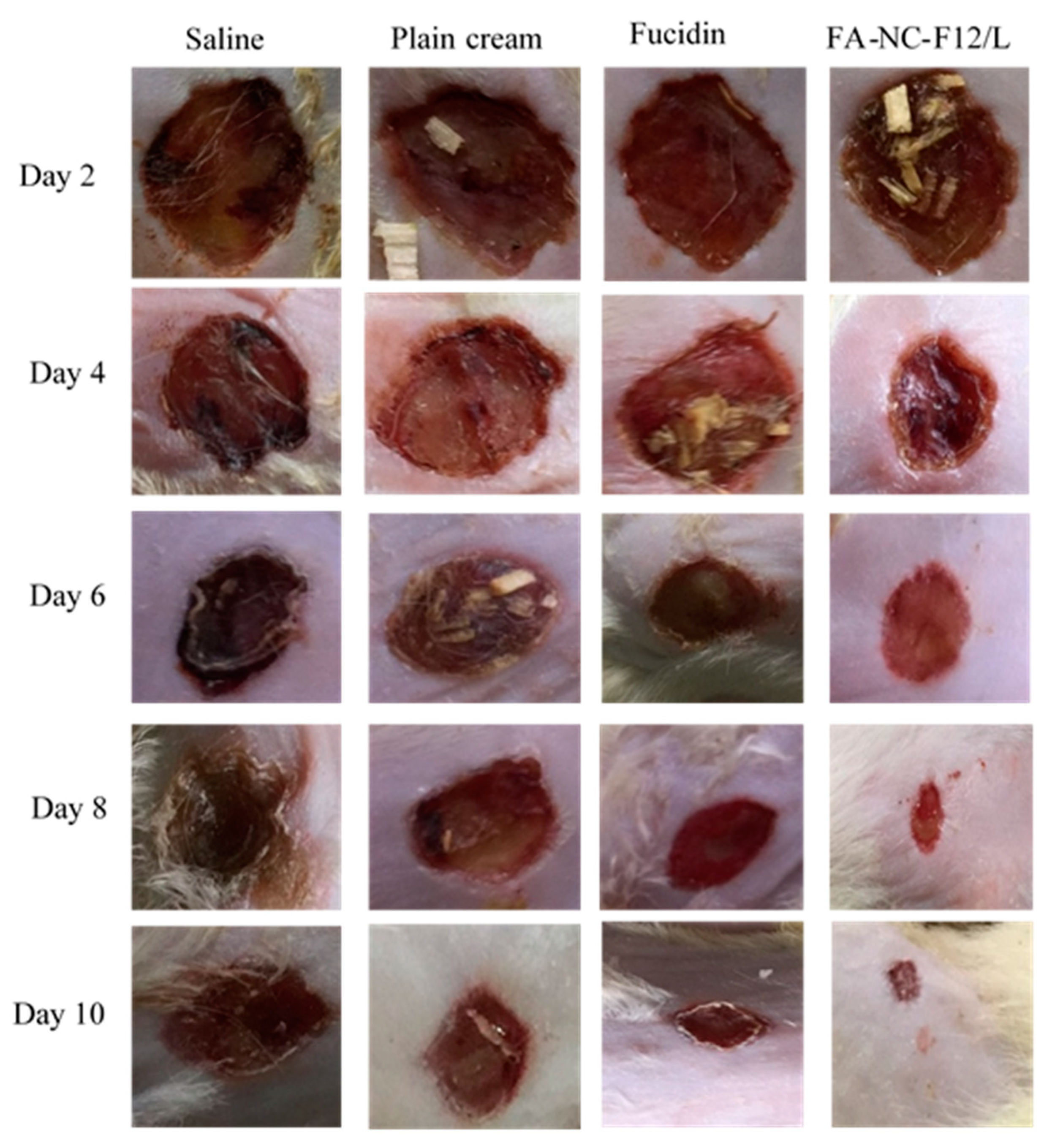
2.12.4. Wound Healing Assessment
2.12.5. Bacterial Load
2.12.6. Macroscopic Examination
2.12.7. Histological Examination
2.13. HPLC Analysis
2.14. Statistical Analysis
3. Results and Discussion
3.1. Optimization of FA-NC Formulation
3.2. Characterization of Optimized FA-NC
3.2.1. Micromeritics of FA-NC
3.2.2. Saturation Solubility and Dissolution Studies
3.2.3. DSC and XRD Studies
3.3. Morphology and Surface Characteristics Studies
3.4. Short-Term Stability Studies
3.5. Skin Permeation Studies
3.5.1. Ex-Vivo Permeation
3.5.2. Retention Studies
3.6. Evaluation of FA-NC in Topical Formulation
3.6.1. In-Vitro Release Studies
3.6.2. Microbiological Studies
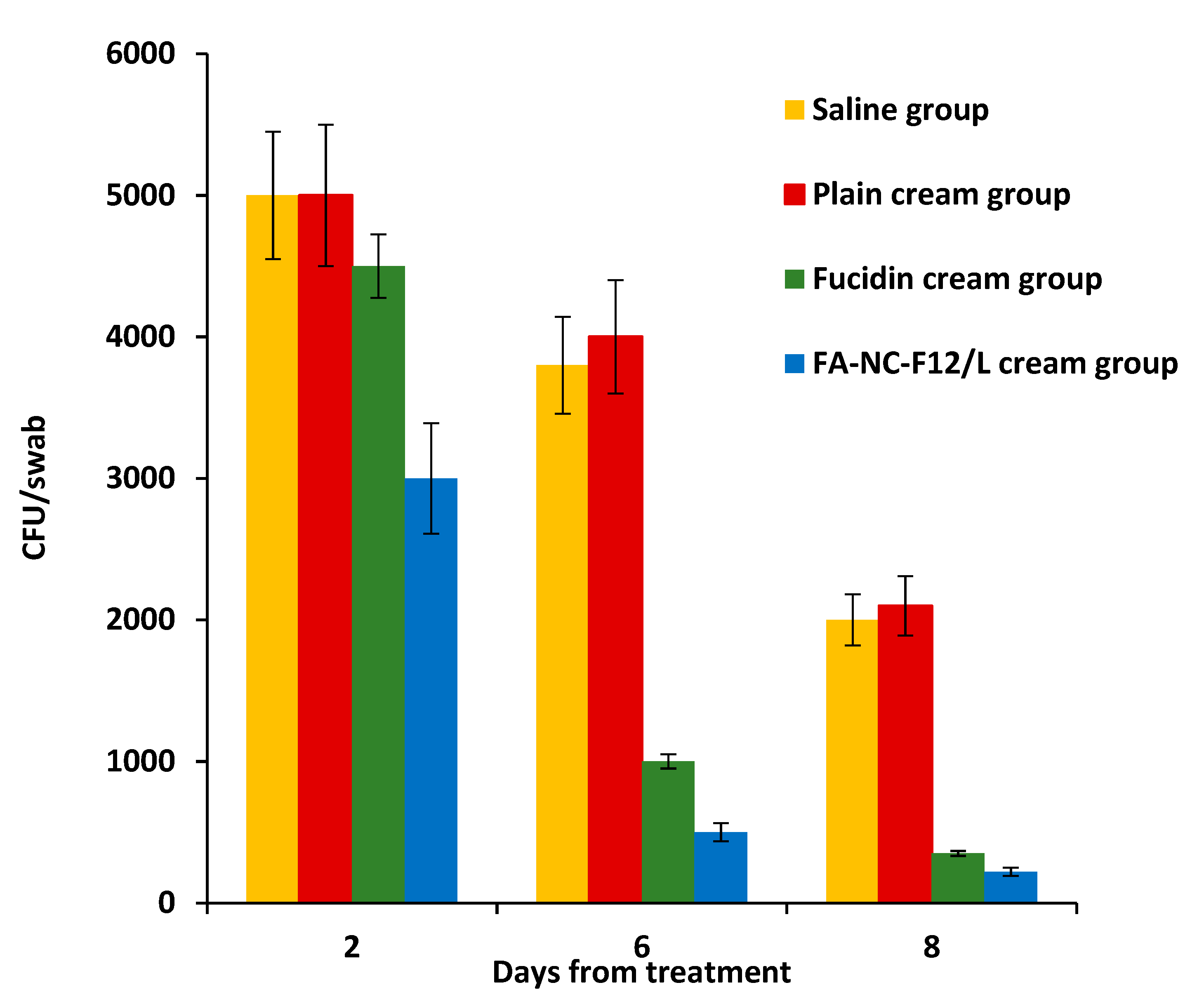
3.6.3. In-Vivo Studies
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lipsky, B.A.; Hoey, C. Topical antimicrobial therapy for treating chronic wounds. Clin. Infect. Dis. 2009, 49, 1541–1549. [Google Scholar] [CrossRef] [Green Version]
- Pachuau, L. Recent developments in novel drug delivery systems for wound healing. Expert Opin. Drug Deliv. 2015, 12, 1895–1909. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Thakur, K.; Raza, K.; Singh, B.; Katare, O.P. Nanostructured Lipid Carriers: A New Paradigm in Topical Delivery for Dermal and Transdermal Applications. Crit. Rev. Ther. Drug Carr. Syst. 2017, 34, 355–386. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Mohanraj, M.; Shah, M. High levels of fusidic acid-resistant Staphylococcus aureus despite restrictions on antibiotic use. Clin. Exp. Dermatol. 2009, 34, 136–139. [Google Scholar] [CrossRef] [PubMed]
- Mason, B.W.; Howard, A.J.; Magee, J.T. Fusidic acid resistance in community isolates of methicillin-susceptible Staphylococcus aureus and fusidic acid prescribing. J. Antimicrob. Chemother. 2003, 51, 1033–1036. [Google Scholar] [CrossRef] [Green Version]
- Farrell, D.J.; Mendes, R.E.; Castanheira, M.; Jones, R.N. Activity of Fusidic Acid Tested against Staphylococci Isolated from Patients in U.S. Medical Centers in 2014. Antimicrob. Agents Chemother. 2016, 60, 3827–3831. [Google Scholar] [CrossRef] [Green Version]
- Craft, J.C.; Moriarty, S.R.; Clark, K.; Scott, D.; Degenhardt, T.P.; Still, J.G.; Corey, G.R.; Das, A.; Fernandes, P. A randomized, double-blind phase 2 study comparing the efficacy and safety of an oral fusidic acid loading-dose regimen to oral linezolid for the treatment of acute bacterial skin and skin structure infections. Clin. Infect. Dis. 2011, 52 (Suppl. S7), S520–S526. [Google Scholar] [CrossRef]
