In-Depth Characterization of EpiIntestinal Microtissue as a Model for Intestinal Drug Absorption and Metabolism in Human


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Material
2.2. Cell Culture
2.3. Bidirectional Permeability Assay
2.4. Measurement of DME Activities in EpiIntestinal and Caco-2
2.5. CES-Mediated Metabolism of Dabigatran Etexilate
2.6. Metabolite Identification
2.7. Measurement of Intestinal First-Pass Availability in EpiIntestinal Microtissues and Caco-2
2.8. Calculation of Fa × Fg in Human
3. Results
3.1. Barrier Function and Transporter Activities
3.2. Drug-Metabolising Enzymes (DME) in EpiIntestinal Microtissues
3.3. Differential Expression of CES1 and CES2 in EpiIntestinal Microtissues and Caco-2 cells
3.4. UGT and SULT Activities in EpiIntestinal Microtissues
3.5. Prediction of Fa × Fg in Human using EpiIntestinal Microtissues
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lennernäs, H.; Palm, K.; Fagerholm, U.; Artursson, P. Comparison between active and passive drug transport in human intestinal epithelial (caco-2) cells in vitro and human jejunum in vivo. Int. J. Pharm. 1996, 127, 103–107. [Google Scholar] [CrossRef]
- Kerns, E.H.; Di, L.; Petusky, S.; Farris, M.; Ley, R.; Jupp, P. Combined Application of Parallel Artificial Membrane Permeability Assay and Caco-2 Permeability Assays in Drug Discovery. J. Pharm. Sci. 2004, 93, 1440–1453. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Kulkarni, K.; Basu, S.; Zhang, S.; Hu, M. First-Pass Metabolism via UDP-Glucuronosyltransferase: A Barrier to Oral Bioavailability of Phenolics. J. Pharm. Sci. 2011, 100, 3655–3681. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, N.; Kishimoto, W.; Volz, A.; Ludwig-Schwellinger, E.; Ebner, T.; Schaefer, O. Impact of Endogenous Esterase Activity on In Vitro P-Glycoprotein Profiling of Dabigatran Etexilate in Caco-2 Monolayers. Drug Metab. Dispos. 2013, 42, 250–256. [Google Scholar] [CrossRef] [Green Version]
- Prueksaritanont, T.; Gorham, L.M.; Hochman, J.H.; O Tran, L.; Vyas, K.P. Comparative studies of drug-metabolizing enzymes in dog, monkey, and human small intestines, and in Caco-2 cells. Drug Metab. Dispos. 1996, 24, 634–642. [Google Scholar]
- Rozehnal, V.; Nakai, D.; Hoepner, U.; Fischer, T.; Kamiyama, E.; Takahashi, M.; Yasuda, S.; Mueller, J. Human small intestinal and colonic tissue mounted in the Ussing chamber as a tool for characterizing the intestinal absorption of drugs. Eur. J. Pharm. Sci. 2012, 46, 367–373. [Google Scholar] [CrossRef]
