Lack of Correlation between In Vitro and In Vivo Studies on the Inhibitory Effects of (‒)-Sophoranone on CYP2C9 Is Attributable to Low Oral Absorption and Extensive Plasma Protein Binding of (‒)-Sophoranone

 ,
,
Abstract
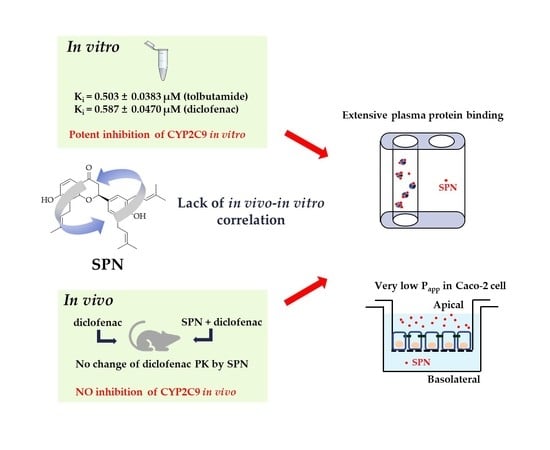
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Reversible Inhibition of (‒)-Sophoranone towards the Nine CYP Isoforms in Human Liver Microsomes
2.3. Determination of the Ki of (‒)-Sophoranone on CYP2C9 Activity in Human Liver Microsomes
2.4. Time-Dependent Inactivation of (‒)-Sophoranone toward the Nine CYP Isoforms in Human Liver Microsomes
2.5. Caco-2 Cell Permeability of (‒)-Sophoranone
2.6. Effects of (‒)-Sophoranone on the Pharmacokinetics of Diclofenac in Rats
2.7. Determination of the Unbound Fraction of (‒)-Sophoranone in Plasma and Human Liver Microsomes
2.8. LC-MS/MS Analysis
2.8.1. In Vitro Samples
2.8.2. In Vivo Samples
2.9. Analysis of Inhibition Kinetics and Pharmacokinetic Parameters
3. Results
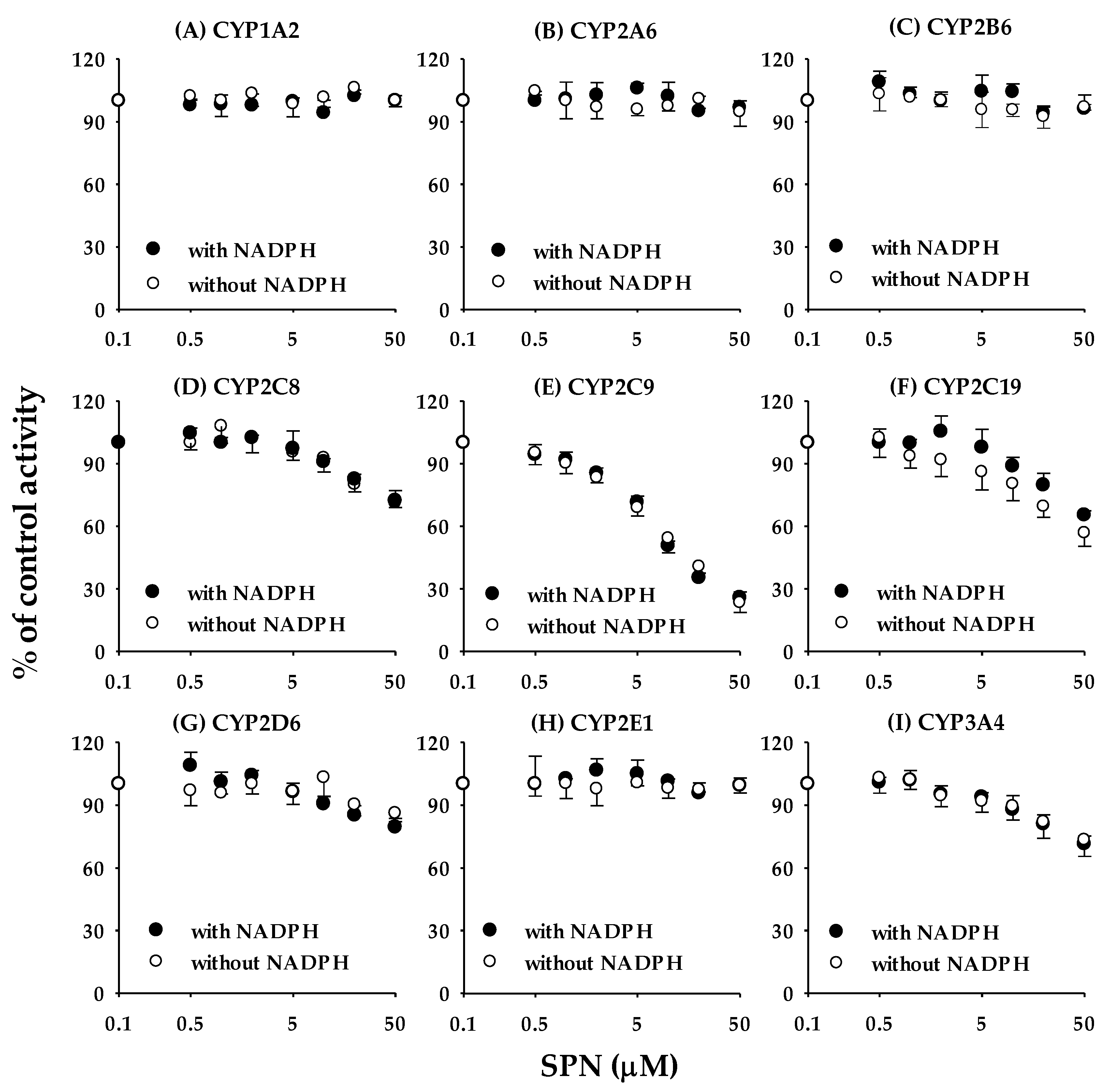
3.1. Reversible Inhibition of (‒)-Sophoranone toward the Nine CYP Isoforms in Human Liver Microsomes
3.2. Determination of the Ki of (‒)-Sophoranone for CYP2C9 Activity
3.3. Time-Dependent Inactivation of (‒)-Sophoranone towards the Nine CYP Isoforms in Human Liver Microsomes
3.4. Caco-2 Cell Permeability of (‒)-Sophoranone
3.5. Effects of (‒)-Sophoranone on the Pharmacokinetics of Diclofenac in Rats
3.6. Determination of the Unbound Fraction of (‒)-Sophoranone in Plasma and Human Liver Microsomes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Previous Presentation of Information
References
- Ding, P.L.; He, C.M.; Cheng, Z.H.; Chen, D.F. Flavonoids rather than alkaloids as the diagnostic constituents to distinguish Sophorae Flavescentis Radix from Sophorae Tonkinensis Radix et Rhizoma: An HPLC fingerprint study. Chin. J. Nat. Med. 2018, 16, 951–960. [Google Scholar] [CrossRef]
- He, C.M.; Cheng, Z.H.; Chen, D.F. Qualitative and quantitative analysis of flavonoids in Sophora tonkinensis by LC/MS and HPLC. Chin. J. Nat. Med. 2013, 11, 690–698. [Google Scholar] [CrossRef]
- Yoo, H.; Kang, M.; Pyo, S.; Chae, H.S.; Ryu, K.H.; Kim, J.; Chin, Y.W. SKI3301, a purified herbal extract from Sophora tonkinensis, inhibited airway inflammation and bronchospasm in allergic asthma animal models in vivo. J. Ethnopharmacol. 2017, 206, 298–305. [Google Scholar] [CrossRef]
- Lee, J.W.; Lee, J.H.; Lee, C.; Jin, Q.; Lee, D.; Kim, Y.; Hong, J.T.; Lee, M.K.; Hwang, B.Y. Inhibitory constituents of Sophora tonkinensis on nitric oxide production in RAW 264.7 macrophages. Bioorg. Med. Chem. Lett. 2015, 25, 960–962. [Google Scholar] [CrossRef]