- Manconi, M.; Valenti, D.; Sinico, C.; Lai, F.; Loy, G.; Fadda, A.M. Niosomes as carriers for tretinoin. II. Influence of vesicular incorporation on tretinoin photostability. Int. J. Pharm. 2003, 260, 261–272. [Google Scholar] [CrossRef]
- Sinico, C.; Manconi, M.; Peppi, M.; Lai, F.; Valenti, D.; Fadda, A.M. Liposomes as carriers for dermal delivery of tretinoin: In vitro evaluation of drug permeation and vesicle-skin interaction. J. Control. Release 2005, 103, 123–136. [Google Scholar] [CrossRef]
- Manconi, M.; Sinico, C.; Valenti, D.; Lai, F.; Fadda, A.M. Niosomes as carriers for tretinoin. III. A study into the in vitro cutaneous delivery of vesicle-incorporated tretinoin. Int. J. Pharm. 2006, 311, 11–19. [Google Scholar] [CrossRef]
- Ourique, A.F.; Melero, A.; de Bona da Silva, C.; Schaefer, U.F.; Pohlmann, A.R.; Guterres, S.S.; Lehr, C.M.; Kostka, K.H.; Beck, R.C. Improved photostability and reduced skin permeation of tretinoin: Development of a semisolid nanomedicine. Eur. J. Pharm. Biopharm. 2011, 79, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, S.; Singh, B.; Sharma, G.; Raza, K.; Katare, O.P. Liposomal fusidic acid as a potential delivery system: A new paradigm in the treatment of chronic plaque psoriasis. Drug Deliv. 2016, 23, 1204–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, A.J.; Kwon, Y.J. “Nanoantibiotics”: A new paradigm for treating infectious diseases using nanomaterials in the antibiotics resistant era. J. Control. Release 2011, 156, 128–145. [Google Scholar] [CrossRef] [PubMed]
- Kalhapure, R.S.; Suleman, N.; Mocktar, C.; Seedat, N.; Govender, T. Nanoengineered drug delivery systems for enhancing antibiotic therapy. J. Pharm. Sci. 2015, 104, 872–905. [Google Scholar] [CrossRef]
- Sharma, G.; Goyal, H.; Thakur, K.; Raza, K.; Katare, O.P. Novel elastic membrane vesicles (EMVs) and ethosomes-mediated effective topical delivery of aceclofenac: A new therapeutic approach for pain and inflammation. Drug Deliv. 2016, 23, 3135–3145. [Google Scholar] [CrossRef] [Green Version]