- Takenaka, T.; Kazuki, K.; Harada, N.; Kuze, J.; Chiba, M.; Iwao, T.; Matsunaga, T.; Abe, S.; Oshimura, M.; Kazuki, Y. Development of Caco-2 cells co-expressing CYP3A4 and NADPH-cytochrome P450 reductase using a human artificial chromosome for the prediction of intestinal extraction ratio of CYP3A4 substrates. Drug Metab. Pharmacokinet. 2017, 32, 61–68. [Google Scholar] [CrossRef]
- Gertz, M.; Harrison, A.; Houston, J.B.; Galetin, A. Prediction of Human Intestinal First-Pass Metabolism of 25 CYP3A Substrates from In Vitro Clearance and Permeability Data. Drug Metab. Dispos. 2010, 38, 1147–1158. [Google Scholar] [CrossRef]
- Bein, A.; Shin, W.; Jalili-Firoozinezhad, S.; Park, M.H.; Sontheimer-Phelps, A.; Tovaglieri, A.; Chalkiadaki, A.; Kim, H.J.; Ingber, D. Microfluidic Organ-on-a-Chip Models of Human Intestine. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Rahmani, S.; Breyner, N.; Su, H.-M.; Verdu, E.F.; Didar, T.F. Intestinal organoids: A new paradigm for engineering intestinal epithelium in vitro. Biomater. 2019, 194, 195–214. [Google Scholar] [CrossRef]
- Ayehunie, S.; Landry, T.; Stevens, Z.; Armento, A.; Hayden, P.; Klausner, M. Human Primary Cell-Based Organotypic Microtissues for Modeling Small Intestinal Drug Absorption. Pharm. Res. 2018, 35, 72. [Google Scholar] [CrossRef] [PubMed]
- Madden, L.R.; Nguyen, T.V.; Garcia-Mojica, S.; Shah, V.; Le, A.V.; Peier, A.; Visconti, R.; Parker, E.M.; Presnell, S.C.; Nguyen, D.G.; et al. Bioprinted 3D Primary Human Intestinal Tissues Model Aspects of Native Physiology and ADME/Tox Functions. iScience 2018, 2, 156–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieger, P.; Cui, Y.; Scheuerer, S. pH-dependent solubility and permeability profiles: A useful tool for prediction of oral bioavailability. Eur. J. Pharm. Sci. 2017, 105, 82–90. [Google Scholar] [CrossRef]
- Cui, Y.; Lotz, R.; Rapp, H.; Klinder, K.; Himstedt, A.; Sauer, A. Muscle to Brain Partitioning as Measure of Transporter-Mediated Efflux at the Rat Blood–Brain Barrier and Its Implementation into Compound Optimization in Drug Discovery. Pharmaceutics 2019, 11, 595. [Google Scholar] [CrossRef] [Green Version]
- Obach, R.S.; Lombardo, F.; Waters, N. Trend Analysis of a Database of Intravenous Pharmacokinetic Parameters in Humans for 670 Drug Compounds. Drug Metab. Dispos. 2008, 36, 1385–1405. [Google Scholar] [CrossRef] [Green Version]
- Varma, M.V.S.; Feng, B.; Obach, R.S.; Troutman, M.D.; Chupka, J.; Miller, H.R.; El-Kattan, A. Physicochemical Determinants of Human Renal Clearance. J. Med. Chem. 2009, 52, 4844–4852. [Google Scholar] [CrossRef]
- Gibson, D.M.; Stern, R.H.; Abel, R.B.; Whitfield, L.R. Absolute bioavailability of atorvastatin in man. Pharm. Res. 1997, 14, S253. [Google Scholar]
- Stern, R.H.; Yang, B.-B.; Horton, M.; Moore, S.; Abel, R.B.; Olson, S.C. Renal dysfunction does not alter the pharmacokinetics or LDL-cholesterol reduction of atorvastatin. J. Clin. Pharmacol. 1997, 37, 816–819. [Google Scholar] [CrossRef]
- Mahmood, I.; Sahajwalla, C. Clinical Pharmacokinetics and Pharmacodynamics of Buspirone, an Anxiolytic Drug. Clin. Pharmacokinet. 1999, 36, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Yeh, K.C.; Stone, J.A.; Carides, A.D.; Rolan, P.; Woolf, E.; Ju, W.D. Simultaneous investigation of indinavir nonlinear pharmacokinetics and bioavailability in healthy volunteers using stable isotope labeling technique: Study design and model-independent data analysis. J. Pharm. Sci. 1999, 88, 568–573. [Google Scholar] [CrossRef]
- Combes, O.; Barré, J.; Duché, J.-C.; Vernillet, L.; Archimbaud, Y.; Marietta, M.P.; Tillement, J.-P.; Urien, S. In vitro binding and partitioning of irinotecan (CPT-11) and its metabolite, SN-38, in human blood. Investig. New Drugs 2000, 18, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, H.; Takanaka, K.; Abe, K.; Fukazawa, I.; Ishizuka, H. Stereoselective pharmacokinetics of oxybutynin and N -desethyloxybutynin in vitro and in vivo. Xenobiotica 2007, 37, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Martin, P. Absolute oral bioavailability of rosuvastatin in healthy white adult male volunteers. Clin. Ther. 2003, 25, 2553–2563. [Google Scholar] [CrossRef]
- Nordt, S.P.; Clark, R.F. Midazolam: A review of therapeutic uses and toxicity. J. Emerg. Med. 1997, 15, 357–365. [Google Scholar] [CrossRef]
- Lee, C.A.; Neul, D.; Clouser-Roche, A.; Dalvie, D.; Wester, M.R.; Jiang, Y.; Jones, J.P.; Freiwald, S.; Zientek, M.; Totah, R.A. Identification of Novel Substrates for Human Cytochrome P450 2J2. Drug Metab. Dispos. 2009, 38, 347–356. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, S.; Hirama, T.; Matsubara, T.; Nagata, K.; Yamazoe, Y. Involvement of CYP2J2 on the intestinal first-pass metabolism of antihistamine drug, astemizole. Drug Metab. Dispos. 2002, 30, 1240–1245. [Google Scholar] [CrossRef]
- Walsky, R.L.; Obach, R.S.; Hyland, R.; Kang, P.; Zhou, S.; West, M.; Geoghegan, K.F.; Helal, C.J.; Walker, G.; Goosen, T.C.; et al. Selective Mechanism-Based Inactivation of CYP3A4 by CYP3cide (PF-04981517) and Its Utility as an In Vitro Tool for Delineating the Relative Roles of CYP3A4 versus CYP3A5 in the Metabolism of Drugs. Drug Metab. Dispos. 2012, 40, 1686–1697. [Google Scholar] [CrossRef] [Green Version]
