Acamprosate Is a Substrate of the Human Organic Anion Transporter (OAT) 1 without OAT3 Inhibitory Properties: Implications for Renal Acamprosate Secretion and Drug–Drug Interactions

 , , ,
, , , 
Abstract
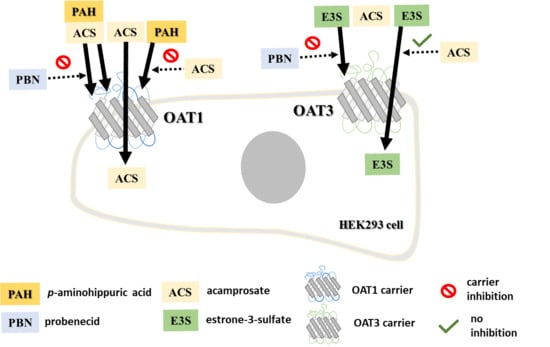
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Establishment of Stable Cell Lines
2.3. LC/MS-MS-Based Protein Quantification
2.4. Cell Cultivation
2.5. Buffers and Applied Solutions
2.6. Uptake Studies in HEK293-Flp-In OAT1, OAT3, and Mock Cells
2.7. Preincubation Assay
2.8. Cell Viability Assay
2.9. Acamprosate Intracellular Uptake and Inhibition of Uptake in HEK293-Flp-In OAT1, OAT3, and Mock Cells
2.10. Analytical Procedure
2.11. Data Treatment
2.12. Statistical Analysis
3. Results
3.1. Establishment and Validation of HEK293-Flp-In OAT1 Transfected Cells
3.2. Establishment and Validation of HEK293-Flp-In OAT3 Transfected Cells
3.3. Inhibition of OAT1 and OAT3 by Acamprosate and Standard Substrates
3.4. Evaluating If Acamprosate Is a Substrate for OAT1 and OAT3
3.5. Evaluation of Cell Toxicity upon Application of Substances
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
LC/MS-MS Method Validation
Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Adm. Route | Dose | Cmax | fu | Cmax,u | p-Aminohippuric Acid (PAH) | Acamprosate | Estrone-3-Sulfate | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 | DDI Index | IC50 | DDI Index | IC50 | DDI Index | ||||||
| mg | µM | µM | µM | µM | µM | ||||||
| Probenecid | Oral | 500 | 124 ± 30 [52] | 0.07 [53] | 8.66 ± 2.09 | 13 | 0.67 * | 11 | 0.79 * | 6 | 1.69 * |
| 1000 | 244 ± 94 [52] | 0.11 [53] | 26.83 ± 10.37 | 2.06 * | 2.44 * | 5.22 * | |||||
| 2000 | 521 ± 166 [52] | 0.17 [53] | 88.53 ± 28.24 | 6.81 * | 8.05 * | 17.23 * | |||||
| Acamprosate | IV bolus | 333 | 153 ± 38 [7] | 1 [5] | 153 ± 38 | 287–1229 | 0.12–0.53 * | - | >>10,000 | N.A. | |
| 666 | 214 ± 10 [6] | 214 ± 10 | 0.17–0.75 * | ||||||||
| 710 | 230 ± 64 [10] | 230 ± 64 | 0.19–0.80 * | ||||||||
| 1420 | 486 ± 106 [10] | 486 ± 106 | 0.40–1.69 * | ||||||||
| 2130 | 768 ± 149 [10] | 768 ± 149 | 0.62–2.68 * | ||||||||
| Oral solution | 333 | 1.8 ± 0.8 [9] | 1.8 ± 0.8 | 0.00–0.01 | |||||||
| 666 | 4.3 ± 1.4 [9] | 4.3 ± 1.4 | 0.00–0.01 | ||||||||
| 1332 | 5.0 ± 1.7 [9] | 5.0 ± 1.7 | 0.00–0.02 | ||||||||
| 2664 | 8.6 ± 4.1 [9] | 8.6 ± 4.1 | 0.01–0.03 | ||||||||
| Oral tablet | 666 | 1.4 ± 1.1 [6,54] | 1.4 ± 1.1 | 0.00–0.00 | |||||||
| 1332 | 1.5 ± 0.1 [55] | 1.5 ± 0.1 | 0.001–0.01 | ||||||||

References
- Mason, B.J.; Heyser, C.J. Acamprosate: A prototypic neuromodulator in the treatment of alcohol dependence. CNS Neurol. Disord. Drug Targets 2010, 9, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Approval Package for Application Number 21-431: Campral Clinical Pharmacology and Biopharmaceutics Review. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2004/21-431_Campral_BioPharmr.pdf (accessed on 13 August 2018).