- Kuchler, S.; Radowski, M.R.; Blaschke, T.; Dathe, M.; Plendl, J.; Haag, R.; Schafer-Korting, M.; Kramer, K.D. Nanoparticles for skin penetration enhancement—A comparison of a dendritic core-multishell-nanotransporter and solid lipid nanoparticles. Eur. J. Pharm. Biopharm. 2009, 71, 243–250. [Google Scholar] [CrossRef]
- Agrawal, U.; Mehra, N.K.; Gupta, U.; Jain, N.K. Hyperbranched dendritic nano-carriers for topical delivery of dithranol. J. Drug Target. 2013, 21, 497–506. [Google Scholar] [CrossRef]
- Junghanns, J.U.; Muller, R.H. Nanocrystal technology, drug delivery and clinical applications. Int. J. Nanomed. 2008, 3, 295–309. [Google Scholar] [CrossRef] [Green Version]
- Muller, R.H.; Gohla, S.; Keck, C.M. State of the art of nanocrystals—Special features, production, nanotoxicology aspects and intracellular delivery. Eur. J. Pharm. Biopharm. 2011, 78, 1–9. [Google Scholar] [CrossRef]
- Mishra, P.R.; Al Shaal, L.; Muller, R.H.; Keck, C.M. Production and characterization of Hesperetin nanosuspensions for dermal delivery. Int. J. Pharm. 2009, 371, 182–189. [Google Scholar] [CrossRef]
- Chhibber, T.; Wadhwa, S.; Chadha, P.; Sharma, G.; Katare, O.P. Phospholipid structured microemulsion as effective carrier system with potential in methicillin sensitive Staphylococcus aureus (MSSA) involved burn wound infection. J. Drug Target. 2015, 23, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Eid, A.M.; Istateyeh, I.; Salhi, N.; Istateyeh, T. Antibacterial Activity of Fusidic Acid and Sodium Fusidate Nanoparticles Incorporated in Pine Oil Nanoemulgel. Int. J. Nanomed. 2019, 14, 9411–9421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, K.; Sharma, G.; Singh, B.; Chhibber, S.; Patil, A.B.; Katare, O.P. Chitosan-tailored lipidic nanoconstructs of Fusidic acid as promising vehicle for wound infections: An explorative study. Int. J. Biol. Macromol. 2018, 115, 1012–1025. [Google Scholar] [CrossRef] [PubMed]
- Thakur, K.; Sharma, G.; Singh, B.; Jain, A.; Tyagi, R.; Chhibber, S.; Katare, O.P. Cationic-bilayered nanoemulsion of fusidic acid: An investigation on eradication of methicillin-resistant Staphylococcus aureus 33591 infection in burn wound. Nanomedicine (Lond.) 2018, 13, 825–847. [Google Scholar] [CrossRef] [PubMed]
- Omolo, C.A.; Kalhapure, R.S.; Agrawal, N.; Rambharose, S.; Mocktar, C.; Govender, T. Formulation and Molecular Dynamics Simulations of a Fusidic Acid Nanosuspension for Simultaneously Enhancing Solubility and Antibacterial Activity. Mol. Pharm. 2018, 15, 3512–3526. [Google Scholar] [CrossRef]