- Kajimoto, S.; Takanashi, N.; Kajimoto, T.; Xu, M.; Cao, J.; Masuda, Y.; Aiuchi, T.; Nakajo, S.; Ida, Y.; Nakaya, K. Sophoranone, extracted from a traditional Chinese medicine Shan Dou Gen, induces apoptosis in human leukemia U937 cells via formation of reactive oxygen species and opening of mitochondrial permeability transition pores. Int. J. Cancer 2002, 99, 879–890. [Google Scholar] [CrossRef]
- Yang, X.; Deng, S.; Huang, M.; Wang, J.; Chen, L.; Xiong, M.; Yang, J.; Zheng, S.; Ma, X.; Zhao, P.; et al. Chemical constituents from Sophora tonkinensis and their glucose transporter 4 translocation activities. Bioorg. Med. Chem. Lett. 2017, 27, 1463–1466. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Atta-Ur-Rahman; Haroone, M.S.; Tareen, R.B.; Mohammed Ahmed Hassan, O.M.; Jan, S.; Abbaskhan, A.; Asif, M.; Gulzar, T.; Al-Majid, A.M.; Yousuf, S.; et al. Secondary metabolites of Sophora mollis subsp. griffithii (Stocks) Ali. Phytochem. Lett. 2012, 5, 613–616. [Google Scholar] [CrossRef]
- Jang, S.M.; Bae, S.H.; Choi, W.K.; Park, J.B.; Kim, D.; Min, J.S.; Yoo, H.; Kang, M.; Ryu, K.H.; Bae, S.K. Pharmacokinetic properties of trifolirhizin, (-)-maackiain, (-)-sophoranone and 2-(2,4-dihydroxyphenyl)-5,6-methylenedioxybenzofuran after intravenous and oral administration of Sophora tonkinensis extract in rats. Xenobiotica 2015, 45, 1092–1104. [Google Scholar] [CrossRef] [PubMed]
- Rekić, D.; Reynolds, K.S.; Zhao, P.; Zhang, L.; Yoshida, K.; Sachar, M.; Piquette Miller, M.; Huang, S.M.; Zineh, I. Clinical drug-drug interaction evaluations to inform drug use and enable drug access. J. Pharm. Sci. 2017, 106, 2214–2218. [Google Scholar] [CrossRef]
- Bjornsson, T.D.; Callaghan, J.T.; Einolf, H.J.; Fischer, V.; Gas, L.; Grimm, S.; Kao, J.; King, S.P.; Miwa, G.; Ni, L.; et al. The conduct of in vitro and in vivo drug-drug interaction studies: A pharmaceutical research and manufactures of America (PhRMA) perspective. Drug Metab. Dispos. 2003, 31, 815–832. [Google Scholar] [CrossRef]
- Lin, J.H.; Lu, A.Y. Inhibition and induction of cytochrome P450 and the clinical implications. Clin. Pharmacokinet. 1998, 35, 361–390. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Wu, H.; Zhang, X.; Zhang, F.; Qi, H.; Zhong, Y.; Wang, Y.; Sang, H.; Wang, G.; Sun, J. A comprehensive assay for nine major cytochrome P450 enzymes activities with 16 probe reactions on human liver microsomes by a single LC/MS/MS run to support reliable in vitro inhibitory drug-drug interaction evaluation. Xenobiotica 2015, 45, 961–977. [Google Scholar] [CrossRef]
