Liposome-Embedding Silicon Microparticle for Oxaliplatin Delivery in Tumor Chemotherapy


 ,
,  ,
,  ,
,  , , , ,
, , , , 
 ,
,  ,
,
Abstract
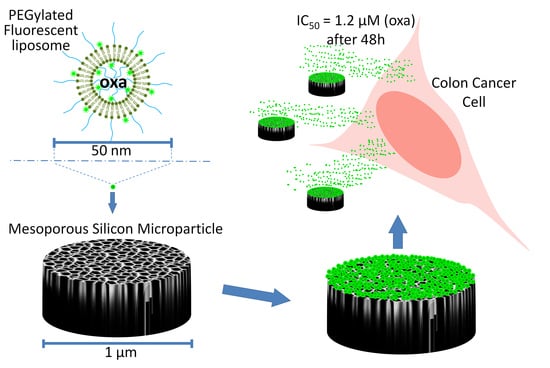
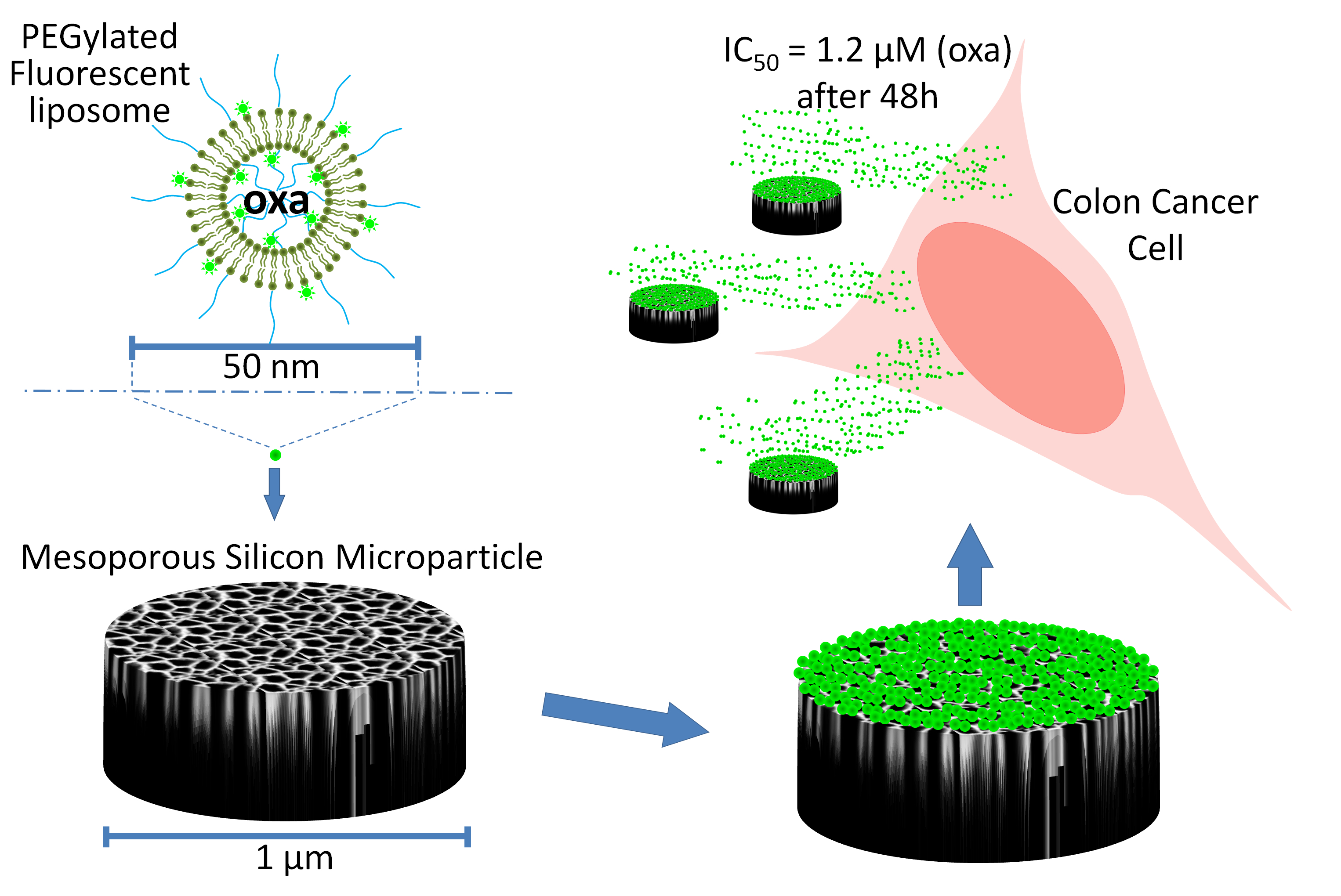
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Equipment
2.3. MSMP Production
2.4. Liposome Production
2.5. Physicochemical Characterization of Liposomes
2.6. Transmission Electron Microscopy (TEM)
2.7. Oxaliplatin Encapsulation Efficiency
2.8. HPLC Analysis and Validation
2.9. Cell Cultures
2.10. MTT Assay
2.11. Cellular Uptake of Liposomes
2.12. Surface Charge Modification of MSMP
2.13. MSV Assembly
2.14. Release Kinetics of Liposomes from MSV
2.15. Size of Released Liposomes
2.16. Confocal Microscopy of Cells Incubated with MSVs.
3. Results
3.1. Design of Particles
3.2. Effect of Liposomal Formulations in Cell Cultures
3.3. Multistage Vector Assembly
3.4. Release of Liposomes from the Multistage Vector
3.5. Effects of the Multistage Vector in Cell Culture
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tang, J.; Zhang, R.; Guo, M.; Shao, L.; Liu, Y.; Zhao, Y.; Zhang, S.-J.; Wu, Y.; Chen, C. Nucleosome-inspired nanocarrier obtains encapsulation efficiency enhancement and side effects reduction in chemotherapy by using fullerenol assembled with doxorubicin. Biomaterials 2018, 167, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, J.H.; Van De Ven, A.; Godin, B.; Blanco, E.; Serda, R.E.; Grattoni, A.; Ziemys, A.; Bouamrani, A.; Hu, T.; Ranganathan, S.; et al. Enabling individualized therapy through nanotechnology. Pharmacol. Res. 2010, 62, 57–89. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef]
- Pasut, G.; Paolino, D.; Celia, C.; Mero, A.; Joseph, A.S.; Wolfram, J.; Cosco, D.; Schiavon, O.; Shen, H.; Fresta, M.; et al. Polyethylene glycol (PEG)-dendron phospholipids as innovative constructs for the preparation of super stealth liposomes for anticancer therapy. J. Control. Release 2015, 199, 106–113. [Google Scholar] [CrossRef]
- D’Apolito, R.; Tomaiuolo, G.; Taraballi, F.; Minardi, S.; Kirui, D.; Liu, X.; Cevenini, A.; Palomba, R.; Ferrari, M.; Salvatore, F.; et al. Red blood cells affect the margination of microparticles in synthetic microcapillaries and intravital microcirculation as a function of their size and shape. J. Control. Release 2015, 217, 263–272. [Google Scholar] [CrossRef]
- Wolfram, J.; Shen, H.; Ferrari, M. Multistage vector (MSV) therapeutics. J. Control. Release 2015, 219, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Venuta, A.; Wolfram, J.; Shen, H.; Ferrari, M. Post-nano strategies for drug delivery: Multistage porous silicon microvectors. J. Mater. Chem. B 2016, 5, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Godin, B.; Chiappini, C.; Srinivasan, S.; Alexander, J.F.; Yokoi, K.; Ferrari, M.; Decuzzi, P.; Liu, X. Discoidal porous silicon particles: Fabrication and biodistribution in breast cancer bearing mice. Adv. Funct. Mater. 2012, 22, 4225–4235. [Google Scholar] [CrossRef]