- Romero, G.B.; Chen, R.; Keck, C.M.; Muller, R.H. Industrial concentrates of dermal hesperidin smartCrystals®—Production, characterization & long-term stability. Int. J. Pharm. 2015, 482, 54–60. [Google Scholar] [CrossRef]
- Xing, M.; Zhong, W.; Xu, X.; Thomson, D. Adhesion force studies of nanofibers and nanoparticles. Langmuir 2010, 26, 11809–11814. [Google Scholar] [CrossRef]
- Sinha, B.; Muller, R.H.; Moschwitzer, J.P. Bottom-up approaches for preparing drug nanocrystals: Formulations and factors affecting particle size. Int. J. Pharm. 2013, 453, 126–141. [Google Scholar] [CrossRef]
- Singh, G.; Pai, R.S.; Devi, V.K. Optimization of pellets containing solid dispersion prepared by extrusion/spheronization using central composite design and desirability function. J. Young Pharm. 2012, 4, 146–156. [Google Scholar] [CrossRef] [Green Version]
- Sharma, G.; Thakur, K.; Raza, K.; Katare, O.P. Stability kinetics of fusidic acid: Development and validation of stability indicating analytical method by employing Analytical Quality by Design approach in medicinal product(s). J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2019, 1120, 113–124. [Google Scholar] [CrossRef]
- Godin, B.; Touitou, E. Transdermal skin delivery: Predictions for humans from in vivo, ex vivo and animal models. Adv. Drug Deliv. Rev. 2007, 59, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Weigmann, H.J.; Lademann, J.; Schanzer, S.; Lindemann, U.; von Pelchrzim, R.; Schaefer, H.; Sterry, W.; Shah, V. Correlation of the local distribution of topically applied substances inside the stratum corneum determined by tape-stripping to differences in bioavailability. Skin Pharmacol. Appl. Skin Physiol. 2001, 14 (Suppl. S1), 98–102. [Google Scholar] [CrossRef]
- Teichmann, A.; Jacobi, U.; Ossadnik, M.; Richter, H.; Koch, S.; Sterry, W.; Lademann, J. Differential stripping: Determination of the amount of topically applied substances penetrated into the hair follicles. J. Investig. Dermatol. 2005, 125, 264–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, R.H.; Hespeler, D.; Jin, N.; Pyo, S.M. Smartpearls—Novel physically stable amorphous delivery system for poorly soluble dermal actives. Int. J. Pharm. 