- Wienkers, L.C.; Heath, T.G. Predicting in vivo drug interactions from in vitro drug discovery data. Nat. Rev. Drug Discov. 2005, 4, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, T.; Wang, J.F.; Wei, D.Q. Advances in human cytochrome P450 and personalized medicine. Curr. Drug Metab. 2011, 12, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Ma, J.; Xu, K.; Gao, G.; Xiang, Y.; Lin, C. Effect of Radix Sophorae Tonkinensis on the activity of cytochrome P450 isoforms in rats. Int. J. Clin. Exp. Med. 2015, 8, 9737–9743. [Google Scholar]
- Li, Y.; Ning, J.; Wang, Y.; Wang, C.; Sun, C.; Huo, X.; Yu, Z.; Feng, L.; Zhang, B.; Tian, X.; et al. Drug interaction study of flavonoids toward CYP3A4 and their quantitative structure activity relationship (QSAR) analysis for predicting potential effects. Toxicol. Lett. 2018, 294, 27–36. [Google Scholar] [CrossRef]
- Cho, D.Y.; Bae, S.H.; Lee, J.K.; Kim, Y.W.; Kim, B.T.; Bae, S.K. Selective inhibition of cytochrome P450 2D6 by Sarpogrelate and its active metabolite, M-1, in human liver microsomes. Drug Metab. Dispos. 2014, 42, 33–39. [Google Scholar] [CrossRef]
- Zheng, Y.F.; Bae, S.H.; Choi, E.J.; Park, J.B.; Kim, S.O.; Jang, M.J.; Park, G.H.; Shin, W.G.; Oh, E.; Bae, S.K. Evaluation of the in vitro/in vivo drug interaction potential of BST204, a purified dry extract of ginseng, and its four bioactive ginsenosides through cytochrome P450 inhibition/induction and UDP-glucuronosyltransferase inhibition. Food Chem. Toxicol. 2014, 68, 117–127. [Google Scholar] [CrossRef]
- Li, G.; Huang, K.; Nikolic, D.; van Breemen, R.B. High-throughput cytochrome P450 cocktail inhibition assay for assessing drug-drug and drug-botanical interactions. Drug Metab. Dispos. 2015, 43, 1670–1678. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, H.; Ji, H.K.; Lee, T.; Liu, K.H. Screening of ten cytochrome P450 enzyme activities with 12 probe substrates in human liver microsomes using cocktail incubation and liquid chromatography-tandem mass spectrometry. Biopharm. Drug Dispos. 2019, 40, 101–111. [Google Scholar] [CrossRef]
- Valicherla, G.R.; Mishra, A.; Lenkalapelly, S.; Jillela, B.; Francis, F.M.; Rajagopalan, L.; Srivastava, P. Investigation of the inhibition of eight major human cytochrome P450 isozymes by a probe substrate cocktail in vitro with emphasis on CYP2E1. Xenobiotica 2019, 49, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Wahlstrom, J.L.; Rock, D.A.; Warren, C.J.; Gorman, L.A.; Tracy, T.S. CYP2C9 inhibition: Impact of probe selection and pharmacogenetics on in vitro inhibition profiles. Drug Metab. Dispos. 2006, 34, 1966–1975. [Google Scholar] [CrossRef] [PubMed]