- Dalvie, D.; Kang, P.; Zientek, M.; Xiang, C.; Zhou, S.; Obach, R.S. Effect of Intestinal Glucuronidation in Limiting Hepatic Exposure and Bioactivation of Raloxifene in Humans and Rats. Chem. Res. Toxicol. 2008, 21, 2260–2271. [Google Scholar] [CrossRef]
- Kemp, D.C.; Fan, P.W.; Stevens, J.C.; Hill, G.; Cihlar, T.; Oo, C.; Ho, E.S.; Prior, K.; Wiltshire, H.; Barrett, J.; et al. Characterization of Raloxifene Glucuronidation in Vitro: Contribution of Intestinal Metabolism to Presystemic Clearance. Drug Metab. Dispos. 2002, 30, 694–700. [Google Scholar] [CrossRef] [Green Version]
- Van Heek, M.; Farley, C.; Compton, D.S.; Hoos, L.; Alton, K.B.; Sybertz, E.J.; Davis, H.R. Comparison of the activity and disposition of the novel cholesterol absorption inhibitor, SCH58235, and its glucuronide, SCH60663. Br. J. Pharmacol. 2000, 129, 1748–1754. [Google Scholar] [CrossRef]
- Allen, J.D.; Van Loevezijn, A.; Lakhai, J.M.; Van Der Valk, M.; Van Tellingen, O.; Reid, G.; Schellens, J.; Koomen, G.-J.; Schinkel, A.H. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol. Cancer Ther. 2002, 1, 417–425. [Google Scholar] [PubMed]
- Luo, F.R.; Paranjpe, P.V.; Guo, A.; Rubin, E.; Sinko, P.J. Intestinal transport of irinotecan in Caco-2 cells and MDCK II cells overexpressing efflux transporters Pgp, cMOAT, and MRP1. Drug Metab. Dispos. 2002, 30, 763–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepard, R.L.; Cao, J.; Starling, J.J.; Dantzig, A.H. Modulation of P-glycoprotein but not MRP1- or BCRP-mediated drug resistance by LY335979. Int. J. Cancer 2002, 103, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, S.; Maeda, K.; Wang, Y.; Sugiyama, Y. Involvement of Multiple Transporters in the Hepatobiliary Transport of Rosuvastatin. Drug Metab. Dispos. 2008, 36, 2014–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brück, S.; Strohmeier, J.; Busch, D.; Droździk, M.; Oswald, S. Caco-2 cells-expression, regulation and function of drug transporters compared with human jejunal tissue. Biopharm. Drug Dispos. 2016, 38, 115–126. [Google Scholar] [CrossRef]
- Nakamura, K.; Hirayama-Kurogi, M.; Ito, S.; Kuno, T.; Yoneyama, T.; Obuchi, W.; Terasaki, T.; Ohtsuki, S. Large-scale multiplex absolute protein quantification of drug-metabolizing enzymes and transporters in human intestine, liver, and kdney microsomes by SWATH-MS: Comparison with MRM/SRM and HR-MRM/PRM. Proteomics 2016, 16, 2106–2117. [Google Scholar] [CrossRef]
- Uchida, Y.; Ohtsuki, S.; Kamiie, J.; Ohmine, K.; Iwase, R.; Terasaki, T. Quantitative targeted absolute proteomics for 28 human transporters in plasma membrane of Caco-2 cell monolayer cultured for 2, 3, and 4 weeks. Drug Metab. Pharmacokinet. 2015, 30, 205–208. [Google Scholar] [CrossRef]