2019, 555, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Pelikh, O.; Stahr, P.L.; Huang, J.; Gerst, M.; Scholz, P.; Dietrich, H.; Geisel, N.; Keck, C.M. Nanocrystals for improved dermal drug delivery. Eur. J. Pharm. Biopharm. 2018, 128, 170–178. [Google Scholar] [CrossRef] [PubMed]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing, Disc Diffusion Supplemental Tables; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2013. [Google Scholar]
- CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, Approved Standard, 10th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2015. [Google Scholar]
- Kahlmeter, G.; Brown, D.F.; Goldstein, F.W.; MacGowan, A.P.; Mouton, J.W.; Odenholt, I.; Rodloff, A.; Soussy, C.J.; Steinbakk, M.; Soriano, F.; et al. European Committee on Antimicrobial Susceptibility Testing (EUCAST) Technical Notes on antimicrobial susceptibility testing. Clin. Microbiol. Infect. 2006, 12, 501–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samy, R.P.; Gopalakrishnakone, P.; Houghton, P.; Ignacimuthu, S. Purification of antibacterial agents from Tragia involucrata—A popular tribal medicine for wound healing. J. Ethnopharmacol. 2006, 107, 99–106. [Google Scholar] [CrossRef]
- Mukherjee, P.K.; Suresh, B. The evaluation of wound-healing potential of Hypericum hookerianum leaf and stem extracts. J. Altern. Complement. Med. 2000, 6, 61–69. [Google Scholar] [CrossRef]
- Levine, N.S.; Lindberg, R.B.; Mason, A.D., Jr.; Pruitt, B.A., Jr. The quantitative swab culture and smear: A quick, simple method for determining the number of viable aerobic bacteria on open wounds. J. Trauma 1976, 16, 89–94. [Google Scholar] [CrossRef]
- Gardner, S.E.; Frantz, R.A. Wound bioburden and infection-related complications in diabetic foot ulcers. Biol. Res. Nurs. 2008, 10, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Bancroft, J.D.; Gamble, M. Theory and Practice of Histological Techniques; Elsevier Health Sciences: Philadelphia, PA, USA, 2008. [Google Scholar]
- Keck, C.M.; Muller, R.H. Drug nanocrystals of poorly soluble drugs produced by high pressure homogenisation. Eur. J. Pharm. Biopharm. 2006, 62, 3–16. [Google Scholar] [CrossRef]