- Rhee, Y.S.; Park, S.; Lee, T.W.; Park, C.W.; Nam, T.Y.; Oh, T.O.; Jeon, J.W.; Lee, D.S.; Park, E.S. Investigation of the relationship between in vitro and in vivo release behaviors of acamprosate from enteric-coated tablets. Arch. Pharm. Res. 2008, 31, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Scott. The Metabolism and Pharmacokinetics of 14C Aota-Ca in Man; NDA: 21-431; U.S. Food and Drug Administration: Silver Spring, MD, USA, 1990; p. 40.
- Chasseaud. Pharmacokinetics and Metabolism of 35-S-AOTA-Ca Following Oral Administration of Single Doses to Two Human Subjects; NDA: 21-431; U.S. Food and Drug Administration: Silver Spring, MD, USA, 1988; p. 39.
- Fourtillan, J.B. Pharmacokinetics of Acetylhomotaurinate (AOTA) in Young Healthy Subjects after Single and Multiple Oral Administration of Doses Equal to 666 mg of Calcium Acetylhomotaurinate (AOTA-Ca); U.S. Food and Drug Administration: Silver Spring, MD, USA, 1990; pp. 47–49.
- Caplain. Pharmacokinetic Study Following Intravenous Infusion of 333 mg of Acamprosate to Assess the Pharmacokinetic and Elimination Parameters of Acamprosate; NDA: 21-431; U.S. Food and Drug Administration: Silver Spring, MD, USA, 1998; pp. 41–42.
- Saivin, S.; Hulot, T.; Chabac, S.; Potgieter, A.; Durbin, P.; Houin, G. Clinical Pharmacokinetics of Acamprosate. Clin. Pharm. 1998, 35, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Dewland, P.M. A Rising Dose Tolerance and Pharmacokinetic Study of Calcium Bis Acetyl Homotaurine Following Single Oral Administration of a Solution at Four Dose Levels; U.S. Food and Drug Administration: Silver Spring, MD, USA, 1991; pp. 43–45.
- Jaillon, P.; Salvat, M. Pharmacokinetics Study of Intravenous Administration of AOTAL (Calcium acetylhomotaurinate) at Single Doses of 10, 20 and 30 mg/kg; NDA: 21-431; U.S. Food and Drug Administration: Silver Spring, MD, USA, 1991; p. 46.
- Theodor, R.A. Double-Blind, Placebo-Controlled, Multiple Rising Dose Bioavailability Study to Determine Tolerability, Safety and Pharmacokinetic Parameters under Steady State Conditions of Four Acamprosate Treatments (300 mg vs. 50 mg vs. 800 mg vs. 1000 mg Acamprosate Administered b.i.d as an Oral Aqueous Solution) in Four Groups of 15 Healthy Male Volunteers Each; U.S. Food and Drug Administration: Silver Spring, MD, USA, 1994; pp. 44–46.
- Zornoza, T.; Guerri, C.; Polache, A.; Granero, L. Disposition of acamprosate in the rat: Influence of probenecid. Biopharm. Drug Dispos. 2002, 23, 283–291. [Google Scholar] [CrossRef]
- FDA. Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. Available online: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers#table1 (accessed on 23 April 2020).