- Martinez, J.O.; Evangelopoulos, M.; Chiappini, C.; Liu, X.; Ferrari, M.; Tasciotti, E. Degradation and biocompatibility of multistage nanovectors in physiological systems. J. Biomed. Mater. Res. Part A 2013, 102, 3540–3549. [Google Scholar] [CrossRef]
- Blanco, E.; Sangai, T.; Hsiao, A.; Ferrati, S.; Bai, L.; Liu, X.; Meric-Bernstam, F.; Ferrari, M. Multistage delivery of chemotherapeutic nanoparticles for breast cancer treatment. Cancer Lett. 2013, 334, 245–252. [Google Scholar] [CrossRef]
- Van De Ven, A.; Kim, P.; Haley, O.; Fakhoury, J.R.; Adriani, G.; Schmulen, J.; Moloney, P.; Hussain, F.; Ferrari, M.; Liu, X.; et al. Rapid tumoritropic accumulation of systemically injected plateloid particles and their biodistribution. J. Control. Release 2011, 158, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Zhang, G.; Mai, J.; Deng, X.; Segura-Ibarra, V.; Wu, S.; Shen, J.; Liu, H.; Hu, Z.; Chen, L.; et al. An injectable nanoparticle generator enhances delivery of cancer therapeutics. Nat. Biotechnol. 2016, 34, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, M. Frontiers in cancer nanomedicine: Directing mass transport through biological barriers. Trends Biotechnol. 2010, 28, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Corbo, C.; Parodi, A.; Evangelopoulos, M.; Engler, D.A.; Matsunami, R.K.; Engler, A.C.; Molinaro, R.; Scaria, S.; Salvatore, F.; Tasciotti, E.; et al. Proteomic profiling of a biomimetic drug delivery platform. Curr. Drug Targets 2015, 16, 1540–1547. [Google Scholar] [CrossRef]
- Palomba, R.; Parodi, A.; Evangelopoulos, M.; Acciardo, S.; Corbo, C.; De Rosa, E.; Yazdi, I.K.; Scaria, S.; Molinaro, R.; Furman, N.E.T.; et al. Biomimetic carriers mimicking leukocyte plasma membrane to increase tumor vasculature permeability. Sci. Rep. 2016, 6, 34422. [Google Scholar] [CrossRef]
- Parodi, A.; Corbo, C.; Cevenini, A.; Molinaro, R.; Palomba, R.; Pandolfi, L.; Agostini, M.; Salvatore, F.; Tasciotti, E. Enabling cytoplasmic delivery and organelle targeting by surface modification of nanocarriers. Nanomedicine 2015, 10, 1923–1940. [Google Scholar] [CrossRef]
- Parodi, A.; Quattrocchi, N.; Van De Ven, A.; Chiappini, C.; Evangelopoulos, M.; Martinez, J.O.; Brown, B.S.; Khaled, S.Z.; Yazdi, I.K.; Enzo, M.V.; et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat. Nanotechnol. 2012, 8, 61–68. [Google Scholar] [CrossRef]
- Shen, H.F.; Rodriguez-Aguayo, C.; Xu, R.; Gonzalez-Villasana, V.; Mai, J.H.; Huang, Y.; Zhang, G.D.; Guo, X.J.; Bai, L.T.; Qin, G.T.; et al. Enhancing chemotherapy response with sustained EphA2 silencing using multistage vector delivery. Clin. Cancer Res. 2013, 19, 1806–1815. [Google Scholar] [CrossRef]
- Haynes, M.T.; Huang, L. Multistage delivery technologies: Multifunctional, interdisciplinary approaches to nanomedicine. Mol. Ther. 2016, 24, 849–851. [Google Scholar] [CrossRef][Green Version]
- Stylianopoulos, T.; Wong, C.; Bawendi, M.G.; Jain, R.K.; Fukumura, D. Multistage nanoparticles for improved delivery into tumor tissue. Nanomed. Cancer Diabetes Cardiovasc. Cent. Nerv. Syst. Pulm. Inflamm. Dis. 2012, 508, 109–130. [Google Scholar] [CrossRef]
- Mi, Y.; Mu, C.; Wolfram, J.; Deng, Z.; Hu, T.; Liu, X.; Blanco, E.; Shen, H.; Ferrari, M. A micro/nano composite for combination treatment of melanoma lung metastasis. Adv. Health Mater. 2016, 5, 936–946. [Google Scholar] [CrossRef]
- Mann, A.P.; Tanaka, T.; Somasunderam, A.; Liu, X.; Gorenstein, D.G.; Ferrari, M. E-selectin-targeted porous silicon particle for nanoparticle delivery to the bone marrow. Adv. Mater. 2011, 23, H278–H282. [Google Scholar] [CrossRef]
- Tanaka, T.; Mangala, L.S.; Vivas-Mejia, P.E.; Nieves-Alicea, R.; Mann, A.P.; Mora, E.; Han, H.-D.; Shahzad, M.M.; Liu, X.; Bhavane, R.; et al. Sustained small interfering RNA delivery by mesoporous silicon particles. Cancer Res. 2010, 70, 3687–3696. [Google Scholar] [CrossRef]
- Maeda, H.; Matsumura, Y. EPR effect based drug design and clinical outlook for enhanced cancer chemotherapy Preface. Adv. Drug Deliv. Rev. 2011, 63, 129–130. [Google Scholar] [CrossRef]
- Torchilin, V.P. Drug targeting. Eur. J. Pharm. Sci. 2000, 11, S81–S91. [Google Scholar] [CrossRef]
- Zhu, M.; Ding, X.; Zhao, R.; Liu, X.; Shen, H.; Cai, C.; Ferrari, M.; Wang, H.Y.; Wang, R.-F. Co-delivery of tumor antigen and dual toll-like receptor ligands into dendritic cell by silicon microparticle enables efficient immunotherapy against melanoma. J. Control. Release 2018, 272, 72–82. [Google Scholar] [CrossRef]
- Shen, J.; Xu, R.; Mai, J.; Kim, H.-C.; Guo, X.; Qin, G.; Yang, Y.; Wolfram, J.; Mu, C.; Xia, X.; et al. High capacity nanoporous silicon carrier for systemic delivery of gene silencing therapeutics. ACS Nano 2013, 7, 9867–9880. [Google Scholar] [CrossRef]
- Shen, J.; Kim, H.-C.; Su, H.; Wang, F.; Wolfram, J.; Kirui, D.; Mai, J.; Mu, C.; Ji, L.-N.; Mao, Z.-W.; et al. Cyclodextrin and polyethylenimine functionalized mesoporous silica nanoparticles for delivery of siRNA cancer therapeutics. Theranostics 2014, 4, 487–497. [Google Scholar] [CrossRef]
- Shen, J.; Liu, H.; Mu, C.; Wolfram, J.; Zhang, W.; Kim, H.-C.; Zhu, G.; Hu, Z.; Ji, L.-N.; Liu, X.; et al. Multi-step encapsulation of chemotherapy and gene silencing agents in functionalized mesoporous silica nanoparticles. Nanoscale 2017, 9, 5329–5341. [Google Scholar] [CrossRef]
- Corbo, C.; Cevenini, A.; Salvatore, F. Biomarker discovery by proteomics-based approaches for early detection and personalized medicine in colorectal cancer. Proteom. Clin. Appl. 2017, 11. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Puccini, A.; Lenz, H.-J. Practice-changing updates in the adjuvant and metastatic setting. Nat. Rev. Clin. Oncol. 2017, 15, 77–78. [Google Scholar] [CrossRef]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef]
- Kagiava, A.; Theophilidis, G.; Sargiannidou, I.; Kyriacou, K.; Kleopa, K.A. Oxaliplatin-induced neurotoxicity is mediated through gap junction channels and hemichannels and can be prevented by octanol. Neuropharmacology 2015, 97, 289–305. [Google Scholar] [CrossRef]
- Godin, B.; Tasciotti, E.; Liu, X.; Serda, R.E.; Ferrari, M. Multistage nanovectors: From concept to novel imaging contrast agents and therapeutics. Acc. Chem. Res. 2011, 44, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Chiappini, C.; Tasciotti, E.; Fakhoury, J.R.; Fine, D.; Pullan, L.; Wang, Y.-C.; Fu, L.; Liu, X.; Ferrari, M. Tailored porous silicon microparticles: Fabrication and properties. ChemPhysChem 2010, 11, 1029–1035. [Google Scholar] [CrossRef]
- Cilurzo, F.; Critello, C.D.; Paolino, D.; Fiorillo, A.S.; Fresta, M.; De Franciscis, S.; Celia, C. Polydocanol foam stabilized by liposomes: Supramolecular nanoconstructs for sclerotherapy. Colloids Surf. B Biointerfaces 2019, 175, 469–476. [Google Scholar] [CrossRef]
- Di Francesco, M.; Primavera, R.; Fiorito, S.; Cristiano, M.; Taddeo, V.; Epifano, F.; Di Marzio, L.; Genovese, S.; Celia, C. Acronychiabaueri analogue derivative-loaded ultradeformable vesicles: Physicochemical characterization and potential applications. Planta Medica 2016, 83, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Cilurzo, F.; Cristiano, M.; Di Marzio, L.; Cosco, D.; Carafa, M.; Ventura, C.; Paolino, D.; Paolino, D. Influence of the supramolecular micro-assembly of multiple emulsions on their biopharmaceutical features and in vivo therapeutic response. Curr. Drug Targets 2015, 16, 1612–1622. [Google Scholar] [CrossRef]
- Malatesta, L.; Cosco, D.; Paolino, D.; Cilurzo, F.; Costa, N.; Di Tullio, A.; Fresta, M.; Celia, C.; Di Marzio, L.; Locatelli, M.; et al. Simultaneous quantification of Gemcitabine and Irinotecan hydrochloride in rat plasma by using high performance liquid chromatography-diode array detector. J. Pharm. Biomed. Anal. 2018, 159, 192–199. [Google Scholar] [CrossRef]
- Primavera, R.; Palumbo, P.; Celia, C.; Cinque, B.; Carata, E.; Carafa, M.; Paolino, D.; Cifone, M.G.; Di Marzio, L. An insight of in vitro transport of PEGylated non-ionic surfactant vesicles (NSVs) across the intestinal polarized enterocyte monolayers. Eur. J. Pharm. Biopharm. 2018, 127, 432–442. [Google Scholar] [CrossRef]
- ICH Harmonised Tripartite Guideline, Validation of Aanalitical Procedures: Text and Methodology, Q2(R1), Step 4 version. In Proceedings of the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, Geneva, Switzerland, 1–13 November 2005.
- Costanzo, M.; Cevenini, A.; Marchese, E.; Imperlini, E.; Raia, M.; Del Vecchio, L.; Caterino, M.; Ruoppolo, M. Label-free quantitative proteomics in a methylmalonyl-CoA mutase-silenced neuroblastoma cell line. Int. J. Mol. Sci. 2018, 19, 3580. [Google Scholar] [CrossRef]
- Di Marzio, L.; Marianecci, C.; Cinque, B.; Nazzarri, M.; Cimini, A.; Cristiano, L.; Cifone, M.G.; Alhaique, F.; Carafa, M. pH-sensitive non-phospholipid vesicle and macrophage-like cells: Binding, uptake and endocytotic pathway. Biochim. Biophys. Acta (BBA)-Biomembr. 2008, 1778, 2749–2756. [Google Scholar] [CrossRef][Green Version]
- Tasciotti, E.; Liu, X.; Bhavane, R.; Plant, K.; Leonard, A.D.; Price, B.K.; Cheng, M.M.-C.; Decuzzi, P.; Tour, J.M.; Robertson, F.; et al. Mesoporous silicon particles as a multistage delivery system for imaging and therapeutic applications. Nat. Nanotechnol. 2008, 3, 151–157. [Google Scholar] [CrossRef]
- Martinez, J.O.; Chiappini, C.; Ziemys, A.; Faust, A.M.; Kojić, M.; Liu, X.; Ferrari, M.; Tasciotti, E. Engineering multi-stage nanovectors for controlled degradation and tunable release kinetics. Biomaterials 2013, 34, 8469–8477. [Google Scholar] [CrossRef]
- Evangelopoulos, M.; Parodi, A.; Martinez, J.O.; Yazdi, I.K.; Cevenini, A.; Van De Ven, A.; Quattrocchi, N.; Boada, C.; Taghipour, N.; Corbo, C.; et al. Cell source determines the immunological impact of biomimetic nanoparticles. Biomaterials 2015, 82, 168–177. [Google Scholar] [CrossRef]
- Mouritsen, O.G. Lipids, curvature, and nano-medicine. Eur. J. Lipid Sci. Technol. 2011, 113, 1174–1187. [Google Scholar] [CrossRef]
- McMahon, H.T.; Gallop, J. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature 2005, 438, 590–596. [Google Scholar] [CrossRef]
- Wakaskar, R.R. General overview of lipid–polymer hybrid nanoparticles, dendrimers, micelles, liposomes, spongosomes and cubosomes. J. Drug Target. 2017, 26, 311–318. [Google Scholar] [CrossRef]
- Dürr, U.H.; Soong, R.; Ramamoorthy, A. When detergent meets bilayer: Birth and coming of age of lipid bicelles. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 69, 1–22. [Google Scholar] [CrossRef]
- Aryal, S.; Hu, C.-M.J.; Zhang, L. Synthesis of Ptsome: A platinum-based liposome-like nanostructure. Chem. Commun. 2012, 48, 2630–2632. [Google Scholar] [CrossRef] [PubMed]
- Ugwu, S.; Zhang, A.; Parmar, M.; Miller, B.; Sardone, T.; Peikov, V.; Ahmad, I. Preparation, characterization, and stability of liposome-based formulations of mitoxantrone. Drug Dev. Ind. Pharm. 2005, 31, 223–229. [Google Scholar] [CrossRef]
- Litzinger, D.C.; Huang, L. Amphipathic poly(ethylene glycol) 5000-stabilized dioleoylphosphatidylethanolamine liposomes accumulate in spleen. Biochim. Biophys. Acta 1992, 1127, 249–254. [Google Scholar] [CrossRef]
- Zhang, H.; Gong, W.; Wang, Z.-Y.; Yuan, S.-J.; Xie, X.; Yang, Y.-F.; Yang, Y.; Wang, S.-S.; Yang, D.; Xuan, Z.; et al. Preparation, characterization, and pharmacodynamics of thermosensitive liposomes containing docetaxel. J. Pharm. Sci. 2014, 103, 2177–2183. [Google Scholar] [CrossRef]
- Jespersen, H.; Andersen, J.H.; Ditzel, H.J.; Mouritsen, O.G. Lipids, curvature stress, and the action of lipid prodrugs: Free fatty acids and lysolipid enhancement of drug transport across liposomal membranes. Biochimie 2012, 94, 2–10. [Google Scholar] [CrossRef]
- Tippayamontri, T.; Kotb, R.; Paquette, B.; Sanche, L. Cellular uptake and cytoplasm / DNA distribution of cisplatin and oxaliplatin and their liposomal formulation in human colorectal cancer cell HCT116. Investig. New Drugs 2010, 29, 1321–1327. [Google Scholar] [CrossRef]
- Arnould, S.; Hennebelle, I.; Canal, P.; Bugat, R.; Guichard, S. Cellular determinants of oxaliplatin sensitivity in colon cancer cell lines. Eur. J. Cancer 2003, 39, 112–119. [Google Scholar] [CrossRef]
- Lila, A.; Kizuki, S.; Doi, Y.; Suzuki, T.; Ishida, T.; Kiwada, H. Oxaliplatin encapsulated in PEG-coated cationic liposomes induces significant tumor growth suppression via a dual-targeting approach in a murine solid tumor model. J. Control. Release 2009, 137, 8–14. [Google Scholar] [CrossRef]
- Pandelidou, M.; Dimas, K.; Georgopoulos, A.; Hatziantoniou, S.; Demetzos, C. Preparation and characterization of lyophilised egg PC liposomes incorporating curcumin and evaluation of its activity against colorectal cancer cell lines. J. Nanosci. Nanotechnol. 2011, 11, 1259–1266. [Google Scholar] [CrossRef]
- Casadó, A.; Sagristá, M.L.; Mora, M. A novel microfluidic liposomal formulation for the delivery of the SN-38 camptothecin: Characterization and in vitro assessment of its cytotoxic effect on two tumor cell lines. Int. J. Nanomed. 2018, 13, 5301–5320. [Google Scholar] [CrossRef] [PubMed]
- Elizondo, E.; Moreno, E.; Cabrera, I.; Cordoba, A.; Sala, S.; Veciana, J.; Ventosa, N. Liposomes and other vesicular systems: Structural characteristics, methods of preparation, and use in nanomedicine. Nanopart. Transl. Sci. Med. 2011, 104, 1–52. [Google Scholar] [CrossRef]
- Calvagno, M.G.; Celia, C.; Paolino, D.; Cosco, D.; Iannone, L.; Castelli, F.; Doldo, P.; Fresta, M. Effects of lipid composition and preparation conditions on physical-chemical properties, technological parameters and in vitro biological activity of gemcitabine-loaded liposomes. Curr. Drug Deliv. 2007, 4, 89–101. [Google Scholar] [CrossRef]
- Castelli, F.; Raudino, A.; Fresta, M. A mechanistic study of the permeation kinetics through biomembrane models: Gemcitabine–phospholipid bilayer interaction. J. Colloid Interface Sci. 2005, 285, 110–117. [Google Scholar] [CrossRef]
- Martinez, J.O.; Boada, C.; Yazdi, I.K.; Evangelopoulos, M.; Brown, B.S.; Liu, X.W.; Ferrari, M.; Tasciotti, E. Short and long term, in vitro and in vivo correlations of cellular and tissue responses to mesoporous silicon nanovectors. Small 2013, 9, 1722–1733. [Google Scholar] [CrossRef]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2016, 17, 20–37. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Form. | Composition | Short Name | Procedure | Tm | Dh | PDI | ZP |
|---|---|---|---|---|---|---|---|
| 1 | DOPC/DSPEmPEG2000 (7/3 molar ratio) | – | Extrusion 50 nm/Sonication | 37 °C | 58.96 ± 1.21 | 0.369 ± 0.02 | −0.57 ± 0.03 |
| 2 | PC/DSPEmPEG2000 (7/6 molar ratio) | – | Extrusion 50 nm/Sonication | 45 °C | 55.20 ± 2.25 | 0.363 ± 0.09 | −1.59 ± 0.20 |
| 3 | DOPC/DSPEmPEG2000 (7/5 molar ratio) | – | Extrusion 50 nm/Sonication | 45 °C | 55.5 ± 2.1 | 0.340 ± 0.03 | −2.15 ± 0.30 |
| 4 | DOPC/DSPEmPEG2000 (7/6 molar ratio) | – | Extrusion 50 nm/Sonication | 37 °C | 45.33 ± 1.2 | 0.476 ± 0.12 | 0.858 ± 0.10 |
| 5 | PC/DSPEmPEG2000 (7/6 molar ratio) | – | Extrusion 50 nm/Sonication | 45 °C | 52.95 ± 2.1 | 0.467 ± 0.10 | −1.59 ± 0.20 |
| 6 | PC/DSPEmPEG2000 (7/5 molar ratio) | – | Extrusion 50 nm/Sonication | 45 °C | 53.46 ± 2.10 | 0.377 ± 0.09 | −4.83 ± 1.10 |
| 7 | DOPC/DSPEmPEG2000 (7/6 molar ratio) | – | Extrusion 50 nm/Sonication | 37 °C | 45.22 ± 1.90 | 0.471 ± 0.12 | −6.87 ± 1.30 |
| 8 | PC/DSPEmPEG2000 (7/6 molar ratio) | – | Extrusion 50 nm/Sonication | 45 °C | 53.03 ± 0.90 | 0.491 ± 0.13 | −8.72 ± 0.90 |
| 9 | PC/DSPEmPEG2000 (7/5 molar ratio) | – | Extrusion 30 nm | 45 °C | 52.93 ± 1.25 | 0.271 ± 0.12 | −9.47 ± 1.70 |
| 10 | PC/DSPEmPEG2000 (7/6 molar ratio) | – | Extrusion 30 nm | 45 °C | 47.76 ± 3.90 | 0.273 ± 0.09 | −15.0 ± 2.10 |
| 11 | DPPC/Chol/PE/DSPEmPEG2000 (7/7/4/1 molar ratio) | – | Extrusion 50 nm/Sonication | 50 °C | 55.58 ± 1.90 | 0.194 ± 0.09 | −12.63 ± 1.23 |
| 12 | PC/Chol/PE/DSPEmPEG2000 (7/7/4/1 molar ratio) | – | Extrusion 50 nm/Sonication | 50 °C | 54.16 ± 2.10 | 0.197 ± 0.08 | −11.06 ± 0.30 |
| 13 | DOPC/DPPC/DSPEmPEG2000 (7/4/3 molar ratio) | – | Extrusion 50 nm/30 nm | 40 °C | 51.5 ± 8.90 | 0.172 ± 0.10 | −5.40 ± 5.0 |
| 14 | DPPC/DPPS/Chol (7/4/7 molar ratio) | – | Extrusion 50 nm | 50 °C | 64.93 ± 1.21 | 0.053 ± 0.02 | −41.94 ± 2.90 |
| 15 | DSPC/DSPEmPEG2000 (7/3 molar ratio) | DS | Extrusion 30 nm | 60 °C | 26.3 ± 1.2 | 0.578 ± 0.02 | −10.51 ± 0.18 |
| 16 | DSPC/DOPC/DSPEmPEG2000 (7/1/2 molar ratio) | DO | Extrusion 30 nm | 60 °C | 54.46 ± 3.03 | 0.209 ± 0.03 | −9.84 ± 0.40 |
| 4 oxa | DOPC/DSPEmPEG2000 (7/6 molar ratio) + oxa | – | Extrusion 30 nm | 42 °C | 64.5 ± 1.1 | 0.239 ± 0.91 | −6.17 ± 1.23 |
| 10 oxa | PC/DSPEmPEG2000 (7/6 molar ratio) + oxa | – | Extrusion 30 nm | 50 °C | 75.2 ± 1.2 | 0.365 ± 0.14 | −4.25 ± 1.60 |
| 13 oxa | DOPC/DPPC/DSPEmPEG2000 (7/4/3 molar ratio) + oxa | – | Extrusion 30 nm | 50 °C | 64.65 ± 5.15 | 0.098 ± 0.01 | −5.35 ± 2.40 |
| 15 oxa | DSPC/DSPEmPEG2000 (7/3 molar ratio) + oxa | DS-oxa | Extrusion 30 nm | 60 °C | 44.25 ± 4.25 | 0.315 ± 0.04 | −10.95 ± 0.4 |
| 16 oxa | DSPC/DOPC/DSPEmPEG2000 (7/1/2 molar ratio) + oxa | DO-oxa | Extrusion 30 nm | 60 °C | 42.60 ± 3.80 | 0.251 ± 0.01 | −9.41 ± 1.95 |
| IC50 | |||
|---|---|---|---|
| Time (h) | Free oxa | DO-oxa | DS-oxa |
| 24 | 1.9 ± 0.7 μM | N.A. | N.A. |
| 48 | 1.3 ± 0.6 μM | 1.6 ± 0.6 μM | 2.2 ± 0.8 μM |
| 72 | 0.8 ± 0.3 μM | 1.6 ± 0.7 μM | 1.9 ± 0.8 μM |
| Time (h) | Treatment | IC50 (μM) | SD (μM) |
|---|---|---|---|
| 48 | Free oxa | 1.3 | ± 0.2 |
| MSV DO-oxa | 1.2 | ±0.1 | |
| MSV DS-oxa | 1.5 | ±0.3 | |
| 72 | Free oxa | 0.7 | ±0.1 |
| MSV DO-oxa | 1.0 | ±0.1 | |
| MSV DS-oxa | 1.5 | ±0.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cevenini, A.; Celia, C.; Orrù, S.; Sarnataro, D.; Raia, M.; Mollo, V.; Locatelli, M.; Imperlini, E.; Peluso, N.; Peltrini, R.; et al. Liposome-Embedding Silicon Microparticle for Oxaliplatin Delivery in Tumor Chemotherapy. Pharmaceutics 2020, 12, 559. https://doi.org/10.3390/pharmaceutics12060559
Cevenini A, Celia C, Orrù S, Sarnataro D, Raia M, Mollo V, Locatelli M, Imperlini E, Peluso N, Peltrini R, et al. Liposome-Embedding Silicon Microparticle for Oxaliplatin Delivery in Tumor Chemotherapy. Pharmaceutics. 2020; 12(6):559. https://doi.org/10.3390/pharmaceutics12060559
Chicago/Turabian StyleCevenini, Armando, Christian Celia, Stefania Orrù, Daniela Sarnataro, Maddalena Raia, Valentina Mollo, Marcello Locatelli, Esther Imperlini, Nicoletta Peluso, Rosa Peltrini, and et al. 2020. "Liposome-Embedding Silicon Microparticle for Oxaliplatin Delivery in Tumor Chemotherapy" Pharmaceutics 12, no. 6: 559. https://doi.org/10.3390/pharmaceutics12060559
APA StyleCevenini, A., Celia, C., Orrù, S., Sarnataro, D., Raia, M., Mollo, V., Locatelli, M., Imperlini, E., Peluso, N., Peltrini, R., De Rosa, E., Parodi, A., Del Vecchio, L., Di Marzio, L., Fresta, M., Netti, P. A., Shen, H., Liu, X., Tasciotti, E., & Salvatore, F. (2020). Liposome-Embedding Silicon Microparticle for Oxaliplatin Delivery in Tumor Chemotherapy. Pharmaceutics, 12(6), 559. https://doi.org/10.3390/pharmaceutics12060559