- de Waard, H.; Hinrichs, W.L.; Frijlink, H.W. A novel bottom-up process to produce drug nanocrystals: Controlled crystallization during freeze-drying. J. Control. Release 2008, 128, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, J.; Watanabe, W. Physical and chemical stability of drug nanoparticles. Adv. Drug Deliv. Rev. 2011, 63, 456–469. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Guan, J.; Sun, Z.; Shen, X.; Li, L.; Jin, L.; Mao, S. Influence of stabilizer type and concentration on the lung deposition and retention of resveratrol nanosuspension-in-microparticles. Int. J. Pharm. 2019, 569, 118562. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, Y.; Hara, K.; Bando, Y.; Huang, C.C.; Kousaka, Y.; Kawashima, Y.; Morishita, R.; Tsujimoto, H. Particle size control of poly(dl-lactide-co-glycolide) nanospheres for sterile applications. Int. J. Pharm. 2009, 370, 196–201. [Google Scholar] [CrossRef]
- Mauludin, R.; Muller, R.H.; Keck, C.M. Kinetic solubility and dissolution velocity of rutin nanocrystals. Eur. J. Pharm. Sci. 2009, 36, 502–510. [Google Scholar] [CrossRef]
- Rabinow, B.E. Nanosuspensions in drug delivery. Nat. Rev. Drug Discov. 2004, 3, 785–796. [Google Scholar] [CrossRef]
- Ahmed, I.S.; El-Hosary, R.; Shalaby, S.; Abd-Rabo, M.M.; Elkhateeb, D.G.; Nour, S. PD-PK evaluation of freeze-dried atorvastatin calcium-loaded poly-epsilon-caprolactone nanoparticles. Int J Pharm 2016, 504, 70–79. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, D.; Chen, M.; Zheng, T.; Wang, S. Preparation and characterization of an oridonin nanosuspension for solubility and dissolution velocity enhancement. Drug Dev. Ind. Pharm. 2007, 33, 1332–1339. [Google Scholar] [CrossRef]
- Shegokar, R.; Muller, R.H. Nanocrystals: Industrially feasible multifunctional formulation technology for poorly soluble actives. Int. J. Pharm. 2010, 399, 129–139. [Google Scholar] [CrossRef]
- Vidlarova, L.; Romero, G.B.; Hanus, J.; Stepanek, F.; Muller, R.H. Nanocrystals for dermal penetration enhancement—Effect of concentration and underlying mechanisms using curcumin as model. Eur. J. Pharm. Biopharm. 2016, 104, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, S.E.; Letchford, K.; Burt, H.M. The solid-state characterization of fusidic acid. Int. J. Pharm. 2012, 422, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Huang, G. Effects of emulsifiers on the controlled release of paclitaxel (Taxol) from nanospheres of biodegradable polymers. J. Control. Release 2001, 71, 53–69. [Google Scholar] [CrossRef]