- Wright, S.H.; Dantzler, W.H. Molecular and cellular physiology of renal organic cation and anion transport. Physiol. Rev. 2004, 84, 987–1049. [Google Scholar] [CrossRef]
- Nozaki, Y.; Kusuhara, H.; Kondo, T.; Hasegawa, M.; Shiroyanagi, Y.; Nakazawa, H.; Okano, T.; Sugiyama, Y. Characterization of the uptake of organic anion transporter (OAT) 1 and OAT3 substrates by human kidney slices. J. Pharmacol. Exp. Ther. 2007, 321, 362–369. [Google Scholar] [CrossRef]
- Hasegawa, M.; Kusuhara, H.; Sugiyama, D.; Ito, K.; Ueda, S.; Endou, H.; Sugiyama, Y. Functional involvement of rat organic anion transporter 3 (rOat3; Slc22a8) in the renal uptake of organic anions. J. Pharmacol. Exp. Ther. 2002, 300, 746–753. [Google Scholar] [CrossRef]
- Ullrich, K.J.; Rumrich, G. Contraluminal Transport-Systems in the Proximal Renal Tubule Involved in Secretion of Organic-Anions. Am. J. Physiol. 1988, 254, F453–F462. [Google Scholar] [CrossRef] [PubMed]
- Antonescu, I.E.; Rasmussen, K.F.; Neuhoff, S.; Frette, X.; Karlgren, M.; Bergstrom, C.A.S.; Nielsen, C.U.; Steffansen, B. The Permeation of Acamprosate Is Predominantly Caused by Paracellular Diffusion across Caco-2 Cell Monolayers: A Paracellular Modeling Approach. Mol. Pharm. 2019, 16, 4636–4650. [Google Scholar] [CrossRef]
- Karlgren, M.; Ahlin, G.; Bergstrom, C.A.; Svensson, R.; Palm, J.; Artursson, P. In vitro and in silico strategies to identify OATP1B1 inhibitors and predict clinical drug-drug interactions. Pharm. Res. 2012, 29, 411–426. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Mann, M. Consecutive proteolytic digestion in an enzyme reactor increases depth of proteomic and phosphoproteomic analysis. Anal. Chem. 2012, 84, 2631–2637. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Gaugaz, F.Z. Fast and sensitive total protein and Peptide assays for proteomic analysis. Anal. Chem. 2015, 87, 4110–4116. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Rakus, D. Multi-enzyme digestion FASP and the ‘Total Protein Approach’-based absolute quantification of the Escherichia coli proteome. J. Proteom. 2014, 109, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Yung-Chi, C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition ( I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- FDA. Vitro Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry; Center for Drug Evaluation and Research, U.S. Department of Health and Human Services Food and Drug Administration: Silver Spring, MD, USA, 2020; pp. 1–43.
- Hosoyamada, M.; Sekine, T.; Kanai, Y.; Endou, H. Molecular cloning and functional expression of a multispecific organic anion transporter from human kidney. Am. J. Physiol. 1999, 276, F122–F128. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.H.; Sekine, T.; Fukushima, J.I.; Kanai, Y.; Kobayashi, Y.; Goya, T.; Endou, H. Identification and characterization of human organic anion transporter 3 expressing predominantly in the kidney. Mol. Pharmacol. 2001, 59, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Tatrai, P.; Schweigler, P.; Poller, B.; Domange, N.; de Wilde, R.; Hanna, I.; Gaborik, Z.; Huth, F. A Systematic In Vitro Investigation of the Inhibitor Preincubation Effect on Multiple Classes of Clinically Relevant Transporters. Drug Metab. Dispos. 2019, 47, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Nigam, S.K.; Bush, K.T.; Martovetsky, G.; Ahn, S.Y.; Liu, H.C.; Richard, E.; Bhatnagar, V.; Wu, W. The organic anion transporter (OAT) family: A systems biology perspective. Physiol. Rev. 2015, 95, 83–123. [Google Scholar] [CrossRef]