- Yasar, U.; Tybring, G.; Hidestrand, M.; Oscarson, M.; Ingelman-Sundberg, M.; Dahl, M.L.; Eliasson, E. Role of CYP2C9 polymorphism in losartan oxidation. Drug Metab. Dispos. 2001, 29, 1051–1056. [Google Scholar] [PubMed]
- Elsby, R.; Surry, D.D.; Smith, V.N.; Gray, A.J. Validation and application of Caco-2 assays for the in vitro evaluation of development candidate drugs as substrates or inhibitors of P-glycoprotein to support regulatory submissions. Xenobiotica 2008, 38, 1140–1164. [Google Scholar] [CrossRef] [PubMed]
- Markowska, M.; Oberle, R.; Juzwin, S.; Hsu, C.P.; Gryszkiewicz, M.; Streeter, A.J. Optimizing Caco-2 cell monolayers to increase throughput in drug intestinal absorption analysis. J. Pharmacol. Toxicol. Methods 2001, 46, 51–55. [Google Scholar] [CrossRef]
- Cho, M.A.; Yoon, J.G.; Kim, V.; Kim, H.; Lee, R.; Lee, M.G.; Kim, D. Functional characterization of pharmcogenetic variants of human cytochrome P450 2C9 in Korean populations. Biomol. Ther. (Seoul) 2019, 27, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.; Ryu, K.H.; Bae, S.K.; Kim, J. Simultaneous determination of trifolirhizin, (-)-maackiain, (-)-sophoranone, and 2-(2,4-dihydroxyphenyl)-5,6-methylenedioxybenzofuran from Sophora tonkinensis in rat plasma by liquid chromatography with tandem mass spectrometry and its application to a pharmacokinetic study. J. Sep. Sci. 2014, 37, 3235–3244. [Google Scholar] [CrossRef]
- Bourrié, M.; Meunier, V.; Berger, Y.; Fabre, G. Cytochrome P450 isoform inhibitors as a tool for the investigation of metabolic reactions catalyzed by human liver microsomes. J. Pharmacol. Exp. Ther. 1996, 277, 321–332. [Google Scholar]
- Obach, R.S.; Walsky, R.L.; Venkatakrishnan, K. Mechanism-based inactivation of human cytochrome P450 enzymes and the prediction of drug-drug interactions. Drug Metab. Dispos. 2006, 35, 246–255. [Google Scholar] [CrossRef]
- Parkinson, A.; Kazmi, F.; Buckley, D.B.; Yerino, P.; Paris, B.L.; Holsapple, J.; Toren, P.; Otradovec, S.M.; Ogilvie, B.W. An evaluation of the dilution method for identifying metabolism-dependent inhibitors of cytochrome P450 enzymes. Drug Metab. Dispos. 2011, 39, 1370–1387. [Google Scholar] [CrossRef]
- Stresser, D.M.; Mao, J.; Kenny, J.R.; Jones, B.C.; Grime, K. Exploring concepts of in vitro time-dependent CYP inhibition assays. Expert Opin. Drug Metab. Toxicol. 2014, 10, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, W. Herbs for clearing heat. In Chinese Materia Medica: Combinations and Applications, 1st ed.; Donica Publishing Ltd.: St. Albans, UK, 2002; p. 115. [Google Scholar]
- US Food and Drug Administration. Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteer. 2005. Available online: https://www.fda.gov/media/72309/download (accessed on 31 March 2020).