- Rampino, A.; Borgogna, M.; Blasi, P.; Bellich, B.; Cesaro, A. Chitosan nanoparticles: Preparation, size evolution and stability. Int. J. Pharm. 2013, 455, 219–228. [Google Scholar] [CrossRef]
- Patzelt, A.; Richter, H.; Knorr, F.; Schafer, U.; Lehr, C.M.; Dahne, L.; Sterry, W.; Lademann, J. Selective follicular targeting by modification of the particle sizes. J. Control. Release 2011, 150, 45–48. [Google Scholar] [CrossRef]
- Blume-Peytavi, U.; Vogt, A. Human hair follicle: Reservoir function and selective targeting. Br. J. Dermatol. 2011, 165 (Suppl S2), 13–17. [Google Scholar] [CrossRef]
- Lademann, J.; Richter, H.; Teichmann, A.; Otberg, N.; Blume-Peytavi, U.; Luengo, J.; Weiss, B.; Schaefer, U.F.; Lehr, C.M.; Wepf, R.; et al. Nanoparticles—An efficient carrier for drug delivery into the hair follicles. Eur. J. Pharm. Biopharm. 2007, 66, 159–164. [Google Scholar] [CrossRef]
- Teichmann, A.; Heuschkel, S.; Jacobi, U.; Presse, G.; Neubert, R.H.; Sterry, W.; Lademann, J. Comparison of stratum corneum penetration and localization of a lipophilic model drug applied in an o/w microemulsion and an amphiphilic cream. Eur. J. Pharm. Biopharm. 2007, 67, 699–706. [Google Scholar] [CrossRef]
- Ahmed, I.S.; Aboul-Einien, M.H. In vitro and in vivo evaluation of a fast-disintegrating lyophilized dry emulsion tablet containing griseofulvin. Eur. J. Pharm. Sci. 2007, 32, 58–68. [Google Scholar] [CrossRef]
- Seedat, N.; Kalhapure, R.S.; Mocktar, C.; Vepuri, S.; Jadhav, M.; Soliman, M.; Govender, T. Co-encapsulation of multi-lipids and polymers enhances the performance of vancomycin in lipid-polymer hybrid nanoparticles: In vitro and in silico studies. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 61, 616–630. [Google Scholar] [CrossRef]
- Romero, G.B.; Arntjen, A.; Keck, C.M.; Muller, R.H. Amorphous cyclosporin a nanoparticles for enhanced dermal bioavailability. Int. J. Pharm. 2016, 498, 217–224. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation Code | Stabilizer Type | D/S ratio | Homogenization Time (min) | PS (nm) | SV |
|---|---|---|---|---|---|
| F1 | PF-68 | 1:2 | 2 | 155 ± 19 | 6.0 ± 1.4 |
| F2 | PF-68 | 1:2 | 5 | 151 ± 17 | 5.0 ± 0.3 |
| F3 | PF-68 | 1:2 | 10 | 154 ± 16 | 3.9 ± 0.1 |
| F4 | PF-68 | 1:4 | 2 | 165 ± 14 | 5.6 ± 0.2 |
| F5 | PF-68 | 1:4 | 5 | 158 ± 11 | 4.8 ± 0.7 |
| F6 | PF-68 | 1:4 | 10 | 142 ± 27 | 4.8 ± 1.5 |
| F7 | PVA 4-88 | 1:2 | 2 | 140 ± 14 | 4.5 ± 0.0 |
