Quality by Design for the Development and Analysis of Enhanced In-Situ Forming Vesicles for the Improvement of the Bioavailability of Fexofenadine HCl In Vitro and In Vivo

 ,
,  ,
,  ,
,  and
and
Abstract
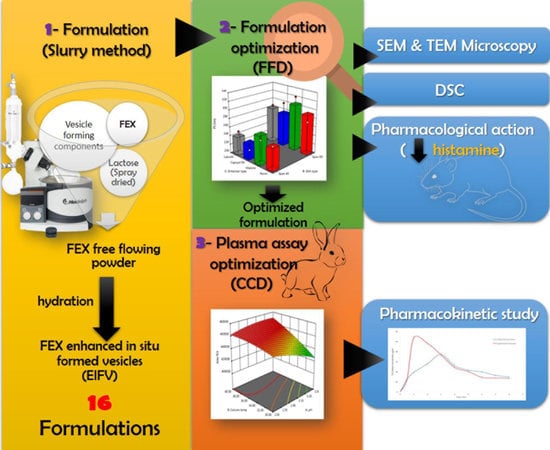
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experimental Design
2.2.1. Full Factorial Design (FFD) for FEX EIFV Powder Optimization
2.2.2 Central Composite Design (CCD) for HPLC Assay Optimization of FEX in Plasma
2.3. Preparation of FEX EIFV Powder
2.4. Micromeritic Properties of the Prepared FEX EIFV Powders
2.5. Formation of the Nanovesicles from FEX-Loaded Provesicular Powders
2.6. Particle Size Analysis and Surface Charge Determination
2.7. Determination of FEX Entrapment Efficiency in the Formed Nanovesicles
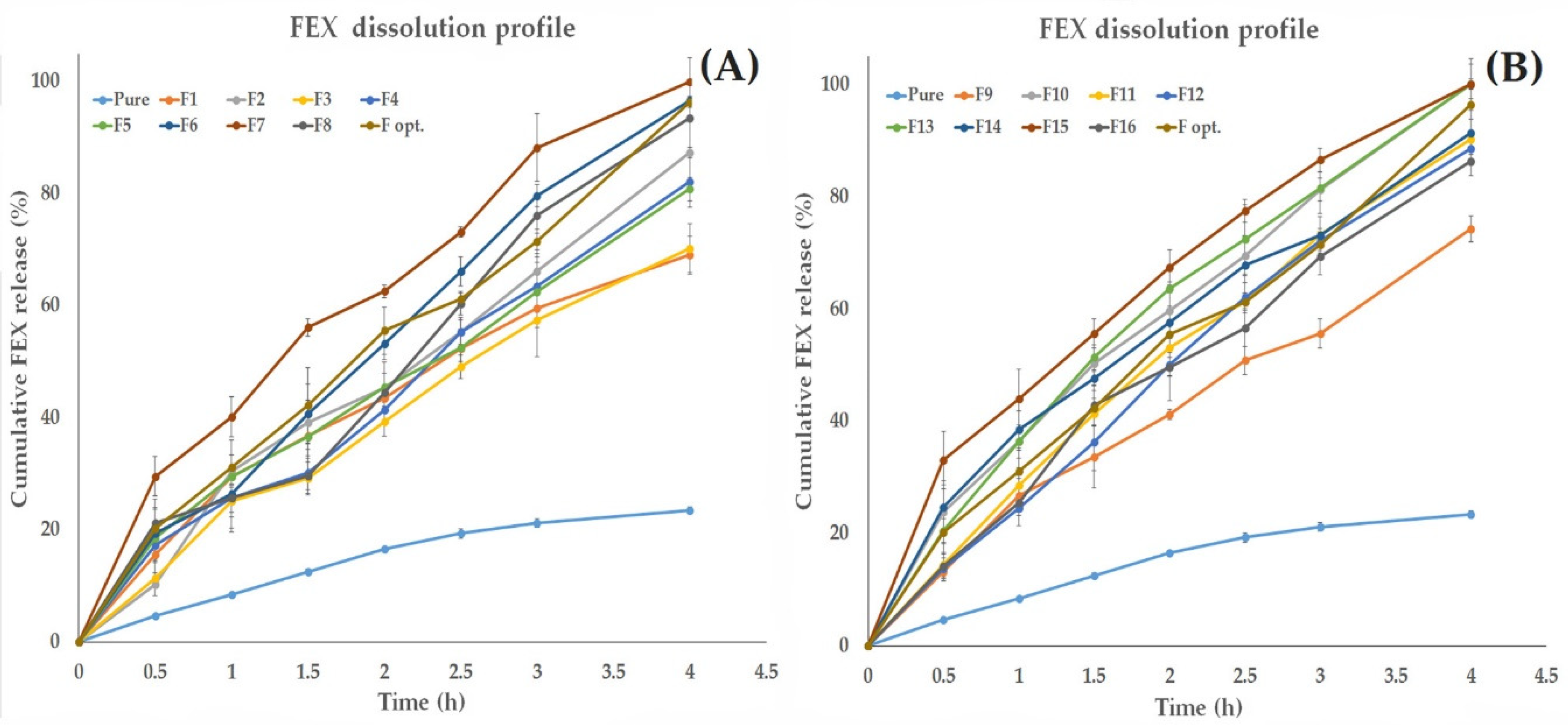
2.8. In Vitro Dissolution study of FEX Release from the Prepared EIFV
2.9. Morphology and Surface Characteristics of the Optimized FEX EIFV Powder and the Formed Nanovesicles
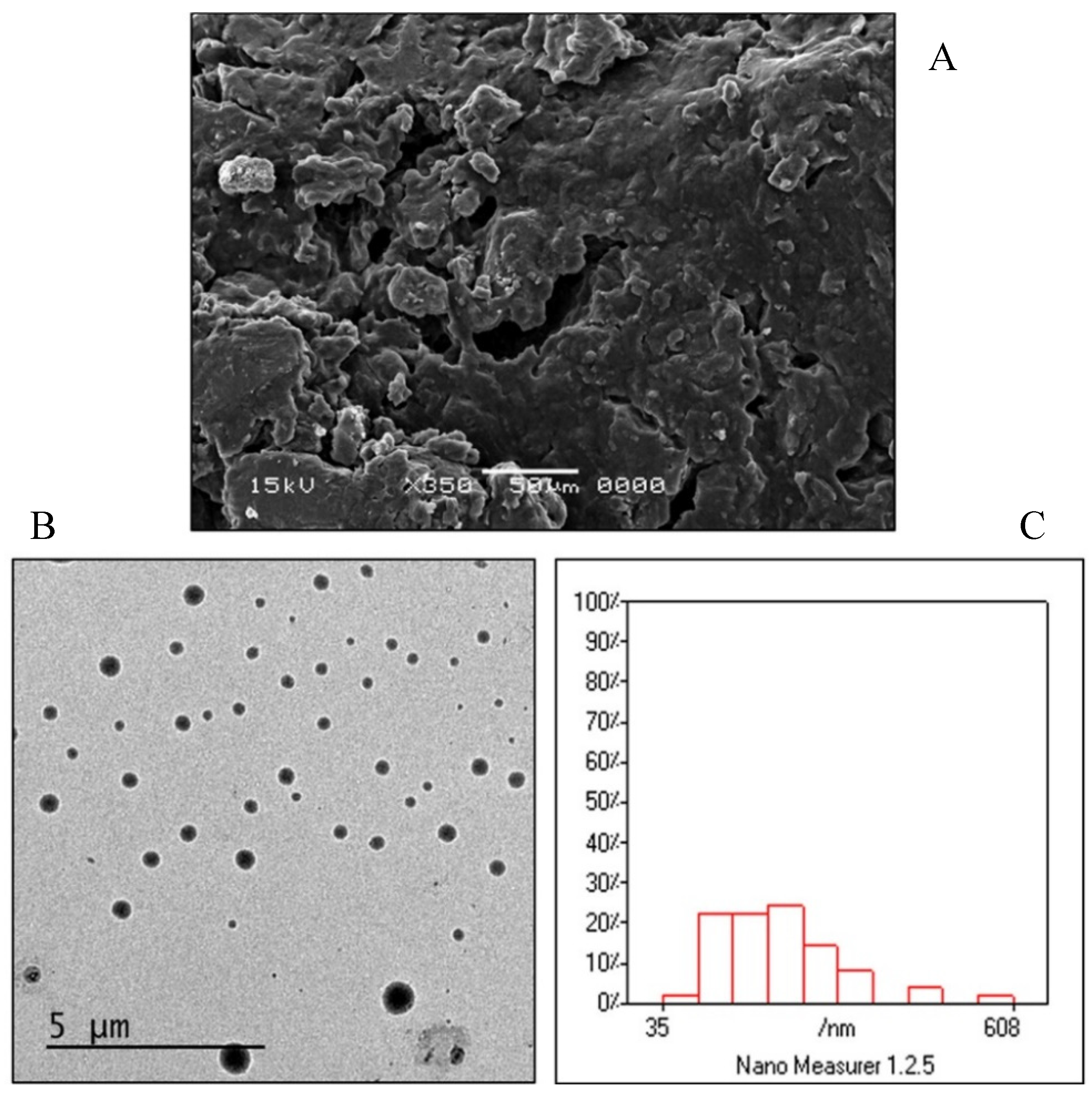
2.9.1. Scanning Electron Microscopy
2.9.2. Transmission Electron Microscopy
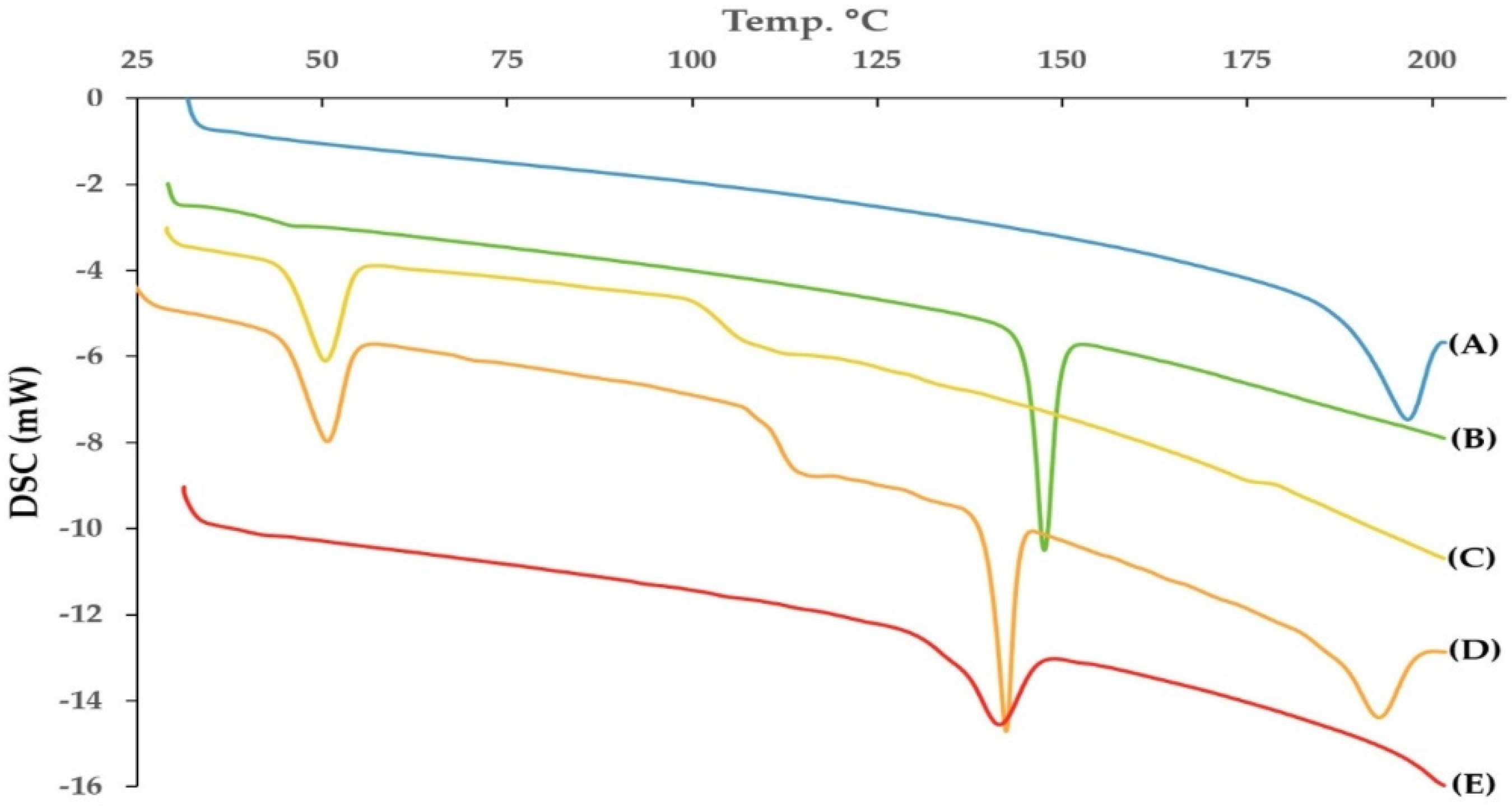
2.10. Thermal Analysis
2.11. Pharmacokinetic Study of FEX in Rabbits
2.11.1. The Design of the Pharmacokinetic Study
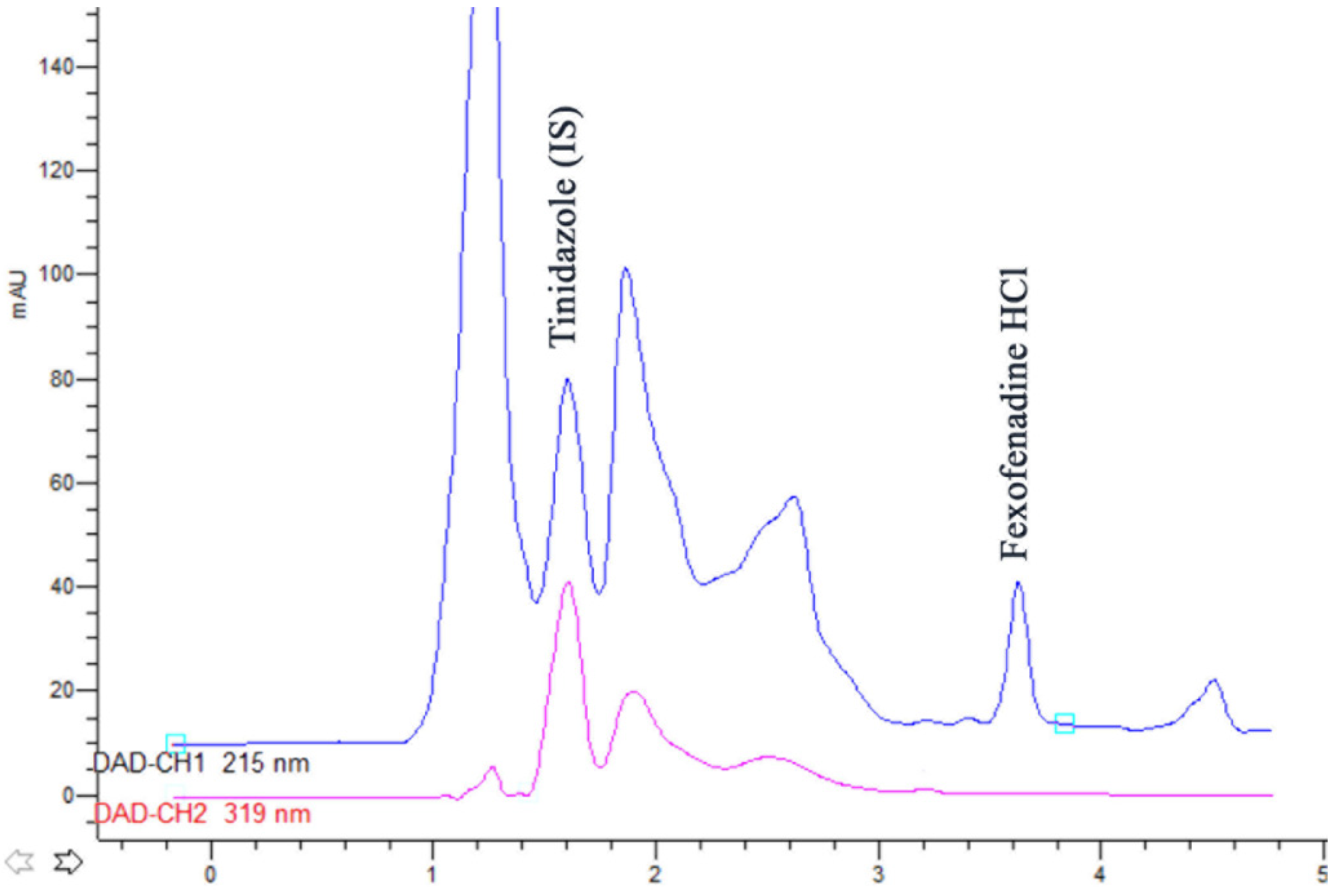
2.11.2. Drug Assay in Plasma
Instrumentation
Chromatographic Parameters
Preparation of Standard and Calibration Solutions
Sample Preparation
Assay Validation
2.11.3. Pharmacokinetic Analysis
2.12. Pharmacological Evaluation of the FEX in the Optimized Formula
2.12.1. Effect of FEX EIFV-Optimized Powder on Compound 48/80-Induced Systemic Anaphylaxis-Like Reactions in Mice
2.12.2. Preparation of Plasma and Histamine Level Determination
2.13. Statistical Analysis
3. Results and Discussion
3.1. Experimental Design
3.1.1. Full Factorial Design (FFD) for FEX EIFV Powder Optimization
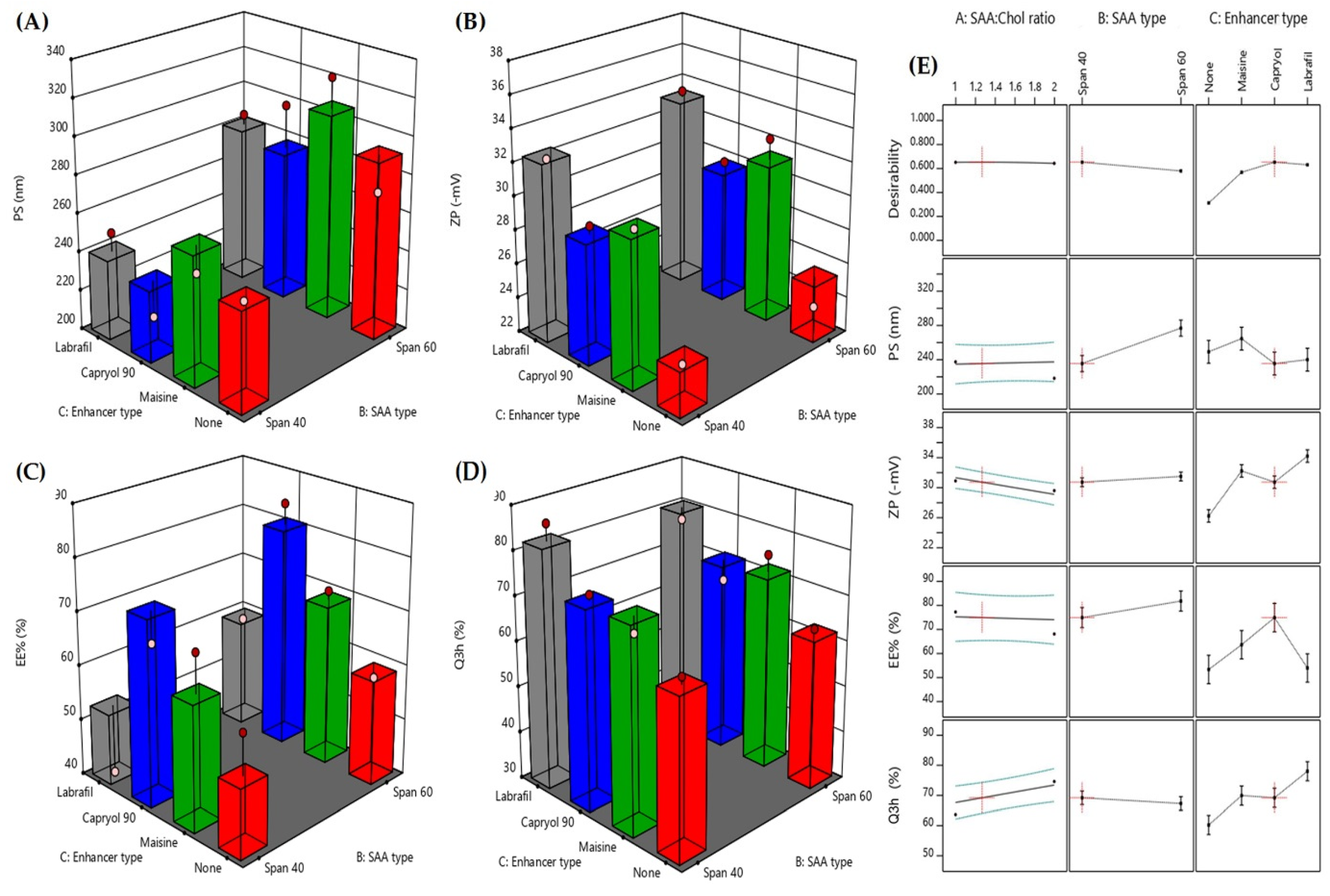
The Effect of Formulation Variables on the PS of the Formed FEX EIFV
The Effect of Formulation Variables on the ZP of the Obtained FEX EIFV
The Effect of Formulation Variables on the EE of the Formed FEX EIFV
The Effect of Formulation Variables on the in Vitro Dissolution Profile of the Formed FEX EIFV
Optimization
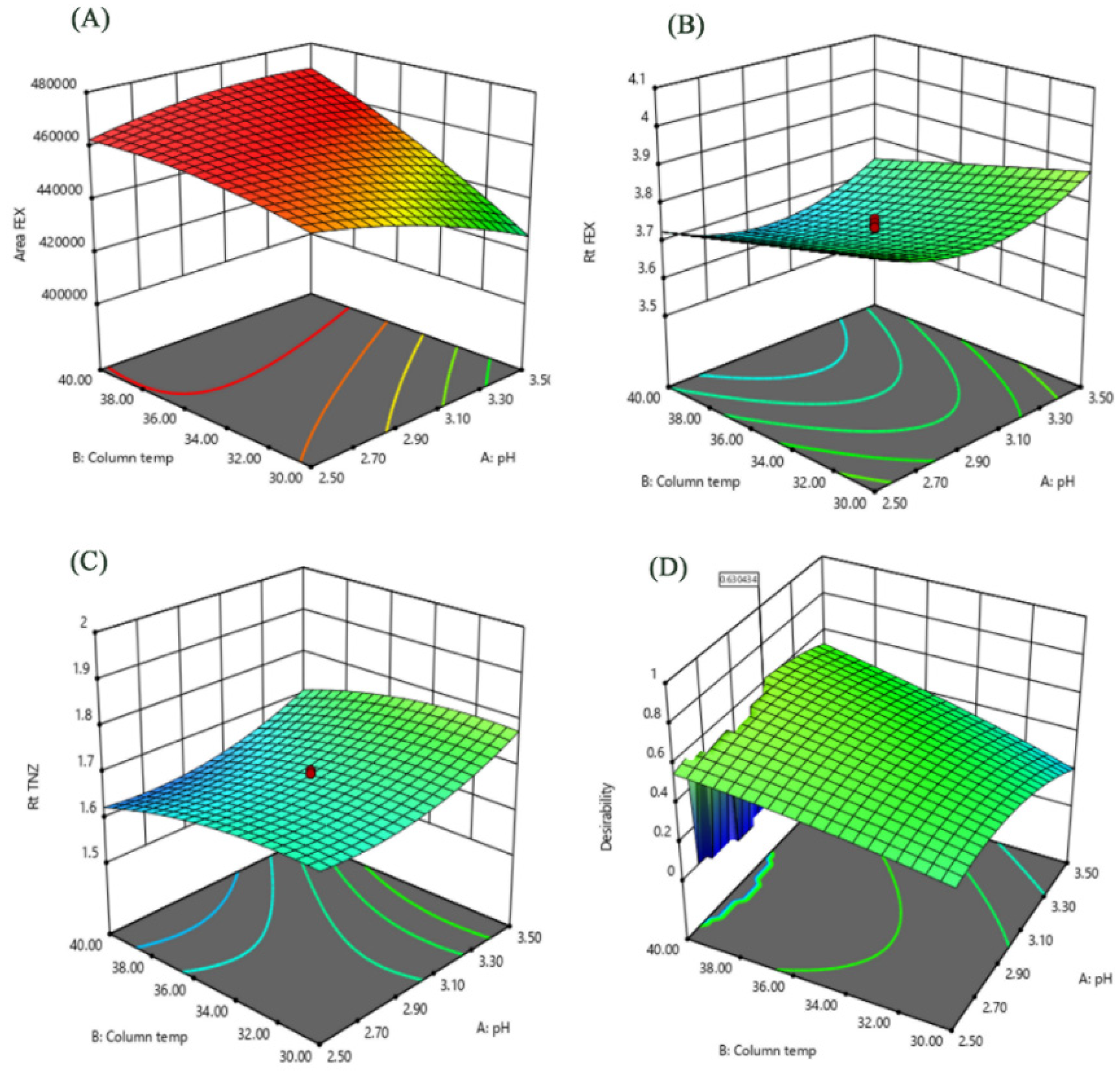
3.1.2. Central Composite Design (CCD) for HPLC Assay Optimization of FEX in Plasma
3.2. Micromeritic Properties of the Prepared FEX EIFV Powders
3.3. Morphological Evaluation of the Optimized FEX EIFV Powder and the Formed Vesicles
3.4. Thermal Analysis
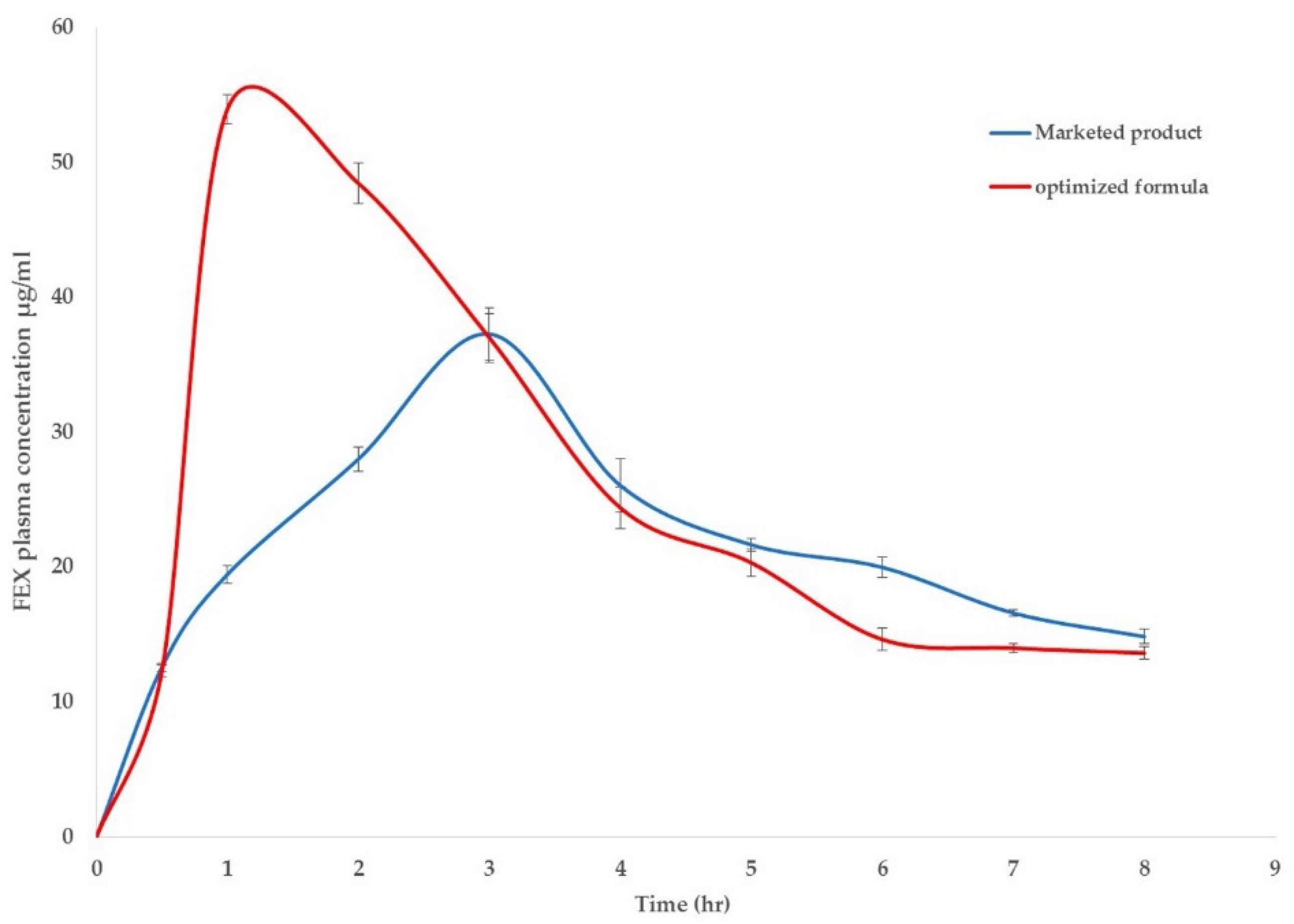
3.5. Pharmacokinetic Study of FEX in Rabbits
3.6. Pharmacological Evaluation of the FEX in the Optimized Formula
Effect of FEX EIFV Optimized Powder on Compound 48/80-Induced Systemic Anaphylaxis-Like Reactions in Mice and Histamine Level in Plasma
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Borges, A.F.; Silva, C.; Coelho, J.F.; Simões, S. Oral films: Current status and future perspectives: I—Galenical development and quality attributes. J. Control. Release 2015, 206, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Cui, Y.; Zhao, Q.; Zhang, Q.; Musetti, S.; Kinghorn, K.A.; Wang, S. Overcoming multiple gastrointestinal barriers by bilayer modified hollow mesoporous silica nanocarriers. Acta Biomater. 2018, 65, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Sammour, O.A.; Hammad, M.A.; Zidan, A.S.; Mowafy, A.G. QbD approach of rapid disintegrating tablets incorporating indomethacin solid dispersion. Pharm. Dev. Technol. 2011, 16, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Massart, D.L.; Vandeginste, B.G.M.; Buydens, L.M.C.; Jong, S.D.; Smeyers-Verbeke, J. Handbook of Chemometrics and Qualimetrics: Part A; Elsevier: Amsterdam, The Netherlands, 1997. [Google Scholar]
- Dejaegher, B.; Heyden, Y.V. Experimental designs and their recent advances in set-up, data interpretation, and analytical applications. J. Pharm. Biomed. Anal. 2011, 56, 141–158. [Google Scholar] [CrossRef] [PubMed]
- Hobson, J.J.; Edwards, S.; Slater, R.A.; Martin, P.; Owen, A.; Rannard, S.P. Branched copolymer-stabilised nanoemulsions as new candidate oral drug delivery systems. RSC Adv. 2018, 8, 12984–12991. [Google Scholar] [CrossRef]
- Bayindir, Z.S.; Yuksel, N. Provesicles as novel drug delivery systems. Curr. Pharm. Biotechnol. 2015, 16, 344–364. [Google Scholar] [CrossRef]
- Mokhtar, M.; Sammour, O.A.; Hammad, M.A.; Megrab, N.A. Effect of some formulation parameters on flurbiprofen encapsulation and release rates of niosomes prepared from proniosomes. Int. J. Pharm. 2008, 361, 104–111. [Google Scholar] [CrossRef]
- Yasam, V.R.; Jakki, S.L.; Natarajan, J.; Kuppusamy, G. A review on novel vesicular drug delivery: Proniosomes. Drug Deliv. 2014, 21, 243–249. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Katare, O.; Singh, B. Optimized self nano-emulsifying systems of ezetimibe with enhanced bioavailability potential using long chain and medium chain triglycerides. Colloids Surf. B Biointerfaces 2012, 100, 50–61. [Google Scholar] [CrossRef]
- Holm, R.; Porter, C.J.; Edwards, G.A.; Müllertz, A.; Kristensen, H.G.; Charman, W.N. Examination of oral absorption and lymphatic transport of halofantrine in a triple-cannulated canine model after administration in self-microemulsifying drug delivery systems (SMEDDS) containing structured triglycerides. Eur. J. Pharm. Sci. 2003, 20, 91–97. [Google Scholar] [CrossRef]
- Han, M.; Fu, S.; Gao, J.-Q.; Fang, X.-L. Evaluation of intestinal absorption of ginsenoside Rg1 incorporated in microemulison using parallel artificial membrane permeability assay. Biol. Pharm. Bull. 2009, 32, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Basalious, E.B.; Shawky, N.; Badr-Eldin, S.M. SNEDDS containing bioenhancers for improvement of dissolution and oral absorption of lacidipine. I: Development and optimization. Int. J. Pharm. 2010, 391, 203–211. [Google Scholar] [CrossRef]
- Eedara, B.B.; Veerareddy, P.R.; Jukanti, R.; Bandari, S. Improved oral bioavailability of fexofenadine hydrochloride using lipid surfactants: Ex vivo, in situ and in vivo studies. Drug Dev. Ind. Pharm. 2014, 40, 1030–1043. [Google Scholar] [CrossRef] [PubMed]
- Drescher, S.; Schaeffeler, E.; Hitzl, M.; Hofmann, U.; Schwab, M.; Brinkmann, U.; Eichelbaum, M.; Fromm, M.F. MDR1 gene polymorphisms and disposition of the P-glycoprotein substrate fexofenadine. Br. J. Clin. Pharmacol. 2002, 53, 526–534. [Google Scholar] [CrossRef]
- Gundogdu, E.; Alvarez, I.G.; Karasulu, E. Improvement of effect of water-in-oil microemulsion as an oral delivery system for fexofenadine: In vitro and in vivo studies. Int. J. Nanomed. 2011, 6, 1631. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, J.; Khaltaev, N.; Cruz, A.A.; Denburg, J.; Fokkens, W.; Togias, A.; Zuberbier, T.; Baena-Cagnani, C.; Canonica, G.; Van Weel, C. Allergic rhinitis and its impact on asthma (ARIA) 2008. Allergy 2008, 63, 8–160. [Google Scholar] [CrossRef]
- Nasr, A.; Qushawy, M.; Swidan, S. Spray Dried Lactose Based Proniosomes as Stable Provesicular Drug Delivery Carriers: Screening, Formulation, and Physicochemical Characterization. Int. J. Appl. Pharm. 2018, 10, 125. [Google Scholar] [CrossRef]
- Kumar, G.P.; Rajeshwarrao, P. Nonionic surfactant vesicular systems for effective drug delivery—An overview. Acta Pharm. Sin. B 2011, 1, 208–219. [Google Scholar] [CrossRef]
- Blazek-Welsh, A.I.; Rhodes, D.G. Maltodextrin-based proniosomes. Aaps Pharmsci 2001, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Al-Hashemi, H.M.B.; Al-Amoudi, O.S.B. A review on the angle of repose of granular materials. Powder Technol. 2018, 330, 397–417. [Google Scholar] [CrossRef]
- Jaimini, M.; Rana, A.; Tanwar, Y. Formulation and evaluation of famotidine floating tablets. Curr. Drug Deliv. 2007, 4, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Veerareddy, P.R.; Bobbala, S.K.R. Enhanced oral bioavailability of isradipine via proniosomal systems. Drug Dev. Ind. Pharm. 2013, 39, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration of the United States (US-FDA); U.S. Department of Health and Human Services (DHHS); Center for Drug Evaluation and Research (CDER); Center for Veterinary Medicine (CVM). Bioanalytical Method Validation. In Guidance for Industry; US-FDA: Rockville, MD, USA, 2018. [Google Scholar]
- Choi, Y.H.; Chai, O.H.; Han, E.-H.; Choi, S.-Y.; Kim, H.T.; Song, C.H. Lipoic acid suppresses compound 48/80-induced anaphylaxis-like reaction. Anat. Cell Biol. 2010, 43, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Shin, T.; Park, J.; Kim, H. Effect of Cryptotympana atrata extract on compound 48/80-induced anaphylactic reactions. J. Ethnopharmacol. 1999, 66, 319–325. [Google Scholar] [CrossRef]
- Shore, P.A. A method for the fluorometric assay of histamine in tissues. J. Pharmacol. Exp. Ther. 1959, 127, 182–186. [Google Scholar] [PubMed]
- Uchegbu, I.F.; Vyas, S.P. Non-ionic surfactant based vesicles (niosomes) in drug delivery. Int. J. Pharm. 1998, 172, 33–70. [Google Scholar] [CrossRef]
- Balakrishnan, P.; Shanmugam, S.; Lee, W.S.; Lee, W.M.; Kim, J.O.; Oh, D.H.; Kim, D.-D.; Kim, J.S.; Yoo, B.K.; Choi, H.-G. Formulation and in vitro assessment of minoxidil niosomes for enhanced skin delivery. Int. J. Pharm. 2009, 377, 1–8. [Google Scholar] [CrossRef]
- Izham, M.; Nadiah, M.; Hussin, Y.; Aziz, M.N.M.; Yeap, S.K.; Rahman, H.S.; Masarudin, M.J.; Mohamad, N.E.; Abdullah, R.; Alitheen, N.B. Preparation and Characterization of Self Nano-Emulsifying Drug Delivery System Loaded with Citraland Its Antiproliferative Effect on Colorectal Cells In Vitro. Nanomaterials 2019, 9, 1028. [Google Scholar] [CrossRef]
- Parmar, N.; Singla, N.; Amin, S.; Kohli, K. Study of cosurfactant effect on nanoemulsifying area and development of lercanidipine loaded (SNEDDS) self nanoemulsifying drug delivery system. Colloids Surf. B Biointerfaces 2011, 86, 327–338. [Google Scholar] [CrossRef]
- Kamboj, S.; Saini, V.; Bala, S. Formulation and characterization of drug loaded nonionic surfactant vesicles (niosomes) for oral bioavailability enhancement. Sci. World J. 2014, 2014, 959741. [Google Scholar] [CrossRef]
- Nasr, M.; Mansour, S.; Mortada, N.D.; Elshamy, A. Vesicular aceclofenac systems: A comparative study between liposomes and niosomes. J. Microencapsul. 2008, 25, 499–512. [Google Scholar] [CrossRef] [PubMed]
- El-Alim, S.A.; Kassem, A.; Basha, M. Proniosomes as a novel drug carrier system for buccal delivery of benzocaine. J. Drug Deliv. Sci. Technol. 2014, 24, 452–458. [Google Scholar] [CrossRef]
- Basiri, L.; Rajabzadeh, G.; Bostan, A. α-Tocopherol-loaded niosome prepared by heating method and its release behavior. Food Chem. 2017, 221, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Guo, R.; Hua, W.; Qiu, J. Structure behaviors of hemoglobin in PEG 6000/Tween 80/Span 80/H2O niosome system. Colloids Surf. A Physicochem. Eng. Asp. 2007, 293, 255–261. [Google Scholar] [CrossRef]
- Ahmed, A.; Ghourab, M.; Shedid, S.; Qushawy, M. Optimization of piroxicam niosomes using central composite design. Int. J. Pharm. Pharm. Sci. 2013, 5, 229–236. [Google Scholar]
- Yoshioka, T.; Sternberg, B.; Florence, A.T. Preparation and properties of vesicles (niosomes) of sorbitan monoesters (Span 20, 40, 60 and 80) and a sorbitan triester (Span 85). Int. J. Pharm. 1994, 105, 1–6. [Google Scholar] [CrossRef]
- Nagarsenker, M.; Londhe, V. Preparation and evaluation of a liposomal formulation of sodium cromoglicate. Int. J. Pharm. 2003, 251, 49–56. [Google Scholar] [CrossRef]
- Fatouros, D.; Hatzidimitriou, K.; Antimisiaris, S. Liposomes encapsulating prednisolone and prednisolone–cyclodextrin complexes: Comparison of membrane integrity and drug release. Eur. J. Pharm. Sci. 2001, 13, 287–296. [Google Scholar] [CrossRef]
- Attia, I.A.; El-Gizawy, S.A.; Fouda, M.A.; Donia, A.M. Influence of a niosomal formulation on the oral bioavailability of acyclovir in rabbits. AAPS PharmSciTech 2007, 8, 206–212. [Google Scholar] [CrossRef]
- Al-mahallawi, A.M.; Khowessah, O.M.; Shoukri, R.A. Nano-transfersomal ciprofloxacin loaded vesicles for non-invasive trans-tympanic ototopical delivery: In-vitro optimization, ex-vivo permeation studies, and in-vivo assessment. Int. J. Pharm. 2014, 472, 304–314. [Google Scholar] [CrossRef]
- Solanki, A.B.; Parikh, J.R.; Parikh, R.H. Formulation and optimization of piroxicam proniosomes by 3-factor, 3-level Box-Behnken design. AAPS PharmSciTech 2007, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Basha, M.; Abd El-Alim, S.H.; Shamma, R.N.; Awad, G.E. Design and optimization of surfactant-based nanovesicles for ocular delivery of Clotrimazole. J. Liposome Res. 2013, 23, 203–210. [Google Scholar] [CrossRef]
- Kumar, L.; Alam, M.S.; Meena, C.L.; Jain, R.; Bansal, A.K. Fexofenadine hydrochloride. In Profiles of Drug Substances, Excipients and Related Methodology; Elsevier: Amsterdam, The Netherlands, 2009; Volume 34, pp. 153–192. [Google Scholar]
- Gurrapu, A.; Jukanti, R.; Bobbala, S.R.; Kanuganti, S.; Jeevana, J.B. Improved oral delivery of valsartan from maltodextrin based proniosome powders. Adv. Powder Technol. 2012, 23, 583–590. [Google Scholar] [CrossRef]
- Aburahma, M.H.; Abdelbary, G.A. Novel diphenyl dimethyl bicarboxylate provesicular powders with enhanced hepatocurative activity: Preparation, optimization, in vitro/in vivo evaluation. Int. J. Pharm. 2012, 422, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Chen, C. Some pharmacokinetic aspects of the lipophilic terfenadine and zwitterionic fexofenadine in humans. Drugs R D 2007, 8, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Wu, H.; Niu, F.; Yan, C.; Yang, X.; Jia, Y. Design of fenofibrate microemulsion for improved bioavailability. Int. J. Pharm. 2011, 420, 251–255. [Google Scholar] [CrossRef]
- Kang, B.K.; Lee, J.S.; Chon, S.K.; Jeong, S.Y.; Yuk, S.H.; Khang, G.; Lee, H.B.; Cho, S.H. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int. J. Pharm. 2004, 274, 65–73. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Form. | X1: Surfactant Type | X2: Enhancer Type | X3: Surfactant: Cholesterol Ratio | Surfactant Weight (mg) | Enhancer Weight (mg) | Cholesterol Weight (mg) |
|---|---|---|---|---|---|---|
| F1 | Span 40 | None | 1 | 500 | 0 | 500 |
| F2 | Span 40 | None | 2 | 666.66 | 0 | 333.33 |
| F3 | Span 60 | None | 1 | 500 | 0 | 500 |
| F4 | Span 60 | None | 2 | 666.66 | 0 | 333.33 |
| F5 | Span 40 | Maisine CC | 1 | 333.33 | 333.33 | 333.33 |
| F6 | Span 40 | Maisine CC | 2 | 500 | 250 | 250 |
| F7 | Span 60 | Maisine CC | 1 | 333.33 | 333.33 | 333.33 |
| F8 | Span 60 | Maisine CC | 2 | 500 | 250 | 250 |
| F9 | Span 40 | Capryol 90 | 1 | 333.33 | 333.33 | 333.33 |
| F10 | Span 40 | Capryol 90 | 2 | 500 | 250 | 250 |
| F11 | Span 60 | Capryol 90 | 1 | 333.33 | 333.33 | 333.33 |
| F12 | Span 60 | Capryol 90 | 2 | 500 | 250 | 250 |
| F13 | Span 40 | Labrafil M 1944 | 1 | 333.33 | 333.33 | 333.33 |
| F14 | Span 40 | Labrafil M 1944 | 2 | 500 | 250 | 250 |
| F15 | Span 60 | Labrafil M 1944 | 1 | 333.33 | 333.33 | 333.33 |
| F16 | Span 60 | Labrafil M 1944 | 2 | 500 | 250 | 250 |
| Formula | PS (nm) | PDI | ZP (mV) | EE (%) | Q3h (%) |
|---|---|---|---|---|---|
| F1 | 276.0 ± 5.279 | 0.380 ± 0.002 | −28.0 ± 0.208 | 55.44 ± 1.25 | 59.55 ± 2.15 |
| F2 | 249.9 ± 2.261 | 0.357 ± 0.010 | −24.3 ± 1.550 | 60.11 ± 0.92 | 66.14 ± 3.24 |
| F3 | 282.9 ± 4.912 | 0.389 ± 0.019 | −28.6 ± 0.902 | 71.63 ± 2.01 | 57.36 ± 4.20 |
| F4 | 272.8 ± 2.261 | 0.306 ± 0.017 | −23.5 ± 1.330 | 61.37 ± 0.55 | 63.53 ± 3.85 |
| F5 | 262.4 ± 3.073 | 0.335 ± 0.039 | −32.9 ± 1.250 | 56.44 ± 0.95 | 62.44 ± 1.11 |
| F6 | 252.1 ± 5.382 | 0.267 ± 0.021 | −30.6 ± 0.954 | 41.32 ± 1.13 | 79.65 ± 6.45 |
| F7 | 304.6 ± 3.213 | 0.360 ± 0.029 | −32.3 ± 0.755 | 44.33 ± 2.15 | 88.19 ± 3.90 |
| F8 | 323.9 ± 3.109 | 0.378 ± 0.018 | −32.6 ± 1.280 | 71.44 ± 1.55 | 76.14 ± 6.54 |
| F9 | 237.3 ± 1.943 | 0.313 ± 0.005 | −30.9 ± 1.440 | 59.67 ± 2.57 | 55.68 ± 1.25 |
| F10 | 218.1 ± 4.729 | 0.281 ± 0.036 | −29.6 ± 0.651 | 62.56 ± 0.63 | 81.23 ± 2.25 |
| F11 | 269.6 ± 5.957 | 0.323 ± 0.018 | −31.7 ± 0.751 | 55.66 ± 2.78 | 73.23 ± 3.40 |
| F12 | 301.5 ± 2.572 | 0.323 ± 0.030 | −30.2 ± 0.416 | 77.60 ± 3.21 | 72.13 ± 1.37 |
| F13 | 235.1 ± 2.325 | 0.317 ± 0.037 | −34.0 ± 1.140 | 44.43 ± 1.78 | 81.52 ± 1.98 |
| F14 | 251.5 ± 4.852 | 0.346 ± 0.016 | −32.4 ± 0.404 | 33.34 ± 0.99 | 73.21 ± 3.14 |
| F15 | 269.6 ± 8.517 | 0.358 ± 0.045 | −36.2 ± 0.874 | 45.82 ± 0.82 | 86.52 ± 1.21 |
| F16 | 288.5 ± 1.457 | 0.384 ± 0.009 | −33.7 ± 1.360 | 59.52 ± 3.15 | 69.33 ± 1.65 |
| Variable | X1:Surfactant Type | X2:Enhancer Type | X3:Surfactant:Chol Ratio | |
| Selected | Span 40 | Capryol 90 | 1.268 | |
| Responses | Y1:PS (nm) | Y2:ZP (mV) | Y3:EE (%) | Y4:Q3h (%) |
| Predicted | 235.3 | ȡ30.7 | 75.0 | 69.72 |
| Observed | 202.6 ± 3.90 | −31.6 ± 0.92 | 73.65 ± 1.68 | 71.5 ± 2.65 |
| Std. | Run | Factors Levels | Responses | ||||
|---|---|---|---|---|---|---|---|
| pH | Column Temperature (°C) | Flow Rate (mL/min) | Rt FEX | Rt TNZ | Peak Area FEX | ||
| 4 | 1 | 2.50 | 30.00 | 0.80 | 3.87 | 1.70 | 426,304 |
| 16 | 2 | 3.50 | 30.00 | 0.90 | 3.75 | 1.68 | 411,926 |
| 12 | 3 | 3.00 | 35.00 | 0.90 | 3.62 | 1.60 | 455,576 |
| 13 | 4 | 2.50 | 40.00 | 0.90 | 3.55 | 1.58 | 454,224 |
| 3 | 5 | 3.50 | 40.00 | 0.90 | 3.65 | 1.61 | 446,531 |
| 5 | 6 | 3.00 | 26.95 | 1.00 | 3.62 | 1.62 | 437,479 |
| 1 | 7 | 2.16 | 35.00 | 1.00 | 3.91 | 1.66 | 458,515 |
| 15 | 8 | 3.00 | 35.00 | 1.00 | 3.75 | 1.69 | 458,882 |
| 8 | 9 | 3.00 | 35.00 | 1.00 | 3.76 | 1.70 | 459,250 |
| 6 | 10 | 3.00 | 35.00 | 1.00 | 3.74 | 1.70 | 458,552 |
| 17 | 11 | 3.00 | 35.00 | 1.00 | 3.74 | 1.70 | 458,735 |
| 19 | 12 | 3.00 | 35.00 | 1.00 | 3.74 | 1.69 | 458,816 |
| 9 | 13 | 3.00 | 35.00 | 1.00 | 3.74 | 1.69 | 458,794 |
| 20 | 14 | 3.00 | 35.00 | 1.00 | 3.74 | 1.69 | 459,213 |
| 14 | 15 | 3.84 | 35.00 | 1.00 | 3.98 | 1.91 | 443,894 |
| 7 | 16 | 2.50 | 30.00 | 1.10 | 4.00 | 1.77 | 426,620 |
| 10 | 17 | 3.50 | 30.00 | 1.10 | 4.07 | 1.87 | 407,547 |
| 11 | 18 | 2.50 | 40.00 | 1.10 | 3.76 | 1.66 | 448,713 |
| 18 | 19 | 3.00 | 43.41 | 1.10 | 3.74 | 1.66 | 461,233 |
| 2 | 20 | 3.50 | 40.00 | 1.20 | 3.87 | 1.81 | 437,156 |
| Formula | Angle of Repose (θ) | Carr’s Index | Hausner Ratio |
|---|---|---|---|
| F1 | 28.15 ± 1.2 | 16.45 ± 1.01 | 1.197 ± 0.04 |
| F2 | 23.45 ± 0.9 | 14.70 ± 0.72 | 1.172 ± 0.03 |
| F3 | 25.89 ± 2.3 | 15.77 ± 0.92 | 1.187 ± 0.05 |
| F4 | 26.17 ± 1.1 | 15.62 ± 0.85 | 1.185 ± 0.03 |
| F5 | 41.08 ± 2.5 | 24.99 ± 0.66 | 1.333 ± 0.03 |
| F6 | 38.19 ±2.8 | 22.53 ± 1.33 | 1.291 ± 0.06 |
| F7 | 38.16 ± 2.1 | 23.00 ± 1.10 | 1.299 ± 0.05 |
| F8 | 40.15 ± 2.5 | 25.36 ± 0.90 | 1.340 ± 0.04 |
| F9 | 28.41 ± 1.6 | 16.51 ± 1.05 | 1.198 ± 0.03 |
| F10 | 27.56 ± 1.2 | 17.17 ± 1.11 | 1.207± 0.04 |
| F11 | 30.05 ± 2.8 | 17.50 ± 1.23 | 1.212 ± 0.04 |
| F12 | 28.48 ± 0.9 | 16.11 ± 0.92 | 1.192 ± 0.03 |
| F13 | 31.54 ± 1.7 | 18.84 ± 1.38 | 1.232 ± 0.06 |
| F14 | 30.78 ± 2.2 | 16.24 ± 1.21 | 1.194 ± 0.04 |
| F15 | 28.88 ± 1.9 | 18.51 ± 1.22 | 1.227 ± 0.05 |
| F16 | 29.58 ± 2.3 | 17.65 ± 1.25 | 1.214 ± 0.05 |
| tmax (h) | t1/2 (h) | Cmax (μg/mL) | AUC(0-8) (μg.h/mL) | MRT (h) | |
|---|---|---|---|---|---|
| Optimized FEX EIFV | 1.00 * | 1.75 * ± 0.41 | 53.94 * ± 6.09 | 212.22 * ± 8.77 | 3.25 * ± 0.33 |
| Marketed product | 3.00 | 2.39 ± 0.36 | 37.28 ± 3.54 | 177.89 ± 8.16 | 3.89 ± 0.24 |
| Treatment | Compound 48/80 (8 mg/kg) | Mortality | Histamine Concentration (ng/mL) | |
|---|---|---|---|---|
| Group 1 | None (PBS) | - | 0% | 111.7 ± 7.9 |
| Group 2 | None (PBS) | + | 100% | 256.8 ± 13.5 |
| Group 3 | FEX powder | + | 62.5% * | 236.4 ± 12.9 |
| Group 4 | Marketed product powder | + | 12.5% * | 201.8 ± 9.2 |
| Group 5 | Optimized FEX EIFV powder | + | 12.5% * | 176.3 ± 11.8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasr, A.M.; Qushawy, M.K.; Elkhoudary, M.M.; Gawish, A.Y.; Elhady, S.S.; Swidan, S.A. Quality by Design for the Development and Analysis of Enhanced In-Situ Forming Vesicles for the Improvement of the Bioavailability of Fexofenadine HCl In Vitro and In Vivo. Pharmaceutics 2020, 12, 409. https://doi.org/10.3390/pharmaceutics12050409
Nasr AM, Qushawy MK, Elkhoudary MM, Gawish AY, Elhady SS, Swidan SA. Quality by Design for the Development and Analysis of Enhanced In-Situ Forming Vesicles for the Improvement of the Bioavailability of Fexofenadine HCl In Vitro and In Vivo. Pharmaceutics. 2020; 12(5):409. https://doi.org/10.3390/pharmaceutics12050409
Chicago/Turabian StyleNasr, Ali M., Mona K. Qushawy, Mahmoud M. Elkhoudary, Aya Y. Gawish, Sameh S. Elhady, and Shady A. Swidan. 2020. "Quality by Design for the Development and Analysis of Enhanced In-Situ Forming Vesicles for the Improvement of the Bioavailability of Fexofenadine HCl In Vitro and In Vivo" Pharmaceutics 12, no. 5: 409. https://doi.org/10.3390/pharmaceutics12050409
APA StyleNasr, A. M., Qushawy, M. K., Elkhoudary, M. M., Gawish, A. Y., Elhady, S. S., & Swidan, S. A. (2020). Quality by Design for the Development and Analysis of Enhanced In-Situ Forming Vesicles for the Improvement of the Bioavailability of Fexofenadine HCl In Vitro and In Vivo. Pharmaceutics, 12(5), 409. https://doi.org/10.3390/pharmaceutics12050409