- Foti, R.S.; Rock, D.A.; Wienkers, L.C.; Wahlstrom, J.L. Selection of alternative CYP3A4 probe substrates for clinical drug interaction studies using in vitro data and in vivo simulation. Drug Metab. Dispos. 2010, 38, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Van Booven, D.; Marsh, S.; McLeod, H.; Carrillo, M.W.; Sangkuhl, K.; Klein, T.E.; Altman, R.B. Cytochrome P450 2C9-CYP2C9. Pharmacogenet. Genom. 2010, 20, 277–281. [Google Scholar] [CrossRef] [PubMed]
- European Medicine Agency. Guideline on the Investigation of Drug Interactions. 2012. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf (accessed on 31 March 2020).
- US Food and Drug Administration. Guidance for industry: In Vitro Drug Interaction Studies-Cytochrome P450 Enzyme-and Transporter-Mediated Drug Interactions. 2020. Available online: https://www.fda.gov/media/134582/download (accessed on 31 March 2020).
- Karjalainen, M.J.; Neuvonen, P.J.; Backman, J.T. Tolfenamic acid is a potent CYP1A2 inhibitor in vitro but does not interact in vivo: Correction for protein binding is needed for data interpretation. Eur. J. Clin. Pharmacol. 2007, 63, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, T.; Backman, J.T.; Neuvonen, M.; Niemi, M.; Neuvonen, P.J. Montelukast and zafirlukast do not affect the pharmacokinetics of the CYP2C8 substrate pioglitazone. Eur. J. Clin. Pharmacol. 2006, 62, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.A.; Park, P.W.; Kim, K.R.; Park, J.Y. Effect of multiple doses of montelukast on the pharmacokinetics of rosiglitazone, a CYP2C8 substrate, in humans. Br. J. Clin. Pharmacol. 2006, 63, 339–345. [Google Scholar] [CrossRef][Green Version]
- Walsky, R.L.; Obach, R.S.; Gaman, E.A.; Gleeson, J.P.; Proctor, W.R. Selective inhibition of human cytochrome P4502C8 by montelukast. Drug Metab. Dispos. 2005, 33, 413–418. [Google Scholar] [CrossRef]
- Press, B.; di Grandi, D. Permeability for intestinal absorption: Caco-2 assay and related issues. Curr. Drug Metab. 2008, 9, 893–900. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CYPs | Probe Substrates | Km (μM) | Metabolite | ESI a | Q1 Ion (m/z) | Q3 Ion (m/z) | Q1 Pre-bias (V) | CE b (eV) | Q3 Pre-bias (V) |
|---|---|---|---|---|---|---|---|---|---|
| 1A2 | Phenacetin | 50 | Acetaminophen | + | 152 | 110.2 | −14 | −12 | −19 |
| 2A6 | Coumarin | 5 | 7-Hydroxycoumarin | + | 163 | 107 | −15 | −35 | −15 |
| 2B6 | Bupropion | 50 | 6-Hydroxybupropion | + | 256 | 238 | −15 | −35 | −15 |
| 2C8 | Rosiglitazone | 10 | p-Hydroxyrosiglitaonze | + | 374 | 151 | −15 | −35 | −15 |
| 2C9 | Tolbutamide | 100 | 4-Hydroxytolbutamide | + | 287 | 87 | −15 | −35 | −15 |
| 2C19 | Omeprazole | 20 | 5-Hydroxyomeprazole | + | 362 | 214 | −13 | −13 | −22 |
| 2D6 | Dextromethorphan | 5 | Dextrorphan | + | 258 | 157 | −15 | −35 | −15 |
| 2E1 | Chlorzoxazone | 50 | 6-Hydroxychlorzoxazone | − | 184 | 119.9 | 18 | 15 | 24 |
| 3A4 | Midazolam | 2 | 1′-Hydroxymidazolam | + | 342 | 203 | −15 | −35 | −15 |
| Chlorpropamide (Internal standard) | 2 | + | 277 | 111 | −15 | −20 | −15 | ||
| − | 275 | 190 | 15 | 35 | 15 | ||||
| CYPs | IC50 Values (μM) | ||
|---|---|---|---|
| Well-Known Inhibitors | SPN | ||
| 1A2 | α-Naphthoflavone | 0.0458 ± 0.00694 | >50 a |
| 2A6 | Tryptamine | 2.98 ± 0.635 | >50 a |
| 2B6 | Ticlopidine | 2.19 ± 0.513 | >50 a |
| 2C8 | Quercetin | 8.51 ± 0.958 | 13.6 ± 3.15 |
| 2C9 | Sulfaphenazole | 0.677 ± 0.109 | 0.966 ± 0.149 |
| 2C19 | S-benzylnirvanol | 0.215 ± 0.0228 | 16.8 ± 3.21 |
| 2D6 | Quinidine | 0.127 ± 0.0192 | >50 |
| 2E1 | Diethyldithiocarbamate | 12.0 ± 3.67 | >50 a |
| 3A4 | Ketoconazole | 0.0404 ± 0.00821 | >50 |
| Parameters | Without SPN (n = 6) | With SPN (n = 6) |
|---|---|---|
| Diclofenac | ||
| AUCt (μg min/mL) a | 63.8 ± 6.28 | 69.4 ± 2.98 |
| AUC∞ (μg min/mL) b | 71.7 ± 9.16 | 80.8 ± 7.78 |
| t1/2 (min) c | 153 ± 60.1 | 173 ± 41.5 |
| Cmax (ng/mL) d | 882 ± 245 | 787 ± 104 |
| Tmax (min) e | 5 (3–5) | 5 (3–5) |
| 4′-hydroxydiclofenac | ||
| AUCt (μg min/mL) | 44.8 ± 6.38 | 44.5 ± 7.24 |
| AUC∞ (μg min/mL) | 68.6 ± 12.1 | 67.6 ± 12.9 |
| t1/2 (min) | 278 ± 70.0 | 296 ± 59.7 |
| Cmax (ng/mL) | 180 ± 55.2 | 173 ± 40.8 |
| Tmax (min) | 10 (10–30) | 15 (10–30) |
| Metabolic conversion ratio f | ||
| AUC∞, 4′-hydroxydiclofenac/AUC∞, diclofenac | 0.904 ± 0.0534 | 0.799 ± 0.167 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Y.F.; Bae, S.H.; Huang, Z.; Chae, S.U.; Jo, S.J.; Shim, H.J.; Lee, C.B.; Kim, D.; Yoo, H.; Bae, S.K. Lack of Correlation between In Vitro and In Vivo Studies on the Inhibitory Effects of (‒)-Sophoranone on CYP2C9 Is Attributable to Low Oral Absorption and Extensive Plasma Protein Binding of (‒)-Sophoranone. Pharmaceutics 2020, 12, 328. https://doi.org/10.3390/pharmaceutics12040328
Zheng YF, Bae SH, Huang Z, Chae SU, Jo SJ, Shim HJ, Lee CB, Kim D, Yoo H, Bae SK. Lack of Correlation between In Vitro and In Vivo Studies on the Inhibitory Effects of (‒)-Sophoranone on CYP2C9 Is Attributable to Low Oral Absorption and Extensive Plasma Protein Binding of (‒)-Sophoranone. Pharmaceutics. 2020; 12(4):328. https://doi.org/10.3390/pharmaceutics12040328
Chicago/Turabian StyleZheng, Yu Fen, Soo Hyeon Bae, Zhouchi Huang, Soon Uk Chae, Seong Jun Jo, Hyung Joon Shim, Chae Bin Lee, Doyun Kim, Hunseung Yoo, and Soo Kyung Bae. 2020. "Lack of Correlation between In Vitro and In Vivo Studies on the Inhibitory Effects of (‒)-Sophoranone on CYP2C9 Is Attributable to Low Oral Absorption and Extensive Plasma Protein Binding of (‒)-Sophoranone" Pharmaceutics 12, no. 4: 328. https://doi.org/10.3390/pharmaceutics12040328
APA StyleZheng, Y. F., Bae, S. H., Huang, Z., Chae, S. U., Jo, S. J., Shim, H. J., Lee, C. B., Kim, D., Yoo, H., & Bae, S. K. (2020). Lack of Correlation between In Vitro and In Vivo Studies on the Inhibitory Effects of (‒)-Sophoranone on CYP2C9 Is Attributable to Low Oral Absorption and Extensive Plasma Protein Binding of (‒)-Sophoranone. Pharmaceutics, 12(4), 328. https://doi.org/10.3390/pharmaceutics12040328