- Elsby, R.; Martin, P.; Surry, D.; Sharma, P.; Fenner, K. Solitary Inhibition of the Breast Cancer Resistance Protein Efflux Transporter Results in a Clinically Significant Drug-Drug Interaction with Rosuvastatin by Causing up to a 2-Fold Increase in Statin Exposure. Drug Metab. Dispos. 2015, 44, 398–408. [Google Scholar] [CrossRef] [Green Version]
- Paine, M.F.; Hart, H.L.; Ludington, S.S.; Haining, R.L.; Rettie, A.E.; Zeldin, D.C. THE HUMAN INTESTINAL CYTOCHROME P450 “PIE”. Drug Metab. Dispos. 2006, 34, 880–886. [Google Scholar] [CrossRef]
- Hui, Y.; Luo, L.; Zhang, L.; Kurogi, K.; Zhou, C.; Sakakibara, Y.; Suiko, M.; Liu, M.-C. Sulfation of afimoxifene, endoxifen, raloxifene, and fulvestrant by the human cytosolic sulfotransferases (SULTs): A systematic analysis. J. Pharmacol. Sci. 2015, 128, 144–149. [Google Scholar] [CrossRef] [Green Version]
- Falany, C.N.; Pilloff, D.E.; Leyh, T.S.; Falany, C.N. Sulfation of raloxifene and 4-hydroxytamoxifen by human cytosolic sulfotransferases. Drug Metab. Dispos. 2005, 34, 361–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, E.J.; Lin, H.; Hu, M. Disposition Mechanisms of Raloxifene in the Human Intestinal Caco-2 Model. J. Pharmacol. Exp. Ther. 2004, 310, 376–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, S.; Lau, A.J. Inhibition of Human Sulfotransferase 2A1-Catalyzed Sulfonation of Lithocholic Acid, Glycolithocholic Acid, and Taurolithocholic Acid by Selective Estrogen Receptor Modulators and Various Analogs and Metabolites. J. Pharmacol. Exp. Ther. 2019, 369, 389–405. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Alam, N.; Amaral, K.; Ho, M.-C.D.; Loretz, C.; Mitchell, W.; Yang, Q. Cryopreserved Human Intestinal Mucosal Epithelium: A Novel In Vitro Experimental System for the Evaluation of Enteric Drug Metabolism, Cytochrome P450 Induction, and Enterotoxicity. Drug Metab. Dispos. 2018, 46, 1562–1571. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.; Doshi, U.; Vuong, P.; Liu, N.; Tey, S.; Le, H.; Kosaka, M.; Kelly, J.R.; Li, A.; Yan, Z. Utility of Pooled Cryopreserved Human Enterocytes as an In vitro Model for Assessing Intestinal Clearance and Drug-Drug Interactions. Drug Metab. Lett. 2018, 12, 3–13. [Google Scholar] [CrossRef]
- Helander, H.F.; Fändriks, L. Surface area of the digestive tract–revisited. Scand. J. Gastroenterol. 2014, 49, 681–689. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | DME/Metabolite | Internal Standard | Drug Concentration (µM) | Solvent |
|---|---|---|---|---|
| Phenacetin | CYP1A2/Acetaminophen | d4-Acetaminophen | 10–100 | 20% ACN |
| Bupropion | CYP2B6/2-OH-Bupropion | d8-OH-bupropion | 15–300 | Aqua bidest. |
| Amodiaquine | CYP2C8/OH-Desethyl-Amodiaquine | d5-Desethylamodiaquine | 20–200 | Aqua bidest. |
| Diclofenac | CYP2C9/4-OH-Diclofenac | (13C6)4′-OH-Diclofenac | 20–200 | 20% ACN |
| S-Mephenytoin | CYP2C19/4-OH-Mephenytoin | d3-OH-Mephenytoin | 20–200 | 40% ACN |
| Testosterone | CYP3A4/6β-OH-Testosterone | d3-6β-OH-Testosterone | 40–400 | ACN/MeOH |
| Midazolam | CYP3A4/1-OH-Midazolam | d4-1-OH-Midazolam | 5–100 | Ready-to-use solution |
| Dextromethorphan | CYP2D6 Dextrorphan | d3-Dextrorphan | 10–100 | Aqua bidest. |
| 7-OH-Coumarin | UGT/7-OH-Coumarin-Glucuronid | α-Naphtylglucuronid | 15–150 | 40% ACN |
| 7-OH-Coumarin | SULT/7-OH-Coumarin-Sulfat | α-Naphtylglucuronid | 15–150 | 40% ACN |
| β-Estradiol | UGT1A1/β-Estradiol-3-Glucuronid | α-Naphtylglucuronid | 20–200 | DMSO |
| Astemizol | CYP2J2/O-Desmethyl-Astemizol | Dextrorphan tartrate | 2–50 | 30% ACN + 10 mM HCl |
| BIBF1120 | CES/BIBF1202 | d8-BIBF1202 | 10–100 | ACN/MeOH |
| Drug | F | RB | CLp (mL/min/kg) | fe |
|---|---|---|---|---|
| Atenolol | 0.5 [8] | 0.95 # | 2.5 [15] | 1 [16] |
| Atorvastatin | 0.14 [17] | 0.85 # | 8.93 [8] | 0.01 [18] |
| Buspirone | 0.05 [8] | 0.81 [8] | 28.3 [8] | 0.45 [19] |
| Felodipine | 0.15 [8] | 0.7 [8] | 11 [15] | 0 [16] |
| Indinavir | 0.6 [8] | 0.84 [8] | 18 [20] | 0.085 [16] |
| Irinotecan | 0.25 [8] | 0.82 [21] | 7 [15] | 0.32 [16] |
| Midazolam | 0.4 [8] | 0.64 # | 5.3 [15] | 0 [16] |
| Nifedipine | 0.9 [8] | 0.67 [8] | 7.3 [15] | 0 [16] |
| Oxybutynin | 0.06 [8] | 0.686 [22] | 5.1 [15] | 0 * |
| Quinidine | 0.9 [8] | 0.87 [8] | 4 [15] | 0.15 [16] |
| Rosuvastatin | 0.2 [8] | 0.75 # | 11 [23] | 0.3 [23] |
| Saquinavir | 0.04 [8] | 0.74 [8] | 13 [15] | 0.01 * |
| Substrate | Inhibitor | Caco-2 | EpiIntestinal | ||
|---|---|---|---|---|---|
| PappAB (10−6 cm/s) | Efflux | PappAB (10−6 cm/s) | Efflux | ||
| Rosuvastatin | None | 0.3 | 21.0 | 0.3 | 100.0 |
| Ko-143 (3 µM) | 0.5 | 5.5 | 2.6 | 3.1 | |
| Zosuqidar (5 µM) | 0.3 | 19.0 | 0.9 | 25 | |
| DME/Substrate | Caco-2 | EpiIntestinal | ||
|---|---|---|---|---|
| Enzyme Activities * (pmol/h/cm2) Mean/SD | >Intracellular Metabolite (% of Total) | Enzyme Activities * (pmol/h/cm2) Mean/SD | Intracellular Metabolite (% of Total) | |
| CYP1A2/Phenacetin | 123.1/4.8 | BLQ | 17.4/3.0 | BLQ |
| CYP2B6/Bupropion | BLQ | BLQ | 2.6/0.9 | BLQ |
| CYP2C8/Amodiaquine | 11.2/1.9 | 36.5 | 107.9/49.1 | 37.5 |
| CYP2C9/Diclofenac | 20.8/1.6 | 12.8 | 28.4/1.6 | 14.7 |