| F8 | PVA 4-88 | 1:2 | 5 | 139 ± 15 | 2.4 ± 1.7 |
| F9 | PVA 4-88 | 1:2 | 10 | 140 ± 21 | 3.9 ± 0.7 |
| F10 | PVA 4-88 | 1:4 | 2 | 137 ± 12 | 3.8 ± 0.1 |
| F11 | PVA 4-88 | 1:4 | 5 | 132 ± 10 | 2.3 ± 1.6 |
| *F12 | PVA 4-88 | 1:4 | 10 | 138 ± 22 | 1.3 ± 0.0 |
| F13 | PVP-K30 | 1:2 | 2 | 188 ± 11 | 5.3 ± 0.1 |
| F14 | PVP-K30 | 1:2 | 5 | 147 ± 12 | 4.6 ± 0.0 |
| F15 | PVP-K30 | 1:2 | 10 | 146 ± 8 | 4.6 ± 0.2 |
| F16 | PVP-K30 | 1:4 | 2 | 181 ± 13 | 3.2 ± 0.2 |
| F17 | PVP-K30 | 1:4 | 5 | 172 ± 11 | 3.3 ± 0.4 |
| F18 | PVP-K30 | 1:4 | 10 | 194 ± 18 | 3.1 ± 0.1 |
| Composition (% w/v) | |
| FA | 0.1 |
| PVA 4-88 | 0.2 |
| Mannitol | 2.0 |
| Responses | |
| PS (nm) | 115.1 ± 13 |
| SV | 1.60 ± 0.00 |
| ZP (mV) | −11.6 ± 3.6 |
| FA-NC | Storage Conditions | PS (nm) | SV |
|---|---|---|---|
| FA-NC-F12 | Fresh | 138 ± 22 | 1.3 ± 0.00 |
| 25 °C | 662.7 ± 42 * | 3.8 ± 0.6 * | |
| 4 °C | 147 ± 15 | 1.4 ± 0.2 | |
| FA-NC-F12/L | Fresh | 115.1 ± 13 | 1.6 ± 0.00 |
| 25 °C | 454.7 ± 21 * | 2.4 ± 0.3 * | |
| 4 °C | 121 ± 14 | 1.7 ± 0.1 |
| Test | FA-NC-F12 | FA-NC-F12/L | Fucidin |
|---|---|---|---|
| Inhibition zone (mm) | |||
| S.aureus | 34.7 ± 1.5 a,* | 37.2 ± 1.2 a,* | 25.3 ± 0.57 |
| S. epidermidis | 38.1 ± 1.4 a,* | 40.2 ± 1.6 a,* | 32.3 ± 0.58 |
| MIC (μg/mL) | |||
| S.aureus | 32 a,* | 32 a,* | >128 |
| S. epidermidis | 16 a,* | 16 a,* | 64 |
| MBC (μg/mL) | |||
| S.aureus | >128 | >128 | >128 |
| S. epidermidis | 128 | 128 | 128 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, I.S.; Elnahas, O.S.; Assar, N.H.; Gad, A.M.; El Hosary, R. Nanocrystals of Fusidic Acid for Dual Enhancement of Dermal Delivery and Antibacterial Activity: In Vitro, Ex Vivo and In Vivo Evaluation. Pharmaceutics 2020, 12, 199. https://doi.org/10.3390/pharmaceutics12030199
Ahmed IS, Elnahas OS, Assar NH, Gad AM, El Hosary R. Nanocrystals of Fusidic Acid for Dual Enhancement of Dermal Delivery and Antibacterial Activity: In Vitro, Ex Vivo and In Vivo Evaluation. Pharmaceutics. 2020; 12(3):199. https://doi.org/10.3390/pharmaceutics12030199
Chicago/Turabian StyleAhmed, Iman S., Osama S. Elnahas, Nouran H. Assar, Amany M. Gad, and Rania El Hosary. 2020. "Nanocrystals of Fusidic Acid for Dual Enhancement of Dermal Delivery and Antibacterial Activity: In Vitro, Ex Vivo and In Vivo Evaluation" Pharmaceutics 12, no. 3: 199. https://doi.org/10.3390/pharmaceutics12030199