- Parvez, M.M.; Kaisar, N.; Shin, H.J.; Jung, J.A.; Shin, J.G. Inhibitory Interaction Potential of 22 Antituberculosis Drugs on Organic Anion and Cation Transporters of the SLC22A Family. Antimicrob. Agents Chemother. 2016, 60, 6558–6567. [Google Scholar] [CrossRef]
- Cheng, Y.; Vapurcuyan, A.; Shahidullah, M.; Aleksunes, L.M.; Pelis, R.M. Expression of organic anion transporter 2 in the human kidney and its potential role in the tubular secretion of guanine-containing antiviral drugs. Drug Metab. Dispos. 2012, 40, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Ueo, H.; Motohashi, H.; Katsura, T.; Inui, K. Human organic anion transporter hOAT3 is a potent transporter of cephalosporin antibiotics, in comparison with hOAT1. Biochem. Pharmacol. 2005, 70, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Tahara, H.; Shono, M.; Kusuhara, H.; Kinoshita, H.; Fuse, E.; Takadate, A.; Otagiri, M.; Sugiyama, Y. Molecular cloning and functional analyses of OAT1 and OAT3 from cynomolgus monkey kidney. Pharm. Res. 2005, 22, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, A.G.; Gao, T.; Khan, U.; Berrigan, L.; Li, M.; Ingraham, L.; Pelis, R.M. Organic Anion Transporter 1 Is Inhibited by Multiple Mechanisms and Shows a Transport Mode Independent of Exchange. Drug Metab. Dispos. 2015, 43, 1847–1854. [Google Scholar] [CrossRef]
- Ho, E.S.; Lin, D.C.; Mendel, D.B.; Cihlar, T. Cytotoxicity of antiviral nucleotides adefovir and cidofovir is induced by the expression of human renal organic anion transporter 1. J. Am. Soc. Nephrol. 2000, 11, 383–393. [Google Scholar]
- Takeda, M.; Narikawa, S.; Hosoyamada, M.; Cha, S.H.; Sekine, T.; Endou, H. Characterization of organic anion transport inhibitors using cells stably expressing human organic anion transporters. Eur. J. Pharmacol. 2001, 419, 113–120. [Google Scholar] [CrossRef]
- Takeda, M.; Hosoyamada, M.; Cha, S.H.; Sekine, T.; Endou, H. Hydrogen peroxide downregulates human organic anion transporters in the basolateral membrane of the proximal tubule. Life Sci. 2000, 68, 679–687. [Google Scholar] [CrossRef]
- Sekine, T.; Watanabe, N.; Hosoyamada, M.; Kanai, Y.; Endou, H. Expression cloning and characterization of a novel multispecific organic anion transporter. J. Biol. Chem. 1997, 272, 18526–18529. [Google Scholar] [CrossRef]
- Shen, H.; Yang, Z.; Zhao, W.; Zhang, Y.; Rodrigues, A.D. Assessment of vandetanib as an inhibitor of various human renal transporters: Inhibition of multidrug and toxin extrusion as a possible mechanism leading to decreased cisplatin and creatinine clearance. Drug Metab. Dispos. 2013, 41, 2095–2103. [Google Scholar] [CrossRef]
- Elsby, R.; Fox, L.; Stresser, D.; Layton, M.; Butters, C.; Sharma, P.; Smith, V.; Surry, D. In vitro risk assessment of AZD9056 perpetrating a transporter-mediated drug-drug interaction with methotrexate. Eur. J. Pharm. Sci. 2011, 43, 41–49. [Google Scholar] [CrossRef]
- Takeda, M.; Khamdang, S.; Narikawa, S.; Kimura, H.; Kobayashi, Y.; Yamamoto, T.; Cha, S.H.; Sekine, T.; Endou, H. Human organic anion transporters and human organic cation transporters mediate renal antiviral transport. J. Pharmacol. Exp. Ther. 2002, 300, 918–924. [Google Scholar] [CrossRef]