| CYP2C19/S-Mephenytoin | 7.1/0.7 | 4.5 | 6.9/0.7 | 4.3 |
| CYP3A4/Testosterone | 26.5/3.6 | BLQ | 176.4/8.0 | 0.9 |
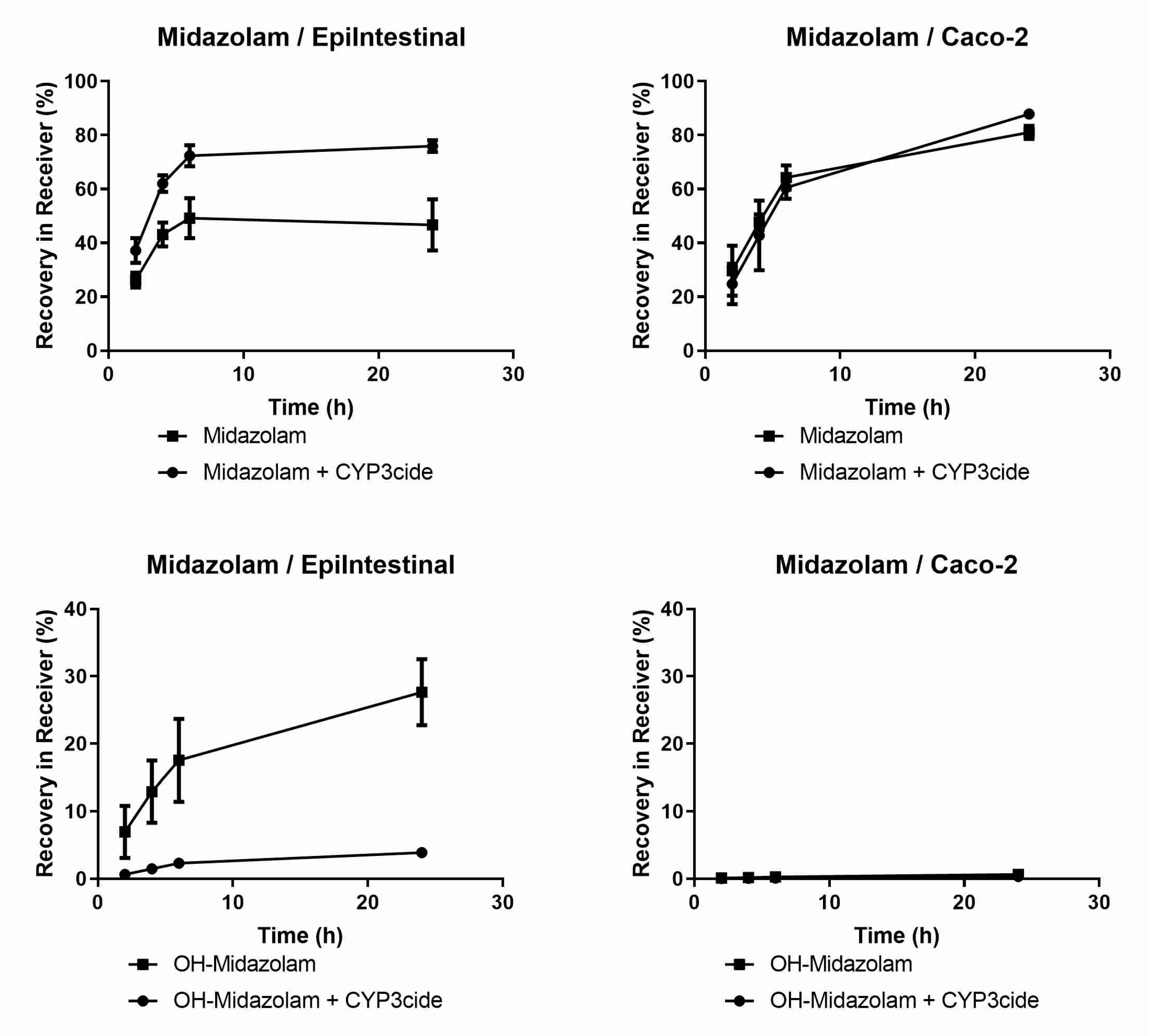
| CYP3A4/Midazolam | BLQ | BLQ | 1.9/0.5 | 14.9 |
| CYP2D6/Dextromethorphan | 10.9/2.7 | BLQ | 9.2/1.3 | BLQ |
| UGT/7-OH-Coumarin | 10,770.5/721.9 | 5.5 | 7583.4/855.2 | 10.0 |
| SULT/7-OH-Coumarin | 508.0/46.1 | BLQ | 1747.4/140.0 | 3.4 |
| UGT1A1/β-Estradiol | 65.3/6.9 | 3.2 | 243.3/6.7 | 2.8 |
| CYP2J2/Astemizole | 4.9/0.3 | 62.9 | 17.7/5.2 | 68.7 |
| CES/BIBF 1120 | 370.9/31.4 | 24.7 | 400.0/12.9 | 13.3 |
| Substrate | EpiIntestinal | Human Intestinal Mucosa (HIM) | ||
|---|---|---|---|---|
| Ezetimibe | Raloxifene | Ezetimibe | Raloxifene | |
| Parent (% of parent drug at T0) | 8.4 | 2.4 | 40.3 | 33.2 |
| Glucuronides (% of parent drug at T0) | 39.1 | 2.2 | 58.6 | 18.7 |
| Sulfates (% of parent drug at T0) | n.d. | 14.1 | n.d. | 2.0 |
| Drug | Recovery in Basal Comp. @ 24h (%) | Fa × Fg in Human (%) | DMEs/Transporters |
|---|---|---|---|
| Atenolol | 86 | 50 | |
| Atorvastatin | 43 | 61 | CYP3A4/BCRP/MRP2 |
| Buspirone | 60 | 70 | CYP3A4 |
| Felodipine | 47 | 62 | CYP3A4 |
| Indinavir | 53 | 100 | CYP3A4 |
| Irinotecan | 62 | 39 | Esterases, CYP3A4 |
| Midazolam | 47 | 59 | CYP3A4 |
| Nifedipine | 110 | 100 | CYP3A4 |
| Oxybutynin | 16 | 9 | Esterases, CYP3A4 |
| Qunidine | 85 | 100 | CYP3A4, etc. |
| Rosuvastatin | 30 | 62 | CYP2C9/BCRP/MRP2 |
| Saquinavir | 18 | 25 | CYP3A4/P-gp |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, Y.; Claus, S.; Schnell, D.; Runge, F.; MacLean, C. In-Depth Characterization of EpiIntestinal Microtissue as a Model for Intestinal Drug Absorption and Metabolism in Human. Pharmaceutics 2020, 12, 405. https://doi.org/10.3390/pharmaceutics12050405
Cui Y, Claus S, Schnell D, Runge F, MacLean C. In-Depth Characterization of EpiIntestinal Microtissue as a Model for Intestinal Drug Absorption and Metabolism in Human. Pharmaceutics. 2020; 12(5):405. https://doi.org/10.3390/pharmaceutics12050405
Chicago/Turabian StyleCui, Yunhai, Stephanie Claus, David Schnell, Frank Runge, and Caroline MacLean. 2020. "In-Depth Characterization of EpiIntestinal Microtissue as a Model for Intestinal Drug Absorption and Metabolism in Human" Pharmaceutics 12, no. 5: 405. https://doi.org/10.3390/pharmaceutics12050405
APA StyleCui, Y., Claus, S., Schnell, D., Runge, F., & MacLean, C. (2020). In-Depth Characterization of EpiIntestinal Microtissue as a Model for Intestinal Drug Absorption and Metabolism in Human. Pharmaceutics, 12(5), 405. https://doi.org/10.3390/pharmaceutics12050405