Physiologically-Based Pharmacokinetic (PBPK) Modeling of Buprenorphine in Adults, Children and Preterm Neonates

 , ,
, , 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Software
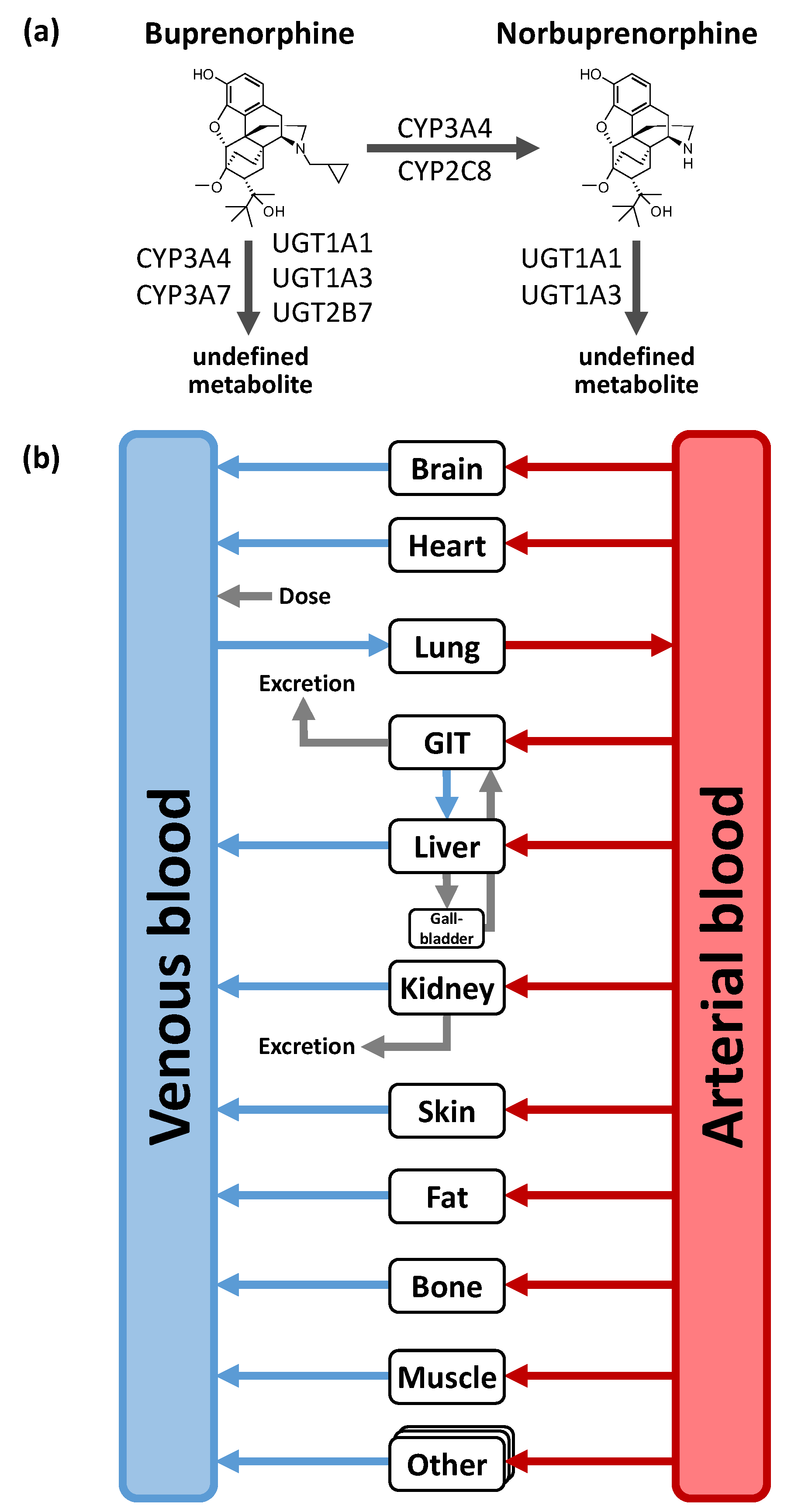
2.2. PBPK Parent-Metabolite Model Building in Adults
2.3. Pediatric Scaling and Model Applications
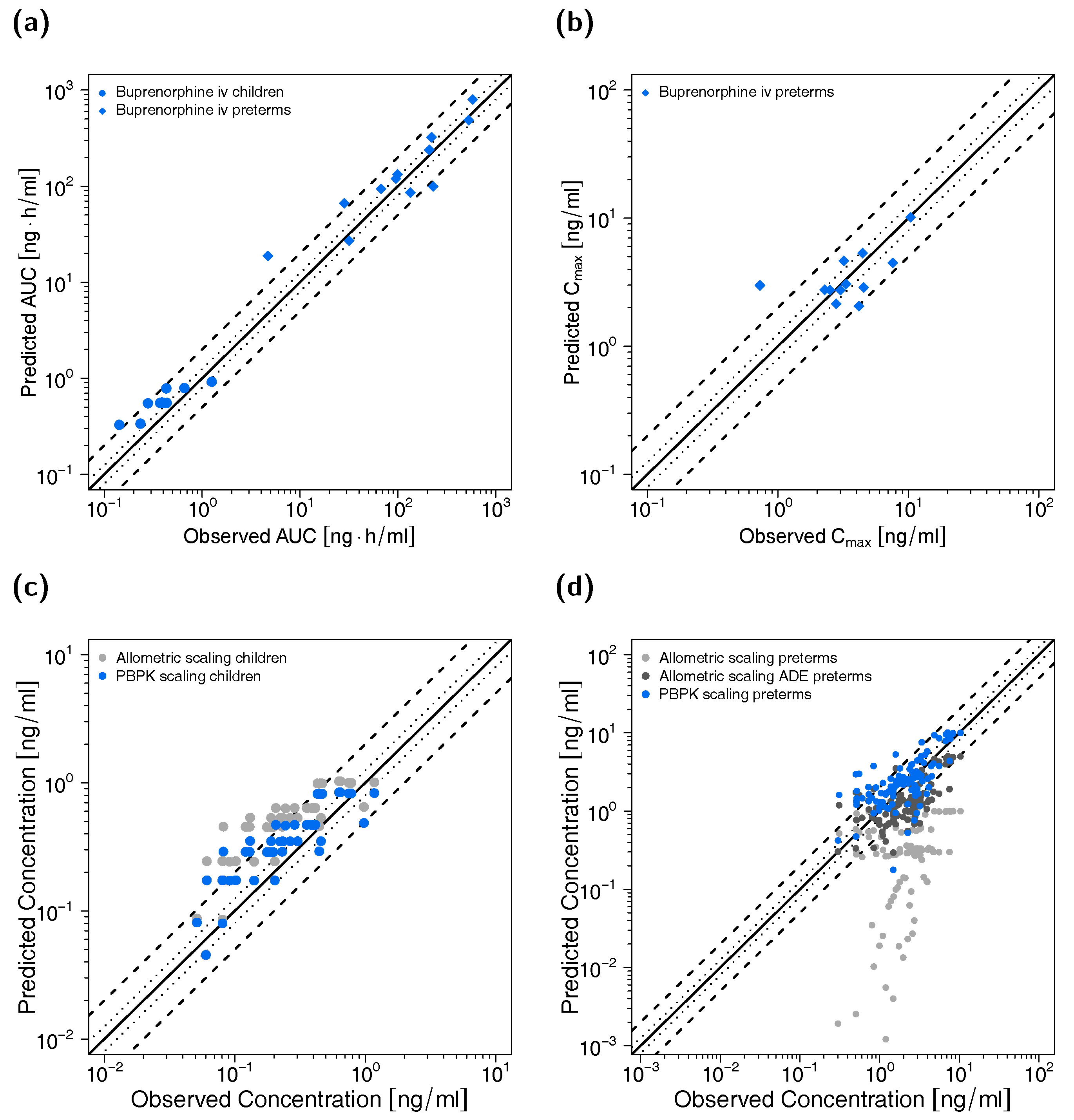
2.4. PBPK Model Evaluation
3. Results
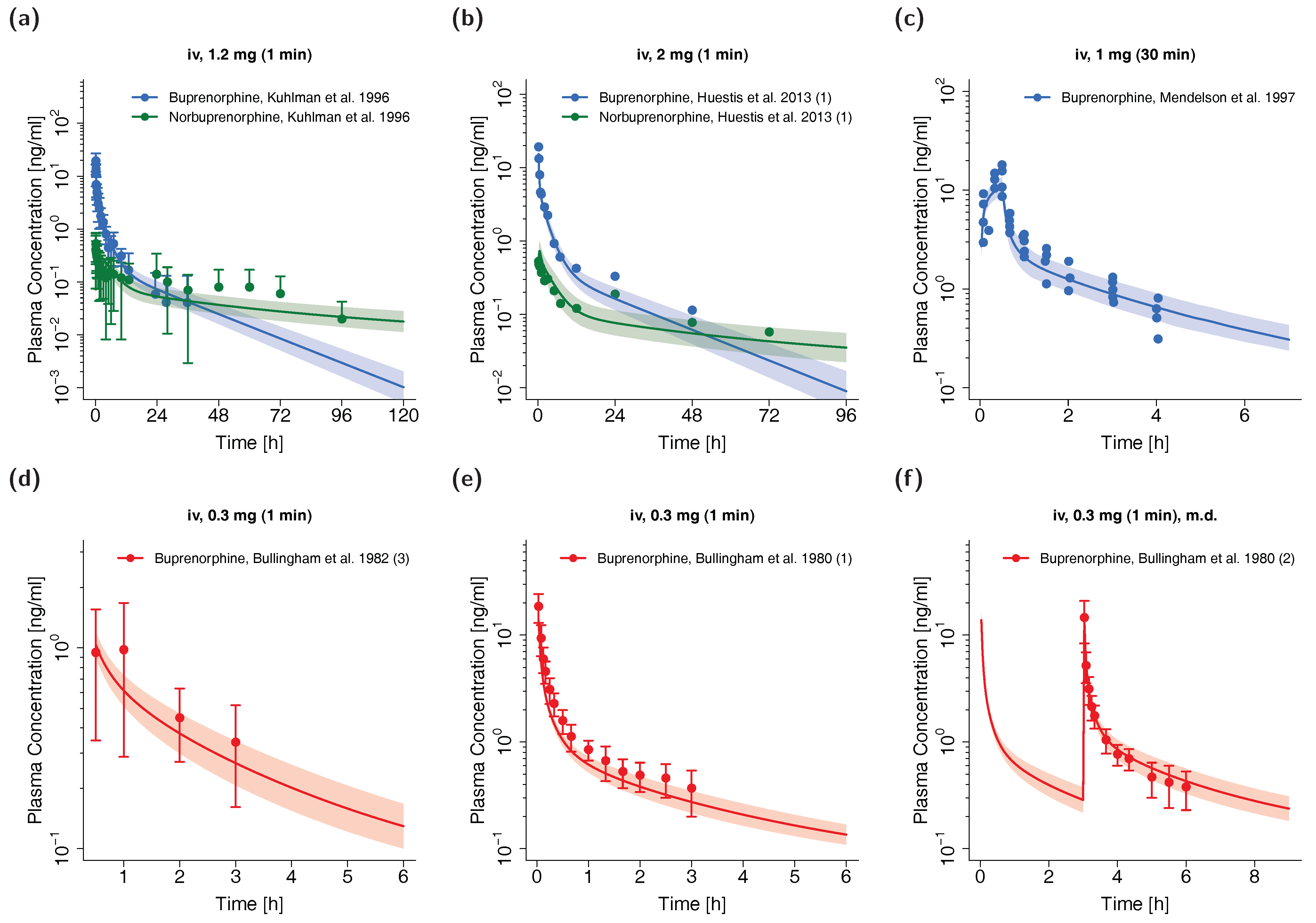
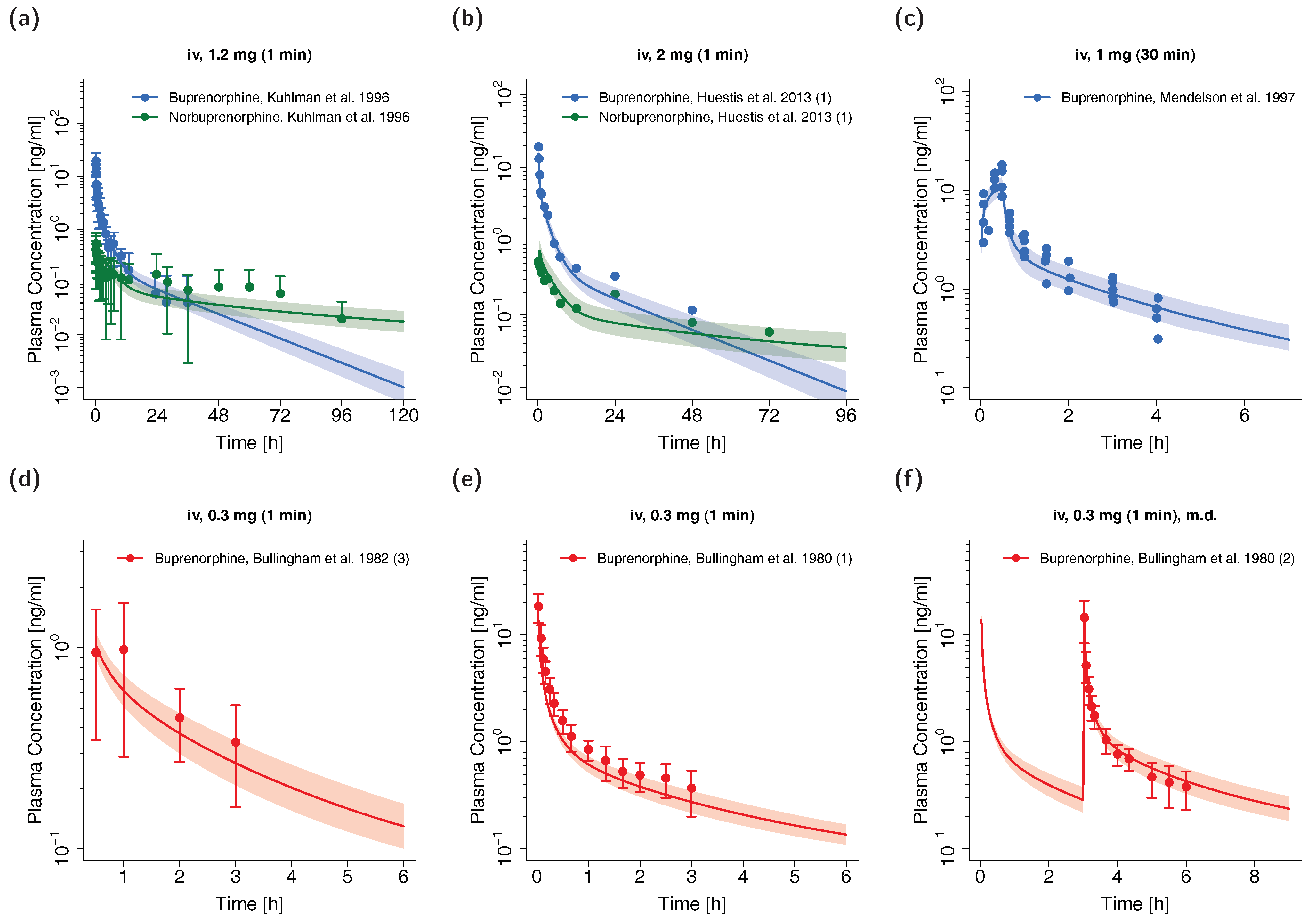
3.1. PK Data for PBPK Model Development and Pediatric Scaling
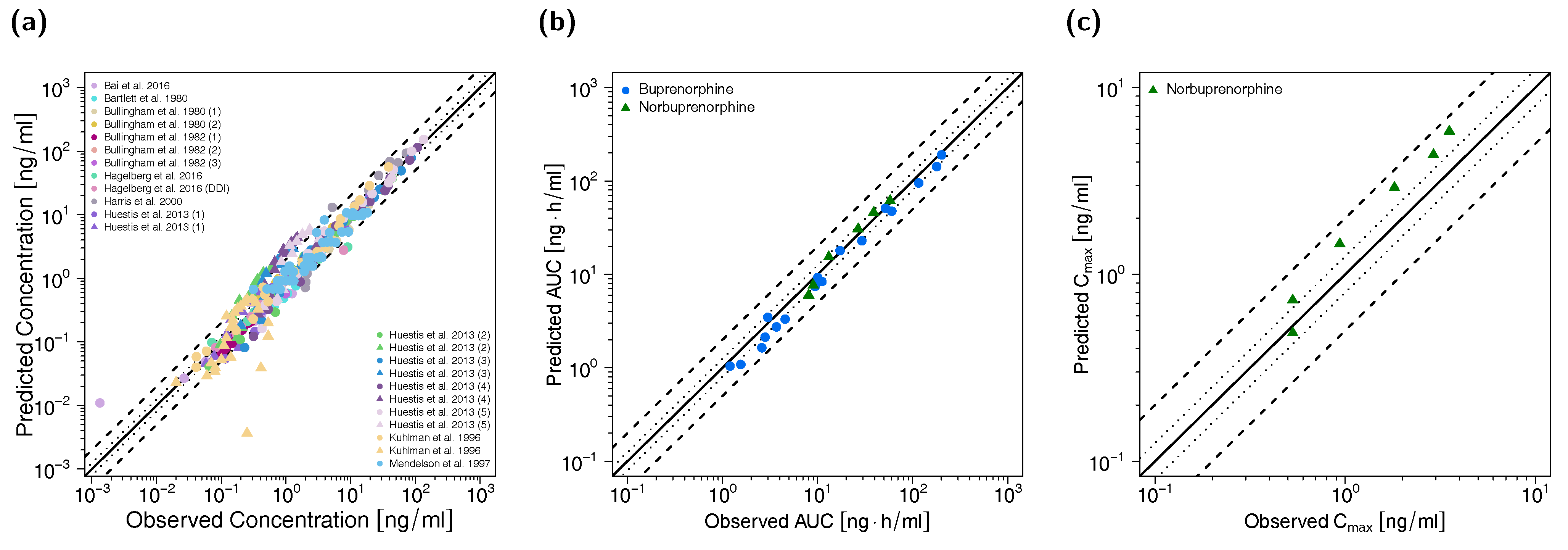
3.2. Adult PBPK Model Building and Evaluation
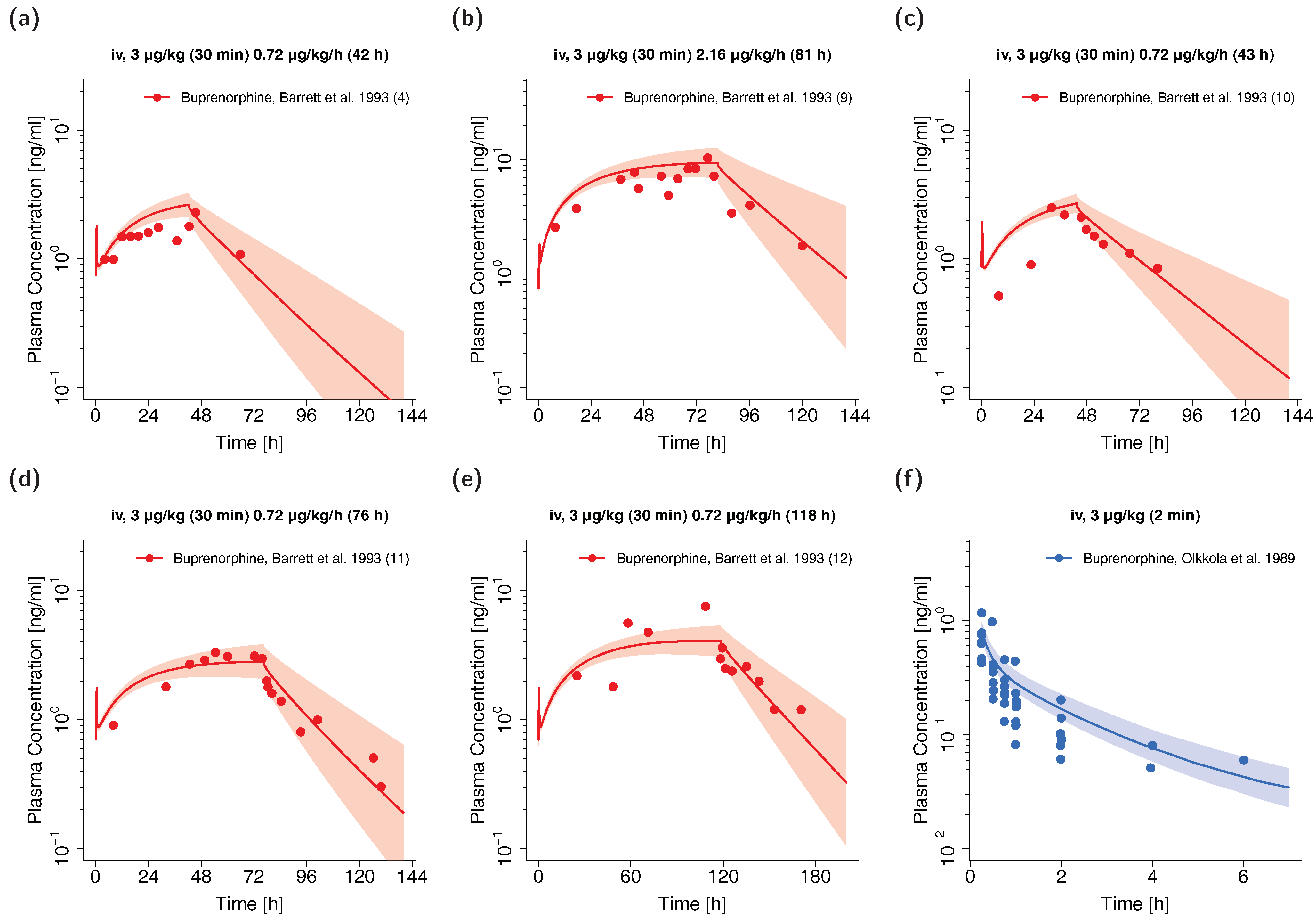
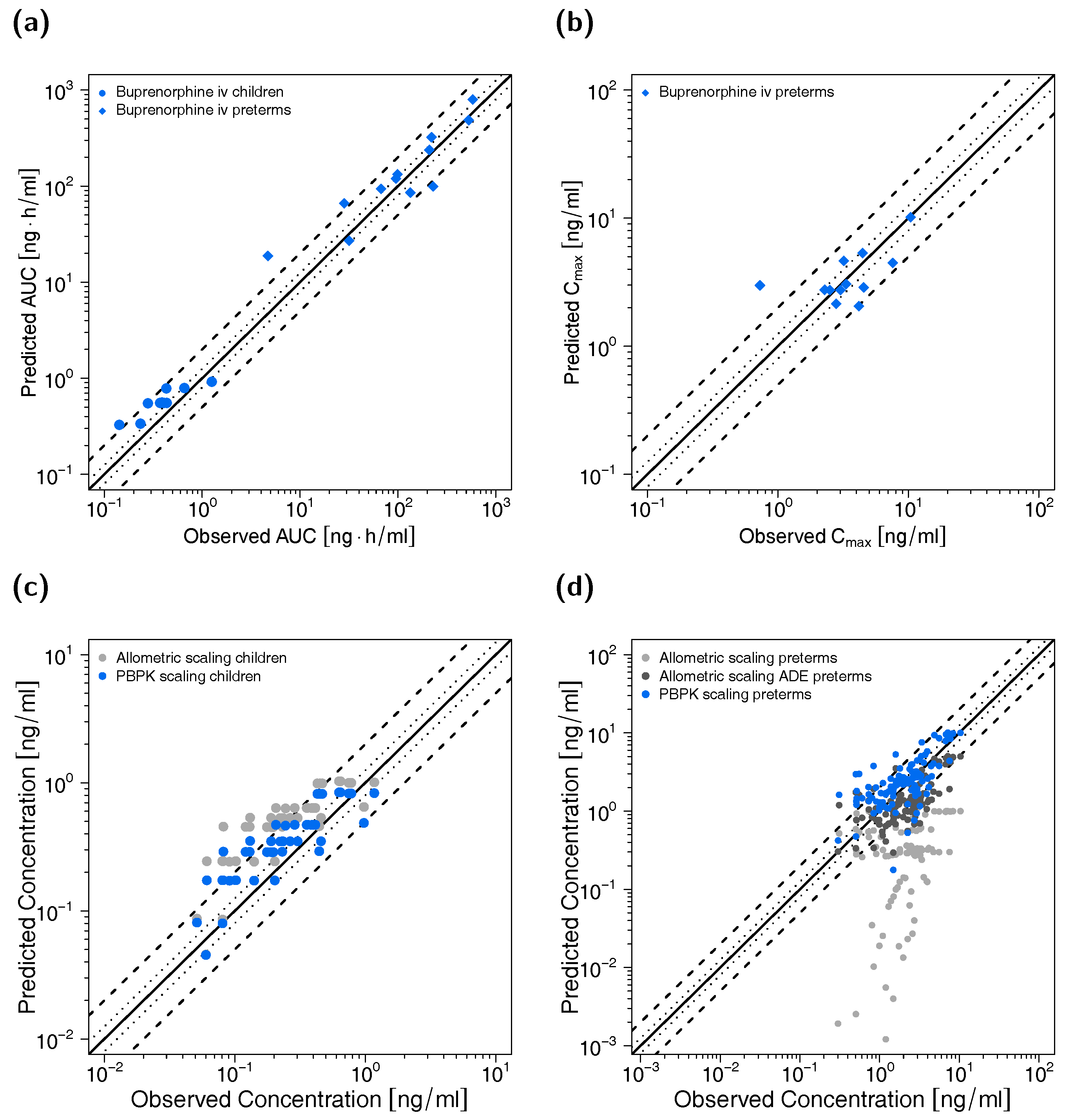
3.3. Pediatric PBPK Model Building and Evaluation
3.4. DDI Evaluation with the Adult PBPK Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Maunuksela, E.-L.; Korpela, R.; Olkkola, K.T. Double-blind, multiple-dose comparison of buprenorphine and morphine in postoperative pain of children. Br. J. Anaesth. 1988, 60, 48–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Guidelines for the Pharmacological and Radiotherapeutic Management of Cancer Pain in Adults and Adolescents; Licence: CC BY-NC-SA 3.0 IGO; World Health Organization: Geneva, Switzerland, 2018; ISBN 978-92-4-155039-0. [Google Scholar]
- Vicencio-Rosas, E.; Pérez-Guillé, M.G.; Flores-Pérez, C.; Flores-Pérez, J.; Trujillo-Jiménez, F.; Chávez-Pacheco, J.L. Buprenorphine and pain treatment in pediatric patients: An update. J. Pain Res. 2018, 11, 549–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, E.; Anderson, B.J.; Zernikow, B. Buprenorphine TTS for children—A review of the drug’s clinical pharmacology. Pediatric Anesth. 2011, 21, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Yassen, A.; Romberg, R.; Sarton, E.; Teppema, L.; Olofsen, E.; Danhof, M. Buprenorphine induces ceiling in respiratory depression but not in analgesia. Br. J. Anaesth. 2006, 96, 627–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhlman, J.J.; Lalani, S.; Magluilo, J.; Levine, B.; Darwin, W.D.; Johnson, R.E.; Cone, E.J. Human Pharmacokinetics of Intravenous, Sublingual, and Buccal Buprenorphine. J. Anal. Toxicol. 1996, 20, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Lo Re, M.; Chaplin, M.; Aronow, B.; Modesto-Lowe, V. Buprenorphine Overdose in Young Children: An Underappreciated Risk. Clin. Pediatric 2019, 58, 613–617. [Google Scholar] [CrossRef]
- Boyer, E.W. Management of Opioid Analgesic Overdose. N. Engl. J. Med. 2012, 367, 146–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, W. Buprenorphine for opioid addiction. Pain Manag. 2012, 2, 345–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Post, S.; Spiller, H.A.; Casavant, M.J.; Chounthirath, T.; Smith, G.A. Buprenorphine Exposures Among Children and Adolescents Reported to US Poison Control Centers. Pediatrics 2018, 142, e20173652. [Google Scholar] [CrossRef]
- Ward, R.M.; Sherwin, C.M.T. Ethics of Drug Studies in the Newborn. Pediatric Drugs 2014, 17, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Leong, R.; Vieira, M.L.T.; Zhao, P.; Mulugeta, Y.; Lee, C.S.; Huang, S.-M.; Burckart, G.J. Regulatory experience with physiologically based pharmacokinetic modeling for pediatric drug trials. Clin. Pharmacol. Ther. 2012, 91, 926–931. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.S.; Della Casa Alberighi, O.; Läer, S.; Meibohm, B. Physiologically Based Pharmacokinetic (PBPK) Modeling in Children. Clin. Pharmacol. Ther. 2012, 92, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Sager, J.E.; Yu, J.; Ragueneau-Majlessi, I.; Isoherranen, N. Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulation Approaches: A Systematic Review of Published Models, Applications, and Model Verification. Drug Metab. Dispos. 2015, 43, 1823–1837. [Google Scholar] [CrossRef] [PubMed]
- Templeton, I.E.; Jones, N.S.; Musib, L. Pediatric Dose Selection and Utility of PBPK in Determining Dose. AAPS J. 2018, 20, 31. [Google Scholar] [CrossRef]
- Strougo, A.; Yassen, A.; Monnereau, C.; Danhof, M.; Freijer, J. Predicting the “First dose in children” of CYP3A-metabolized drugs: Evaluation of scaling approaches and insights into the CYP3A7-CYP3A4 switch at young ages. J. Clin. Pharmacol. 2014, 54, 1006–1015. [Google Scholar] [CrossRef]
- Yokell, M.A.; