Interpretation of Drug Interaction Using Systemic and Local Tissue Exposure Changes


Abstract
:1. Introduction
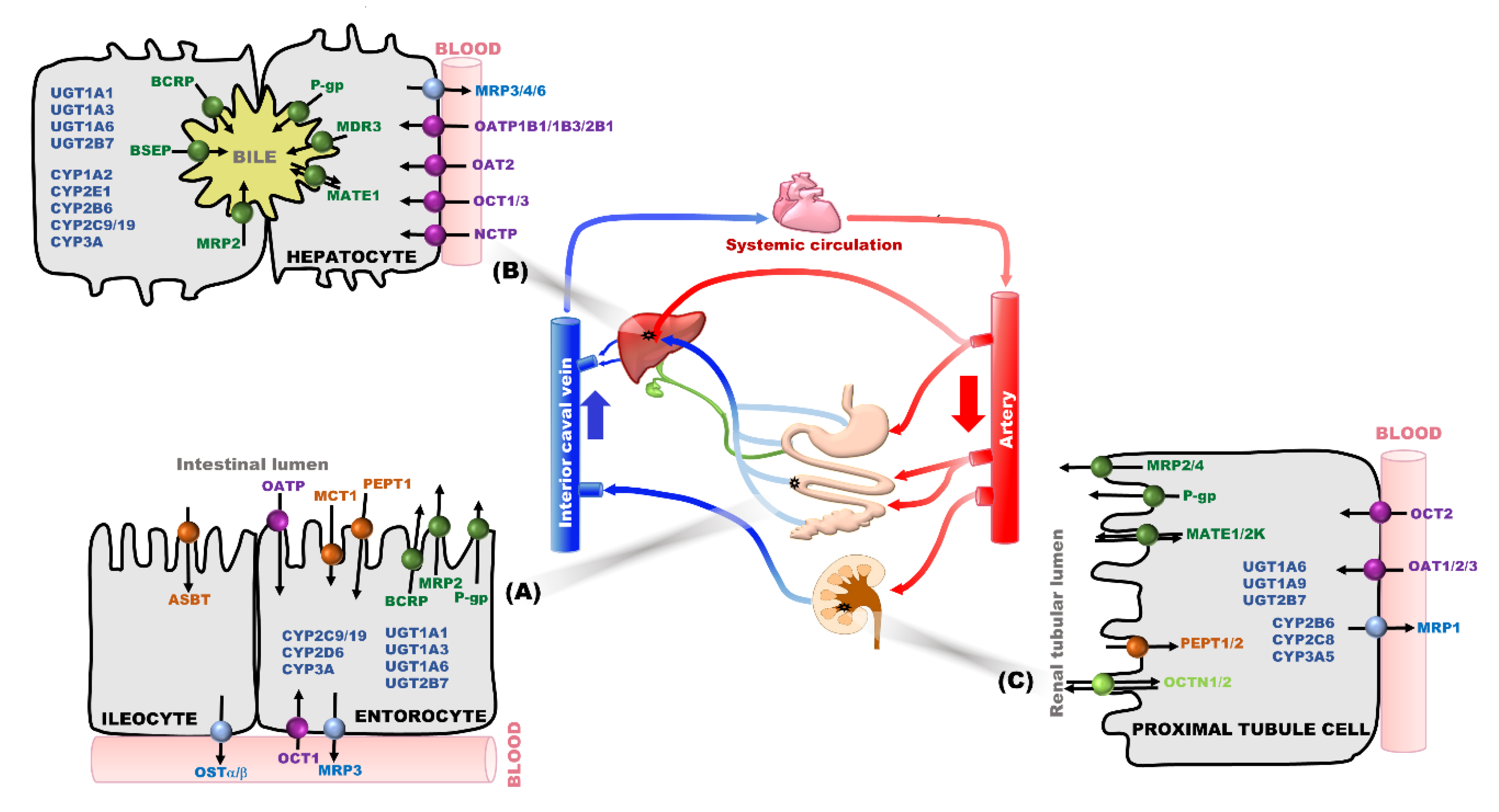
2. Transporter and/or Metabolic Enzymes as Main Determinant Factors in PK-Based DDIs
3. Effects of Transporter- or Metabolic Enzyme Mediated Changes of Systemic Exposure and/or Local Tissue Concentration on PD Effects
3.1. Changed Systemic Exposure of a Victim Drug Affecting the PD Effect
3.2. Changed Local Tissue Concentration of a Victim Drug Affecting the PD Effect
3.3. Additional Factors Affecting PD Effects with Changes of Systemic Exposure or Local Tissue Concentration of a Victim Drug
4. Challenging Experimental Approaches for Exploring the Transporter- or Metabolic Enzyme- Mediated Ddis
5. Concluding Remarks
Funding
Conflicts of Interest
References
- Wiggins, B.S.; Saseen, J.J.; Page, R.L., II; Reed, B.N.; Sneed, K.; Kostis, J.B.; Lanfear, D.; Virani, S.; Morris, P.B. Recommendations for management of clinically significant drug-drug interactions with statins and select agents used in patients with cardiovascular disease. Circulations 2016, 134, e468–e495. [Google Scholar]
- Dechanont, S.; Maphanta, S.; Butthum, B.; Kongkaew, C. Hospital admissions/visits associated with drug-drug interactions: A systemic review and meta-analysis. Pharmacoepidemiol. Drug Saf. 2014, 23, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Strong, J.M.; Zhang, L.; Reynolds, K.S.; Nallani, S.; Temple, R.; Abraham, S.; Habet, S.A.; Baweja, R.K.; Burchart, G.J.; et al. New era in drug interaction evaluations: US Food and Drug Administration update on CYP enzymes, transporters, and guideline process. J. Clin. Pharmacol. 2008, 48, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhou, Z.; Tay-Sontheimer, J.; Levy, R.H.; Ragueneau-Majlessi, I. Risk of clinically relevant pharmacokinetic-based drug-drug interactions with drugs approved by the U.S. Food and Drug Administration between 2013 and 2016. Drug Metab. Dispos. 2018, 46, 835–845. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Iwatsubo, T.; Kanamitsu, S.; Ueda, K.; Suzuki, H.; Sugiyama, Y. Prediction of pharmacokinetic alterations caused by drug-drug interactions: Metabolic interaction in the liver. Pharmacol. Rev. 1998, 50, 387–411. [Google Scholar]
- Zhang, D.; Hop, C.E.; Patilea-Vrana, G.; Gampa, G.; Seneviratne, H.K.; Unadkat, J.D.; Kenny, J.R.; Nagapudi, K.; Di, L.; Zhou, L.; et al. Drug concentration asymmetry in tissues and plasma for small molecule–related therapeutic modalities. Drug Metab. Dispos. 2019, 47, 1122–1135. [Google Scholar] [CrossRef]
- Mager, D.E.; Jusko, W.J. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J. Pharmacokinet. Pharmacodyn. 2001, 28, 507–532. [Google Scholar] [CrossRef]
- Rankovic, Z. CNS drug design: Balancing physicochemical properties for optimal brain exposure. J. Med. Chem. 2015, 58, 2584–2608. [Google Scholar] [CrossRef]
- Smith, D.A.; Di, L.; Kerns, E.H. The effect of plasma protein binding on in vivo efficacy: Misconceptions in drug discovery. Nat. Rev. Drug Discov. 2010, 9, 929–939. [Google Scholar] [CrossRef]
- Di, L.; Umland, J.P.; Trapa, P.E.; Maurer, T.S. Impact of recovery on fraction unbound using equilibrium dialysis. J. Pharm. Sci. 2012, 101, 1327–1335. [Google Scholar] [CrossRef]
- Di, L.; Breen, C.; Chambers, R.; Eckley, S.T.; Fricke, R.; Ghosh, A.; Harradine, P.; Kalvass, J.C.; Ho, S.; Lee, C.A.; et al. Industry perspective on contemporary protein-binding methodologies: Considerations for regulatory drug-drug interaction and related guidelines on highly bound drugs. J. Pharm. Sci. 2017, 106, 3442–3452. [Google Scholar] [CrossRef]
- Gabrielsson, J.; Peletier, I.A. Pharmacokinetic steady-states highlight interesting target-mediated disposition properties. AAPS J. 2017, 19, 772–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Waterschoot, R.A.; Parrott, N.J.; Olivares-Moralres, A.; Lave, T.; Rowland, M.; Smith, D.A. Impact of target interactions on small-molecule drug disposition: An overlooked area. Nat. Rev. Drug Discov. 2018, 17, 299–301. [Google Scholar] [CrossRef]
- Li, R.; Kimoto, E.; Niosi, M.; Tess, D.A.; Lin, J.; Tremaine, L.M.; Di, L. A study on pharmacokinetics of bosentan with systems modeling, part 2: Prospectively predicting systemic and liver exposure in healthy subjects. Drug Metab. Dispos. 2018, 46, 357–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Niosi, M.; Johnson, N.; Tess, D.A.; Kimoto, E.; Lin, J.; Yang, X.; Riccardi, K.A.; Ryu, S.; El-Kattan, A.F.; et al. A study on pharmacokinetics of bosentan with systems modeling, part 1: Translating systemic plasma concentration to liver exposure in healthy subjects. Drug Metab. Dispos. 2018, 46, 346–356. [Google Scholar] [CrossRef] [Green Version]
- Tsamandouras, N.; Dickinson, G.; Guo, Y.; Hall, S.; Rostami-Hodjegan, A.; Galetin, A.; Aarons, L. Development and application of a mechanistic pharmacokinetic model for simvastatin and its active metabolite simvastatin acid using an integrated population PBPK approach. Pharm. Res. 2015, 32, 1864–1883. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kusuhara, H.; Maeda, K.; Shitara, Y.; Sugiyama, Y. Physiologically based pharmacokinetic modeling to predict transporter-mediated clearance and distribution of pravastatin in humans. J. Pharmacol. Exp. Ther. 2009, 328, 652–662. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.K.; Kim, C.O.; Park, E.S.; Chung, J.Y. Verapamil decreases the glucose-lowering effect of metformin in healthy volunteers. Br. J. Clin. Pharmacol. 2014, 78, 1426–1432. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.K.; Yoo, J.S.; Lee, M.G.; Lee, D.H.; Lim, L.A.; Park, K.; Park, M.S.; Chung, J.Y. Rifampin enhances the glucose-lowering effect of metformin and increases OCT1 mRNA levels in healthy participants. Clin. Pharmacol. Ther. 2011, 89, 416–421. [Google Scholar] [CrossRef]
- Li, F.; Zhang, M.; Xu, D.; Liu, C.; Zhong, Z.Y.; Jia, L.L.; Hu, M.Y.; Yang, Y.; Liu, L.; Liu, X.D. Co-administration of paroxetine and pravastatin causes deregulation of glucose homeostasis in diabetic rats via enhanced paroxetine exposure. Acta. Pharmacol. Sin. 2014, 35, 792–805. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, Y.; Kishimoto, S.; Shibatani, N.; Inotsume, N.; Takeuchi, Y.; Fukushima, S. The disposition of pravastatin in a rat model of streptozotocin-induced diabetes and organic anion transporting polypeptide 2 and multidrug resistance associated protein 2 expression in the liver. Biol. Pharm. Bull. 2010, 33, 153–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Medicines Agency. Guideline on the Investigation of Drug Interactions. 2012. Available online: https://www.ema.europa.eu/documents/scientific-guideline/guideline-investigation-drug-interactions-en/pdf (accessed on 15 February 2019).
- US Food and Drug Administration, Center for Drug Evaluation and Research. Clinical Drug Interaction Studies-Study Design, Data Analysis, and Clinical Implications Guideline for Industry. 2017. Available online: https://www.fda.gov/downloads/drugs/guidances/ucm292362.pdf (accessed on 27 February 2020).
- Yu, J.; Petrie, I.D.; Levy, R.H.; Ragueneau-Majlessi, I. Mechanisms and clinical significance of pharmacokinetic-based drug-drug interactions with drug approved by the U.S. Food and Drug Administration in 2017. Drug Metab. Dispos. 2019, 47, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Tornio, A.; Filppula, A.M.; Niemi, M.; Backman, J.T. Clinical studies on drug-drug interactions involving metabolism and transport: Methodology, pitfalls, and interpretation. Clin. Pharmacol. Ther. 2019, 105, 1345–1361. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.P.; Tan, Z.R.; Huang, Z.; Ou-Yang, D.S.; Zhou, H.H. Isozyme-specific induction of low-dose aspirin on cytochrome P450 in healthy subjects. Clin. Pharmacol. Ther. 2003, 73, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Li, M.-P.; Tang, J.; Zhang, Z.-L.; Chen, X.-P. Induction of both P-glycoprotein and specific cytochrome P450 by aspirin eventually does not alter the antithrombotic effect of clopidogrel. Clin. Pharmacol. Ther. 2015, 97, 324. [Google Scholar] [CrossRef] [PubMed]
- Morgan, P.; Brown, D.G.; Lennard, S.; Anderton, M.J.; Barrett, J.C.; Eriksson, U.; Fidock, M.; Hamren, B.; Johnson, A.; March, R.E.; et al. Impact of a five-dimensional framework on R&D productivity at AstraZeneca. Nat. Rev. Drug Dispos. 2018, 17, 167–181. [Google Scholar]
- Oh, J.; Shin, D.; Lim, K.S.; Lee, S.; Jung, K.-H.; Chu, K.; Hong, K.S.; Shin, K.-H.; Cho, J.-Y.; Yoon, S.H.; et al. Aspirin decreases systemic exposure to clopidogrel through modulation of P-glycoprotein but does not alter its antithrombotic activity. Clin. Pharmacol. Ther. 2014, 95, 608–616. [Google Scholar] [CrossRef]
- Poulin, P. A paradigm shift in pharmacokinetic-pharmacodynamic (PKPD) modeling: Rule of thumb for estimating free drug level in tissue compared with plasma to guide drug design. J. Pharm. Sci. 2015, 104, 2359–2368. [Google Scholar] [CrossRef]
- Poulin, P.; Haddad, S. Advancing prediction of tissue distribution and volume of distribution of highly lipophilic compounds from simplified tissue composition-based models as a mechanistic animal alternative model. J. Pharm. Sci. 2012, 101, 2250–2261. [Google Scholar] [CrossRef]
- Rurak, C.D.; Hack, C.E.; Robinson, P.J.; Mahle, D.A.; Gearheart, M. Predicting passive and active tissue:plasma partition coefficient: Interindividual and interspecies variability. J. Pharm. Sci. 2014, 103, 2189–2198. [Google Scholar] [CrossRef]
- Shirata, Y.; Hoire, T.; Sigiyama, Y. Transporters as a determinant of drug clearance and tissue distribution. Eur. J. Pharm. Sci. 2006, 27, 425–446. [Google Scholar]
- Shitara, Y.; Maeda, K.; Ikejiri, K.; Yoshida, K.; Horie, T.; Sugiyama, Y. Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: Their roles in hepatic clearance and intestinal absorption. Biopharm. Drug Dispos. 2013, 34, 45–78. [Google Scholar] [CrossRef]
- Bosilkovska, M.; Samer, C.; Déglon, J.; Thomas, A.; Walder, B.; Desmeules, J.; Daali, Y. Evaluation of mutual drug-drug interaction within Geneva cocktail for cytochrome P450 phenotying using innovative dried blood sampling method. Basic Clin. Pharmacol. Ther. 2016, 119, 284–290. [Google Scholar] [CrossRef] [Green Version]
- Fuhr, U.; Hsin, C.H.; Li, X.; Jabrane, W.; Sorgel, F. Assessment of pharmacokinetic drug-drug interactions in humans: In vivo probe substrates for drug metabolism and drug transporter revised. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 507–536. [Google Scholar] [CrossRef]
- Yoshida, K.; Zhao, P.; Zhang, L.; Abernethy, D.R.; Rekic, D.; Reynolds, K.S.; Galetin, A.; Huang, S.M. In vitro-ion vivo extrapolation of metabolism- and transporter-mediated drug-drug interactions-overview of basic prediction methods. J. Pharm. Sci. 2017, 106, 2209–2213. [Google Scholar] [CrossRef] [Green Version]
- Gessnet, A.; Konig, J.; Fromm, M.F. Clinical aspects of transporter-mediated drug-drug interactions. Clin. Pharmacol. Ther. 2019, 105, 1386–1394. [Google Scholar] [CrossRef]
- Müller, F.; Fromm, M.F. Transporter-mediated drug-drug interactions. Pharmacogenomics 2011, 12, 1017–1037. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, S.M.; Lesko, L.J. Transporter-mediated drug-drug interactions. Clin. Pharmacol. Ther. 2011, 89, 481–484. [Google Scholar] [CrossRef]
- Morrissey, K.M.; Stocker, S.L.; Wittwer, M.B.; Xu, L.; Giacomini, K.M. Renal transporters in drug development. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 503–529. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Maeda, K.; Sugiyama, Y. Hepatic and intestinal drug transporters: Prediction of pharmacokinetic effects caused by drug-drug interactions and genetic polymorphisms. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 581–612. [Google Scholar] [CrossRef]
- König, J.; Fabian Müller, F.; Fromm, M.F. Transporters and drug-drug interactions: Important determinants of drug disposition and effects. Pharmacol. Rev. 2013, 65, 944–966. [Google Scholar] [CrossRef] [Green Version]
- Zientek, M.A.; Youdim, K. Reaction phenotyping: Advances in the experimental strategies used to characterize the contribution of drug-metabolizing enzymes. Drug Metab. Dispos. 2015, 43, 163–181. [Google Scholar] [CrossRef] [Green Version]
- Almazroo, O.A.; Miah, M.K.; Venkataramanan, R. Drug metabolism in the liver. Clin. Liver Dis. 2017, 21, 1–20. [Google Scholar] [CrossRef]
- Mittal, B.; Tulsyan, S.; Kumar, S.; Mittal, R.D.; Agarwal, G. Cytochrome P450 in cancer susceptibility and treatment. Adv. Clin. Chem. 2015, 71, 77–139. [Google Scholar]
- Olsen, L.; Oostenbrink, C.; Jorgensen, F.S. Prediction of cytochrome P450 mediated metabolism. Adv. Drug Deliv. Rev. 2015, 86, 61–71. [Google Scholar] [CrossRef]
- Achour, B.; Barber, J.; Rostami-Hodjegan, A. Expression of hepatic drug metabolizing cytochrome p450 enzymes and their intercorrelations: A meta-analysis. Drug Metab. Dispos. 2014, 42, 1349–1356. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Jia, R.; Ye, C.; Garcia, M.; Li, J.; Hidalgo, I.J. Glucuronidation and sulfation of 7-hydroxycoumarin in liver matrices from human, dog, monkey, rat, and mouse. In Vitro Cell Dev. Biol. Anim. 2005, 41, 97–103. [Google Scholar] [CrossRef]
- Terada, T.; Hira, D. Intestinal and hepatic drug transporters: Pharmacokinetic, pathophysiological, and pharmacogenetic roles. J. Gastroenterol. 2015, 50, 508–519. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Kusuhara, H.; Yokochi, M.; Toyoshima, J.; Inoue, K.; Yuasa, H.; Sugiyama, Y. Competitive inhibition of the luminal efflux by multidrug and toxin extrusions, but not basolateral uptake by organic cation transporter 2, is the likely mechanism underlying the pharmacokinetic drug-drug interactions caused by cimetidine in the kidney. J. Pharmacol. Exp. Ther. 2012, 340, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Kalliokoski, A.; Niemi, M. Impact of OATP transporters on pharmacokinetics. Br. J. Pharmacol. 2009, 158, 693–705. [Google Scholar]
- Chang, J.H.; Ly, J.; Plise, E.; Zhang, X.; Messick, K.; Wright, M.; Cheong, J. Differential effects of rifampin and ketoconazole on the blood and liver concentration of atorvastatin in wild-type and Cyp3a and Oatp1a/b knockout mice. Drug Metab. Dispos. 2014, 42, 1067–1073. [Google Scholar]
- Lepist, E.I.; Ray, A.S. Renal drug-drug interactions: What we have learned and where we are going. Exp. Opin. Drug Metab. Toxicol. 2012, 8, 433–448. [Google Scholar]
- Lepist, E.-I.; Ray, A.S. Renal transporter-mediated drug-drug interactions: Are they clinically relevant? J. Clin. Pharmacol. 2016, 56, S73–S81. [Google Scholar]
- Liang, X.; Giacomini, K.M. Transporters involved in metformin pharmacokinetics and treatment response. J. Pharm. Sci. 2017, 106, 2245–2250. [Google Scholar]
- Stieger, B.; Mahdi, Z.M.; Jager, W. Intestinal and hepatocellular transporters: Therapeutic effects and drug interactions of herbal supplements. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 399–416. [Google Scholar]
- Benet, L.Z.; Hoener, B.A. Changes in plasma protein binding have little clinical relevance. Clin. Pharmacol. Ther. 2002, 71, 115–121. [Google Scholar]
- Liu, X.; Wright, M.; Hop, C.E. Rational use of plasma protein and tissue binding data in drug design. J. Med. Chem. 2014, 57, 8238–8248. [Google Scholar]
- Hochman, J.; Tang, C.; Prueksaritanont, T. Drug-drug interactions related to altered absorption and plasma protein binding: Theoretical and regulatory considerations, and an industry perspective. J. Pharm. Sci. 2015, 104, 916–929. [Google Scholar]
- Kim, D.H. Gut microbiota-mediated drug-antibiotic interactions. Drug Metab. Dispos. 2015, 43, 1581–1589. [Google Scholar]
- Kehrer, D.F.; Sparreboom, A.; Verweij, J.; de Bruijn, P.; Nierop, C.A.; van de Schraaf, J.; Ruijgrok, E.J.; de Jonge, M.J. Modulation of irinotecan-induced diarrhea by cotreatment with neomycin in cancer patients. Clin. Cancer Res. 2001, 7, 1136–1141. [Google Scholar]
- Chiba, M.; Ishii, Y.; Sugiyama, Y. Prediction of hepatic clearance in human from in vitro data for successful drug development. AAPS J. 2009, 11, 262–276. [Google Scholar]
- Watanabe, T.; Kusuhara, H.; Sugiyama, Y. Application of physiologically based pharmacokinetic modeling and clearance concept to drugs showing transporter-mediated distribution and clearance in humans. J. Pharmacokinet. Pharmacodyn. 2010, 37, 575–590. [Google Scholar]
- Motohashi, H.; Inui, K. Multidrug and toxin extrusion family SLC47: Physiological, pharmacokinetic and toxicokinetic importance of MATE1 and MATE2-k. Mol. Aspects Med. 2013, 34, 661–668. [Google Scholar]
- You, B.H.; Chin, Y.-W.; Kim, H.; Choi, H.S.; Choi, Y.H. Houttuynia cordata extract increased systemic exposure and liver concentrations of metformin through OCTs and MATEs in rats. Phytother. Res. 2018, 32, 1004–1013. [Google Scholar]
- Han, S.Y.; Chae, H.-S.; You, B.H.; Chin, Y.-W.; Kim, H.; Choi, H.S.; Choi, Y.H. Lonicera japonica extract increases metformin distribution in the liver without change of systemic exposed metformin in rats. Phytother. Res. 2019, 32, 1004–1013. [Google Scholar]
- Manohar, S.; Leung, N. Cisplatin nephrotoxicity: A review of the literature. J. Nephrol. 2018, 31, 15–25. [Google Scholar]
- Sprowl, J.A.; van Doorn, L.; Hu, S.; van Gerven, L.; de Bruijm, P.; Li, L.; Gibson, A.A.; Mathijssen, R.H.; Sparreboom, A. Conjunctive therapy of cisplatin with the OCT2 inhibitor cimetidine: Influence on antitumor efficacy and systemic clearance. Clin. Pharmacol. Ther. 2013, 94, 585–592. [Google Scholar]
- Frost, C.E.; Byon, W.; Song, Y.; Wang, J.; Schuster, A.E.; Boyd, R.A.; Zhang, D.; Yu, C.; Dias, C.; Shenker, A.; et al. Effect of ketoconazole and diltiazem on the pharmacokinetics of apixaban, an oral direct factor Xa inhibitor. Br. J. Clin. Pharmacol. 2015, 79, 838–846. [Google Scholar]
- Hartter, S.; Koenen-Bergmann, M.; Sharma, A.; Nehmiz, G.; Lemke, U.; Timmer, W.; Reilly, P.A. Decrease in the oral bioavailability of dabigatran etexilate after co-medication with rifampicin. Br. J. Clin. Pharmacol. 2012, 74, 490–500. [Google Scholar]
- Greiner, B.; Eichelbaum, M.; Fritz, P.; Kreichgauer, H.-P.; von Richter, O.; Zundler, J.; Kroemer, H.K. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J. Clin. Invest. 1999, 104, 147–153. [Google Scholar]
- Sadeque, A.J.; Wandel, C.; He, H.; Shah, S.; Wood, A.J. Increased drug delivery to the brain by P-glycoprotein inhibition. Clin. Pharmacol. Ther. 2000, 68, 231–237. [Google Scholar]
- Allred, A.J.; Bowe, C.J.; Park, J.W.; Peng, B.; Williams, D.D.; Wire, M.B.; Lee, E. Eltrombopag increases plasma rosuvastatin exposure in healthy volunteers. Br. J. Clin. Pharmacol. 2011, 72, 321–329. [Google Scholar]
- Elsby, R.; Martin, P.; Surry, D.; Sharma, P.; Fenner, K. Solitary inhibition of the breast cancer resistance protein efflux transporter results in a clinically significant drug-drug interaction with rosuvastatin by causing up to a 2-fold increase in statin exposure. Drug Metab. Dispos. 2016, 44, 398–408. [Google Scholar]
- Rengelshausen, J.; Goggelmann, C.; Burhenne, J.; Riedel, K.D.; Ludwig, J.; Weiss, J.; Mikus, G.; Walter-Sack, I.; Haefeli, W.E. Contribution of increased oral bioavailability and reduced nonglomerular renal clearance of digoxin to the digoxin-clarithromycin interaction. Br. J. Clin. Pharmacol. 2003, 56, 32–38. [Google Scholar]
- Asberg, A.; Hartmann, A.; Fjeldså, E.; Bergan, S.; Holdaas, H. Bilateral pharmacokinetic interaction between cyclosporine A and atorvastatin in renal transplant recipients. Am. J. Transplant. 2001, 1, 382–386. [Google Scholar]
- Asberg, A. Interactions between cyclosporin and lipid-lowering drugs: Implications for organ transplant recipients. Drugs 2003, 63, 367–378. [Google Scholar]
- Maeda, K.; Ikeda, Y.; Fujita, T.; Yoshida, K.; Azuma, Y.; Haruyama, Y.; Yamane, N.; Kumagai, Y.; Sugiyama, Y. Identification of the rate-determining process in the hepatic clearance of atorvastatin in a clinical cassette microdosing study. Clin. Pharmacol. Ther. 2011, 90, 575–581. [Google Scholar]
- Mazzu, A.L.; Lasseter, K.C.; Shamblen, E.C.; Agarwal, V.; Lettieri, J.; Sundaresen, P. Itraconazole alters the pharmacokinetics of atorvastatin to a greater extent than either cerivastatin or pravastatin. Clin. Pharmacokinet. Ther. 2000, 68, 391–400. [Google Scholar]
- Markert, C.; Schweizer, Y.; Hellwig, R.; Wirsching, T.; Riedel, K.D.; Burhenne, J.; Weiss, J.; Mikus, G.; Haefeli, W.E. Clarithromycin substantially increases steady-state bosentan exposure in healthy volunteers. Br. J. Clin. Pharmacol. 2014, 77, 141–148. [Google Scholar]
- Fattinger, K.; Funk, C.; Pantze, M.; Weber, C.; Reichen, J.; Stieger, B.; Meier, P.J. The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: A potential mechanism for hepatic adverse reactions. Clin. Pharmacol. Ther. 2001, 69, 223–231. [Google Scholar]
- Preston, C.L. Stockley’s Drug Interactions, 11th ed.; Pharmaceutical Press: London, UK, 2016. [Google Scholar]
- Prueksaritanont, T.; Chu, X.; Evers, R.; Klopfer, S.O.; Caro, L.; Kothare, P.A.; Dempsey, C.; Rasmussen, S.; Houle, R.; Chan, G.; et al. Pitavastatin is a more sensitive and selective organic anion-transporting polypeptide 1B clinical probe than rosuvastatin. Br. J. Clin. Pharmacol. 2014, 78, 587–598. [Google Scholar]
- Jacobson, T.A. Comparative pharmacokinetic interaction profiles of pravastatin, simvastatin, and atorvastatin when coadministered with cytochrome P450 inhibitors. Am. J. Cardiol. 2004, 94, 1140–1146. [Google Scholar]
- Simonson, S.G.; Raza, A.; Martin, P.D.; Mitchell, P.D.; Jarcho, J.A.; Brown, C.D.; Windass, A.S.; Schneck, D.W. Rosuvastatin pharmacokinetics in heart transplant recipients administered an antirejection regimen including cyclosporine. Clin. Pharmacol. Ther. 2004, 76, 167–177. [Google Scholar]
- He, J.; Yu, Y.; Prasad, B.; Link, J.; Miyaoka, R.S.; Chen, X.; Unadkat, J.D. PET imaging of Oatp mediated hepatobiliary transport of [(11)C] rosuvastatin in the rat. Mol. Pharm. 2014, 11, 2745–2754. [Google Scholar]
- Schneck, D.W.; Birmingham, B.K.; Zalikowski, J.A.; Mitchell, P.D.; Wang, Y.; Martin, P.D.; Lasseter, K.C.; Brown, C.D.; Windass, A.S.; Raza, A. The effect of gemfibrozil on the pharmacokinetics of rosuvastatin. Clin. Pharmacol. Ther. 2004, 75, 455–463. [Google Scholar]
- Maeda, K.; Tian, Y.; Fujita, T.; Ikeda, Y.; Kumagai, Y.; Kondo, T.; Tanabe, K.; Nakayama, H.; Horita, S.; Kusuhara, H.; et al. Inhibitory effects of p-aminohippurate and probenecid on the renal clearance of adefovir and benzylpenicillin as probe drugs for organic anion transporter (OAT) 1 and OAT3 in humans. Eur. J. Pharm. Sci. 2014, 59, 94–103. [Google Scholar]
- Leahey, E.B.; Bigger, J.T.; Butler, V.P.; Reiffel, J.A.; O’Connell, G.C.; Scaffidi, L.E.; Rottman, J.N. Quinidine-digoxin interaction: Time course and pharmacokinetics. Am. J. Cardiol. 1981, 48, 1141–1146. [Google Scholar]
- Moore, K.H.; Yuen, G.J.; Raasch, R.H.; Eron, J.J.; Martin, D.; Mydlow, P.K.; Hussey, E.K. Pharmacokinetics of lamivudine administered alone and with trimethoprim-sulfamethoxazole. Clin. Pharmacol. Ther. 1996, 59, 550–558. [Google Scholar]
- Müller, F.; Konig, J.; Hoier, E.; Mandery, K.; Fromm, M.F. Role of organic cation transporter OCT2 and multidrug and toxin extrusion proteins MATE1 and MATE2-K for transport and drug interactions of the antiviral lamivudine. Biochem. Pharmacol. 2013, 86, 808–815. [Google Scholar]
- Grun, B.; Kiessling, M.K.; Burhenne, J.; Riedel, K.D.; Weiss, J.; Rauch, G.; Haefeli, W.E.; Czock, D. Trimethoprim-metformin interaction and its genetic modulation by OCT2 and MATE1 transporters. Br. J. Clin. Pharmacol. 2013, 76, 787–796. [Google Scholar]
- Müller, F.; Pontones, C.A.; Renner, B.; Mieth, M.; Hoier, E.; Auge, D.; Maas, R.; Zolk, O.; Fromm, M.F. N1-methylnicotinamide as an endogenous probe for drug interactions by renal cation transporters: Studies on the metformin-trimethoprim interaction. Eur. J. Clin. Pharmacol. 2015, 71, 85–94. [Google Scholar]
- Song, I.H.; Zong, J.; Borland, J.; Jerva, F.; Wynne, B.; Zamek-Gilszczynski, M.J.; Humphreys, J.E.; Bowers, G.D.; Choukour, M. The effect of dolutegravir on the pharmacokinetics of metformin in healthy subjects. J. Acquir. Immune. Defic. Syndr. 2016, 72, 400–407. [Google Scholar]
- Li, F.; Xu, D.; Shu, N.; Zhong, Z.; Zhang, M.; Liu, C.; Ling, Z.; Liu, L.; Liu, X. Co-administration of paroxetine increased the systemic exposure of pravastatin in diabetic rats due to the decrease in liver distribution. Xenobiotica 2015, 45, 794–802. [Google Scholar]
- Grube, M.; Köck, K.; Oswald, S.; Draber, K.; Meissner, K.; Eckel, L.; Böhm, M.; Felix, S.B.; Vogelgesang, S.; Jedlitschky, G.; et al. Organic anion transporting polypeptide 2B1 is a high-affinity transporter for atorvastatin and is expressed in the human heart. Clin. Pharmacol. Ther. 2006, 80, 607–620. [Google Scholar]
- Shin, E.; Shin, N.; Oh, J.-H.; Lee, Y.-J. High-dose metformin may increase the concentration of atorvastatin in the liver by inhibition of multidrug resistance-associated protein 2. J. Pharm. Sci. 2017, 106, 961–967. [Google Scholar]
- Li, L.; Lei, H.; Wang, W.; Du, W.; Yuan, J.; Yuan, J.; Tu, M.; Zhou, H.; Zeng, S.; Jiang, H. Co-administration of nuciferine reduced the concentration of metformin in liver via differential inhibition of hepatic drug transporter OCT1 and MATE1. Biopharm. Drug Dispos. 2018, 39, 411–419. [Google Scholar]
- Somogyi, A.; Stockley, C.; Keal, J.; Rolan, P.; Bochner, F. Reduction of metformin renal tubular secretion by cimetidine in man. Br. J. Clin. Pharmacol. 1987, 23, 545–551. [Google Scholar]
- Stocker, S.L.; Morrissey, K.M.; Yee, S.W.; Castro, R.A.; Xu, L.; Dahlin, A.; Ramirez, A.H.; Roden, D.M.; Wilke, R.A.; McCarty, C.A.; et al. The effect of novel promoter variants in MATE1 and MATE2 on the pharmacokinetics and pharmacodynamics of metformin. Clin. Pharmacol. Ther. 2013, 93, 186–194. [Google Scholar]
- Kusuhara, H.; Ito, S.; Kumagai, Y.; Jiang, M.; Shiroshita, T.; Moriyama, Y.; Inoue, K.; Yuasa, H.; Sugiuama, Y. Effects of a MATE protein inhibitor, pyrimethamine, on the renal elimination of metformin at oral microdose and at therapeutic dose in healthy subjects. Clin. Pharmacol. Ther. 2011, 89, 837–844. [Google Scholar]
- Ito, S.; Kusuhara, H.; Kuroiwa, Y.; Wu, C.; Moriyama, Y.; Inoue, K.; Kondo, T.; Yuasa, H.; Nakayama, H.; Horita, S.; et al. Potent and specific inhibition of mMate1-mediated efflux of type 1 organic cations in the liver and kidney by pyrimethamine. J. Pharmakos. Exp. Ther. 2010, 333, 341–350. [Google Scholar]
- Choi, Y.H.; Lee, U.; Lee, B.K.; Lee, M.G. Pharmacokinetic interaction between itraconazole and metformin in rats: Competitive inhibition of metabolism of each drug by each other via hepatic and intestinal CYP3A1/2. Br. J. Pharmacol. 2010, 161, 815–829. [Google Scholar]
- Bechmann, K.A.; Lewis, J.D. Predicting inhibitory drug–drug interactions and evaluating drug interaction reports using inhibition constants. Ann. Pharmacother. 2005, 39, 1064–1072. [Google Scholar]
- Ma, Y.; Khojasteh, S.C.; Hop, C.E.; Erickson, H.K.; Polson, A.; Pillow, T.H.; Yu, S.F.; Wang, H.; Dragovich, P.S.; Zhang, D. Antibody drug conjugates differentiate uptake and DNA alkylation of pyrrolobenzodiazepines in tumors from organs of xenograft mice. Drug Metab. Dispos. 2016, 44, 1958–1962. [Google Scholar]
- Zhang, D.; Yu, S.F.; Ma, Y.; Xu, K.; Dragovich, P.S.; Pillow, T.H.; Liu, L.; Del Rosario, G.; He, J.; Pei, Z.; et al. Chemical structure and concentration of intratumor catabolites determine efficacy of antibody drug conjugates. Drug Metab. Dispos. 2016, 44, 1517–1523. [Google Scholar]
- Yamazaki, M.; Suzuki, H.; Sugiyama, Y. Recent advances in carrier-mediated hepatic uptake and biliary excretion of xenobiotics. Pharm. Res. 1996, 13, 497–513. [Google Scholar]
- Shitara, Y.; Sugiyama, Y. Pharmacokinetic and pharmacodynamic alterations of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors: Drug–drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacol. Ther. 2006, 112, 71–105. [Google Scholar]
- Anitha, N.; Rao, J.; Kavimani, S.; Himabindu, V. Pharmacodynamic drug interaction of metformin with statins in rats. J. Pharmacol. Toxicol. 2008, 3, 409–413. [Google Scholar]
- Bjornsson, E.; Jacobsen, E.I.; Kalaitzakis, E. Hepatotoxicity associated with statins: Reports of idiosyncratic liver injury post-marketing. J. Hepatol. 2012, 56, 374–380. [Google Scholar]
- Rose, R.H.; Neuhoff, S.; Abduljalil, K.; Chetty, M.; Rostami-Hodjegan, A.; Jamei, M. Application of a physiologically based pharmacokinetic model to predict OATP1B1-related variability in pharmacodynamics of rosuvastatin. CPT Pharmacomet. Syst. Pharmacol. 2014, 3, e124. [Google Scholar]
- Sundelin, E.; Gormsen, L.C.; Jensen, J.B.; Vendelbo, M.H.; Jakobsen, S.; Munk, O.L.; Christensen, M.; Brøsen, K.; Frøkiaer, J.; Jessen, N. Genetic polymorphisms in organic cation transporter 1 attenuates hepatic metformin exposure in humans. Clin. Pharmacol. Ther. 2017, 102, 841–848. [Google Scholar]
- Jensen, J.B.; Sundelin, E.I.; Jakobsen, S.; Gormsen, L.C.; Munk, O.L.; Frokier, J.; Jessen, N. [11C]-labeled metformin distribution in the liver and small intestine using dynamic positron emission tomography in mice demonstrates tissue-specific transporter dependency. Diabetes 2016, 65, 1724–1730. [Google Scholar]
- Shu, Y.; Sheardown, S.A.; Brown, C.; Owen, R.P.; Zhang, S.; Castro, R.A.; Ianculescu, A.G.; Yue, L.; Lo, J.C.; Burchard, E.G.; et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J. Clin. Invest. 2007, 117, 1422–1431. [Google Scholar]
- Vildhede, A.; Nguyen, C.; Erickson, B.K.; Kunz, R.C.; Jones, R.; Kimoto, E. Comparison of proteomic quantification approaches for hepatic drug transporters: Multiplexed global quantitation correlates with targeted proteomic quantitation. Drug Metab. Dispos. 2018, 46, 692–696. [Google Scholar]
- Couto, N.; Al-Majdoub, Z.M.; Gibson, S.; Davies, P.J.; Achour, B.; Harwood, M.D.; Carlson, G.; Barber, J.; Rostami-Hodjegan, A.; Warhurst, G. Quantitative proteomics of clinically relevant drug-metabolizing enzymes and drug transporters and their inter-correlations in the human small intestine. Drug Metab. Dispos. 2020, 48, 407. [Google Scholar]
- Pena, A.; Liu, P.; Derendorf, H. Microdialysis in peripheral tissues. Adv. Drug Deliv. Rev. 2000, 45, 189–216. [Google Scholar]
- Dahyot, C.; Marchand, S.; Bodin, M.; Debeane, B.; Mimoz, O.; Couet, W. Application of basic pharmacokinetic concepts to analysis of microdialysis data: Illustration with imipenem muscle distribution. Clin. Pharmacokinet. 2008, 47, 181–189. [Google Scholar]
- Tuntland, T.; Ethell, B.; Kosaka, T.; Blasco, F.; Zhang, R.X.; Jain, M.; Gould, T.; Hoffmaster, K. Implementation of pharmacokinetic and pharmacodynamic strategies in early research phases of drug discovery and development at Novartis Institute of Biomedical Research. Front. Pharmacol. 2014, 28, 174. [Google Scholar]
- Guo, Y.; Chu, X.; Parrott, N.J.; Brouwer, K.L.R.; Hsu, V.; Nagar, S.; Matsson, P.; Sharma, P.; Snoeys, J.; Sugiyama, Y.; et al. Advancing predictions of tissue and intracellular drug concentrations using in vitro, imaging and physiologically based pharmacokinetic modeling approaches. Clin. Pharmacol. Ther. 2018, 104, 865–889. [Google Scholar]

{kind=link}
| Protein | Location | Direction | Ref. |
|---|---|---|---|
| P-gp (MDR1) | Apical membrane in enterocyte | efflux | [43,51] |
| Canalicular membrane in hepatocyte | efflux | ||
| 1 Luminal membrane in renal proximal tubule cell | efflux | ||
| MDR3 | Canalicular membrane in hepatocyte | efflux | [43] |
| BSEP | Canalicular membrane in hepatocyte | efflux | [43] |
| BCRP | Apical membrane in enterocyte | efflux | [43] |
| Canalicular membrane in hepatocyte | efflux | ||
| 1 Luminal membrane in renal proximal tubule cell | efflux | ||
| MRP1 | Basolateral membrane in renal proximal tubule cell | efflux | [6,43] |
| MRP2 | Apical membrane in enterocyte | efflux | |
| Canalicular membrane in hepatocyte | efflux | ||
| 1 Luminal membrane in renal proximal tubule cell | efflux | ||
| MRP3 | Basolateral membrane in enterocyte | uptake | |
| Basolateral membrane in hepatocyte | efflux | ||
| Basolateral membrane in renal proximal tubule cell | efflux | ||
| MRP4 | Basolateral membrane in hepatocyte | efflux | [38,43] |
| 1 Luminal membrane in renal proximal tubule cell | efflux | ||
| MRP5,6 | Basolateral membrane in hepatocyte | efflux | [43] |
| OATP1B1 | Basolateral membrane in enterocyte | uptake | [43,52] |
| Basolateral membrane in hepatocyte | uptake | ||
| OATP1B3 | Basolateral membrane in enterocyte | uptake | |
| Basolateral membrane in hepatocyte | uptake | ||
| OATP2B1 | Basolateral membrane in enterocyte | uptake | [43,53] |
| Basolateral membrane in hepatocyte | uptake | ||
| OAT1 | Basolateral membrane in renal proximal tubule cell | uptake | [43] |
| OAT2 | Basolateral membrane in hepatocyte | uptake | [38,43] |
| Basolateral membrane in renal proximal tubule cell | uptake | ||
| OAT3 | Basolateral membrane in renal proximal tubule cell | uptake | [43] |
| OAT4 | 1 Luminal membrane in renal proximal tubule cell | efflux/uptake 2 | |
| OCT1 | Basolateral membrane in enterocyte | uptake | [43,54,55,56] |
| Basolateral membrane in hepatocyte | uptake | ||
| OCT2 | Basolateral membrane in renal proximal tubule cell | uptake | |
| OCT3 | Basolateral membrane in enterocyte | uptake | |
| Canalicular membrane in hepatocyte | efflux/uptake | ||
| Basolateral membrane in renal proximal tubule cell | uptake | ||
| MATE1 | Canalicular membrane in hepatocyte | efflux/uptake | [43,51] |
| 1 Luminal membrane in renal proximal tubule cell | efflux/uptake 2 | ||
| MATE2-K | 1 Luminal membrane in renal proximal tubule cell | efflux/uptake 2 | |
| PEPT1 | Apical membrane in enterocyte | uptake | [43,57] |
| 1 Luminal membrane in renal proximal tubule cell | uptake 2 | ||
| PEPT2 | 1 Luminal membrane in renal proximal tubule cell | uptake 2 |
| Victim Drug | Perpetrator Drug | Underlying Mechanism 1 | PK Change of a Victim Drug | PD Change of a Victim Drug | Ref. |
|---|---|---|---|---|---|
| Apixaban | Ketoconazole | (−) P-gp in enterocyte | AUC↑ | ADR↑ (bleeding risk) | [70] |
| Dabigatran | Rifampin | (+) P-gp in enterocyte | AUC↓ | TR  , safety , safety | [71] |
| Digoxin | Rifampin | (+) P-gp in enterocyte | AUC↓ | TR↓ | [72] |
| Loperamide | Quinidine | (−) P-gp in enterocyte or brain | AUC↑ | ADR↑(respiratory depression) by P-gp inhibition in brain (not enterocyte) | [73] |
| Rosuvastatin | Eltrombopag, Fostamatinib | (−) BCRP in enterocyte | AUC↑ | ADR↑ (myopathy), TR↑ | [43,74,75] |
| Clopidogrel | Aspirin | (+) P-gp in enterocyte (+) CYP2C9 in hepatocyte | AUC↓, F↓, H4 (active metabolite)↑ | Platelet inhibition effect | [29] |
| Digoxin | Clarithromycin | (−) P-gp in enterocyte (−) CYP2D6 or 3A4 inhibition in hepatocyte | AUC↑, CL↓, CLR↓ (by non-glomerular renal clearance) | ADR↑ (digoxin toxicity) | [76] |
| Atorvastatin | Cyclosporine | (−) OATP1B1, 1B3, 2B1 in hepatocyte (−) CYP3A in hepatocyte | AUC↑, hepatic uptake | Muscle-related toxicity↑ | [1,43,77,78] |
| Itraconazole | (−) CYP3A in hepatocyte | AUC↑, hepatic uptake | - | [62,79,80] | |
| Bosentan | Clarithromycin | (−) OATP1B1, 1B3 in hepatocyte | AUC↑ | ADR↑ (cholestatic liver injury) | [81,82] |
| Pitavastatin | Cyclosporine, rifampin | (−) OATP1B1, 1B3, 2B1 in hepatocyte | AUC↑ | ADR↑ | [83,84] |
| Atrovastatin, simvastatin | Itraconazole, mibefradil, verapamil | (−) CYP3A4 in hepatocyte | AUC↑, Cmax↑ | ADR↑ (myopathy, fatal rhadomyolysis) | [85] |
| Atrovastatin, pravastatin, simvastatin | Clarithromycin | (−) OATP1B1, 1B3, 2B1 in hepatocyte (−) CYP3A4 in hepatocyte | AUC↑, Cmax↑ | ADR↑ (myopathy, fatal rhadomyolysis) | [85] |
| Rosuvastatin | Cyclosporine | (−) OATP1B1, 1B3, 2B1 in hepatocyte | AUC↑, hepatic cons 2 | ADR↑ | [86,87] |
| Gemfibrozil | (−) OATP2B1 in hepatocyte | AUC↑, hepatic cons 2 | ADR↑ | [87,88] | |
| Simvastatin | Cyclosporine | (−) OATP1B1 in hepatocyte (−) CYP3A4 in hepatocyte | AUC↑ | ADR↑ (myopathy) | [43] |
| Adefovir | Probenecid | (−) OAT1 in proximal tubule cell | AUC↑, CLR↓ | ADR↑ (nephrotoxicity) | [43,89] |
| Benzylpenicillin | Probenecid | (−) OAT3 in proximal tubule cell | AUC↑, CLR↓ | ADR↑ | [89] |
| Digoxin | Quinidine | (−) CYP2D6 or 3A4 inhibition in hepatocyte (−) P-gp in proximal tubule cell | AUC↑, CL↓, CLNR↓, CLR↓ | ADR↑ (digoxin toxicity) | [83,90] |
| Lamivudine | Trimethoprim /sulfamethoxazole | (−) OCT2, MATE1, MATE2-K in proximal tubule cell | AUC↑, CLR↓ | ADR↑ (hepatotoxicity) | [91,92] |
| Metformin | Trimethoprim | (−) OCT2, MATE1 in proximal tubule cell | Cmax↑, AUC↑, CL/F↓, CLR↓ | ADR↑ (plasma lactate↑, lactic acidosis especially in renal dysfunction patients) | [93] |
| (−) OCT2, MATE1, MATE2-K in proximal tubule cell | Cmax↑, AUC↑, CLR↓ | ADR↑ (plasma lactate↑, lactic acidosis) | [94] | ||
| Dolutegravir | (−) OCT2 in proximal tubule cell | Cmax↑, AUC↑ | ADR↑ (plasma lactate↑, lactic acidosis) | [95] | |
| Pravastatin | Paroxetine | (−) Mrp2 in enterocyte; (−) Oatp2 in enterocyte/hepatocyte | Intestinal absorption↑, AUC↑, hepatic uptake↓, hepatic cons↓ | Lipid-lowing effect ↓ | [20,21,96] |
| Atorvastatin | Rifampin | (−) OATPs in hepatocyte | AUC↑, hepatic uptake↓ | Lipid-lowing effect↓ | [52,53,79,97] |
| Atorvastatin | Metformin | (−) MRP2 in hepatocyte | Biliary excretion↓, hepatic cons 2↑ | Lipid-lowing effect↑ | [98] |
| Metformin | Rifampin | (+) mRNA of OCT1 in blood cells | AUC↑ (probable hepatic cons 2↑) | Glucose-lowering effect↑ | [19] |
| Metformin | Lonicera japonica extract | (−) MATE1 in hepatocyte | AUC , hepatic cons 2↑ | Glucose tolerance effect↑ | [67] |
| Metformin | Nuciferine | (−) OCT1 and MATE1 in hepatocyte | Hepatic cons 2↓ | Glucose-lowering effect↓ | [99] |
| Rosuvastatin | Rifampin | (−) OATP1B1, 1B3, 2B1 in hepatocyte | AUC↑, hepatic cons 2↓, renal cons 2↓, hepatic biliary excretion | ADR↑ | [87] |
| Metformin | Houttuynia cordata extract | (−) MATE1 in hepatocyte (−) OCT2 in proximal tubule cell | AUC↑, CLR↓, hepatic cons 2↑ | Glucose tolerance effect↑ | [66] |
| Metformin | Cimetidine | (−) MATE1 in hepatocyte; (−) MATE1, MATE2-K in proximal tubule cell | AUC↑, hepatic cons 2↑, (biliary excretion↓), renal cons 2↑, CLR↓ | Glucose-lowering effect↑ | [51,55,56,100,101] |
| Metformin | Pyrimethamine | (−) MATE1 in hepatocyte; (−) MATE1 in proximal renal tubule | AUC↑, Cmax↑, CLR↓, CLCR↓, SCR 3↑, hepatic cons 2↑ | Glucose lowering effect↑ | [101,102,103] |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, Y.H. Interpretation of Drug Interaction Using Systemic and Local Tissue Exposure Changes. Pharmaceutics 2020, 12, 417. https://doi.org/10.3390/pharmaceutics12050417
Choi YH. Interpretation of Drug Interaction Using Systemic and Local Tissue Exposure Changes. Pharmaceutics. 2020; 12(5):417. https://doi.org/10.3390/pharmaceutics12050417
Chicago/Turabian StyleChoi, Young Hee. 2020. "Interpretation of Drug Interaction Using Systemic and Local Tissue Exposure Changes" Pharmaceutics 12, no. 5: 417. https://doi.org/10.3390/pharmaceutics12050417
APA StyleChoi, Y. H. (2020). Interpretation of Drug Interaction Using Systemic and Local Tissue Exposure Changes. Pharmaceutics, 12(5), 417. https://doi.org/10.3390/pharmaceutics12050417