- Cutler, M.J.; Urquhart, B.L.; Velenosi, T.J.; Meyer Zu Schwabedissen, H.E.; Dresser, G.K.; Leake, B.F.; Tirona, R.G.; Kim, R.B.; Freeman, D.J. In vitro and in vivo assessment of renal drug transporters in the disposition of mesna and dimesna. J. Clin. Pharmacol. 2012, 52, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Uwai, Y.; Taniguchi, R.; Motohashi, H.; Saito, H.; Okuda, M.; Inui, K.-i. Methotrexate-Loxoprofen Interaction: Involvement of Human Organic Anion Transporters hOAT1 and hOAT3. Drug Metab. Pharmacokinet. 2004, 19, 369–374. [Google Scholar] [CrossRef]
- Lee, S.C.; Arya, V.; Yang, X.; Volpe, D.A.; Zhang, L. Evaluation of transporters in drug development: Current status and contemporary issues. Adv. Drug Deliv. Rev. 2017, 116, 100–118. [Google Scholar] [CrossRef]
- Prasad, B.; Johnson, K.; Billington, S.; Lee, C.; Chung, G.W.; Brown, C.D.; Kelly, E.J.; Himmelfarb, J.; Unadkat, J.D. Abundance of Drug Transporters in the Human Kidney Cortex as Quantified by Quantitative Targeted Proteomics. Drug Metab. Dispos. 2016, 44, 1920–1924. [Google Scholar] [CrossRef]
- Sharma, P.; Atari, I.M.; Elsby, R.; Thomas, S.; Stahl, S.; Hilgendorf, C.; Fenner, K. Chapter 7. The Characteristics, Validation and Applications of In silico and In vitro Models of Drug Transporters. In Drug Transporters: Volume 1: Role and Importance in ADME and Drug Development; Nichols, G., Youdim, K., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2016; Volume 1, pp. 231–297. [Google Scholar]
- Khamdang, S.; Takeda, M.; Noshiro, R.; Narikawa, S.; Enomoto, A.; Anzai, N.; Piyachaturawat, P.; Endou, H. Interactions of human organic anion transporters and human organic cation transporters with nonsteroidal anti-inflammatory drugs. J. Pharmacol. Exp. Ther. 2002, 303, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Stecula, A.; Gupta, A.; Prasad, B.; Chien, H.C.; Yee, S.W.; Wang, L.; Unadkat, J.D.; Stahl, S.H.; Fenner, K.S.; et al. Molecular Mechanisms for Species Differences in Organic Anion Transporter 1, OAT1: Implications for Renal Drug Toxicity. Mol. Pharmacol. 2018, 94, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, K.M.; Stocker, S.L.; Wittwer, M.B.; Xu, L.; Giacomini, K.M. Renal transporters in drug development. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 503–529. [Google Scholar] [CrossRef]
- Morrissey, K.M.; Wen, C.C.; Johns, S.J.; Zhang, L.; Huang, S.M.; Giacomini, K.M. The UCSF-FDA TransPortal: A public drug transporter database. Clin. Pharmacol. Ther. 2012, 92, 545–546. [Google Scholar] [CrossRef]
- FDA. Bioanalytical Method Validation Guidance for Industry; Center for Drug Evaluation and Research, U.S. Department of Health and Human Services: Silver Spring, MD, USA, 2018; pp. 1–41.
- Selen, A.; Amidon, G.L.; Welling, P.G. Pharmacokinetics of probenecid following oral doses to human volunteers. J. Pharm. Sci. 1982, 71, 1238–1242. [Google Scholar] [CrossRef]
- Dayton, P.G.; Yu, T.F.; Chen, W.; Berger, L.; West, L.A.; Gutman, A.B. The physiological disposition of probenecid, including renal clearance, in man, studied by an improved method for its estimation in biological material. J. Pharmacol. Exp. Ther. 1963, 140, 278–286. [Google Scholar] [PubMed]
- Sennesael, J. Acamprosate Pharmacokinetic Study after Single Oral Administration of Two Acamprosate Tablets (2 × 333 mg) to Subject with Normal or Impaired Renal Function; U.S. Food and Drug Administration: Silver Spring, MD, USA, 1991; pp. 63–64.
- Dewland, P.M. Report of an Investigation of the Effect of Ethanol upon the Pharmacokinetics of Acamprosate; NDA: 21-431; U.S. Food and Drug Administration: Silver Spring, MD, USA, 1990; pp. 68–69.








| Substrate | p-Aminohippuric Acid 2 | Acamprosate 1 | Estrone-3-Sulfate 2 |
|---|---|---|---|
| Carrier | OAT1 | OAT1 | OAT3 |
| Km (µM) | 43 ± 13 | 698 ± 192 | 26 ± 6 |
| Vmax (pmol∙mg protein−1∙min−1) | 429 ± 39 | 1028 ± 99 | |
| Vmax (fmol∙cm−2∙min−1) | 8.8 ± 0.7 | ||
| CLT (cm/min) | 5.6 × 10−9 | ||
| CLT (µL∙mg protein−1∙min−1) | 10 | 1.5 | |
| P (µL∙mg protein−1∙min−1) | 0.35 ± 0.04 |
| Substrate | p-Aminohippuric Acid | Acamprosate | Estrone-3-Sulfate | ||
|---|---|---|---|---|---|
| Solute Carrier | OAT1 | OAT1 | OAT3 | ||
| Inhibitor | probenecid | IC50 (µM) (log IC50 ± log S.E.M.) | 16 1 (1.20 ± 0.26) | 14 1 (1.14 ± 0.27) | 6 2 (0.77 ± 0.22) |
| Ki (µM) | 13 1 | 11 1 | 6 2 | ||
| DDI index | 0.67–6.81 a | 0.79–8.05 a | 1.69–17.23 a | ||
| acamprosate | IC50 (µM) (log IC50 ± log S.E.M.) IC50 (µM) (log IC50 ± log S.E.M.) IC50 (µM), 3 h (log IC50 ± log S.E.M.) | 1391 1 (3.1 ± 0.1) 638 2 (2.8 ± 0.2) 582 2 (2.8 ± 0.2) | - | >10,000 1,2 | |
| Ki (µM) Ki (µM) Ki (µM) | 1140 1 315 2 287 2 | - | - | ||
| DDI index | 0.001–0.008 a 0.14–0.70 b | - | <<0.02 a, b | ||
| Substrate—PAH | 0 (µM) Acamprosate | 625 (µM) Acamprosate | 1250 (µM) Acamprosate | 25,000 (µM) Acamprosate |
|---|---|---|---|---|
| Km, app (µM) | 5.6 ± 1.4 | 11.9 ± 3.2 | 17.9 ± 3.2 | 36.5 ± 7.4 * |
| Vmax, app (pmol∙cm−2∙min−1) | 21.1 ± 1.1 | 23.4 ± 1.7ns | 24.4 ± 1.3ns | 27.6 ± 2.1ns |
| Km (µM) | 7.7 ± 1.3 | |||
| Vmax (pmol∙cm−2∙min−1) | 23.2 ± 0.7 | |||
| Ki (µM) | 1229 ± 341 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antonescu, I.E.; Karlgren, M.; Pedersen, M.L.; Simoff, I.; Bergström, C.A.S.; Neuhoff, S.; Artursson, P.; Steffansen, B.; Nielsen, C.U. Acamprosate Is a Substrate of the Human Organic Anion Transporter (OAT) 1 without OAT3 Inhibitory Properties: Implications for Renal Acamprosate Secretion and Drug–Drug Interactions. Pharmaceutics 2020, 12, 390. https://doi.org/10.3390/pharmaceutics12040390
Antonescu IE, Karlgren M, Pedersen ML, Simoff I, Bergström CAS, Neuhoff S, Artursson P, Steffansen B, Nielsen CU. Acamprosate Is a Substrate of the Human Organic Anion Transporter (OAT) 1 without OAT3 Inhibitory Properties: Implications for Renal Acamprosate Secretion and Drug–Drug Interactions. Pharmaceutics. 2020; 12(4):390. https://doi.org/10.3390/pharmaceutics12040390
Chicago/Turabian StyleAntonescu, Irina E., Maria Karlgren, Maria L. Pedersen, Ivailo Simoff, Christel A. S. Bergström, Sibylle Neuhoff, Per Artursson, Bente Steffansen, and Carsten Uhd Nielsen. 2020. "Acamprosate Is a Substrate of the Human Organic Anion Transporter (OAT) 1 without OAT3 Inhibitory Properties: Implications for Renal Acamprosate Secretion and Drug–Drug Interactions" Pharmaceutics 12, no. 4: 390. https://doi.org/10.3390/pharmaceutics12040390
APA StyleAntonescu, I. E., Karlgren, M., Pedersen, M. L., Simoff, I., Bergström, C. A. S., Neuhoff, S., Artursson, P., Steffansen, B., & Nielsen, C. U. (2020). Acamprosate Is a Substrate of the Human Organic Anion Transporter (OAT) 1 without OAT3 Inhibitory Properties: Implications for Renal Acamprosate Secretion and Drug–Drug Interactions. Pharmaceutics, 12(4), 390. https://doi.org/10.3390/pharmaceutics12040390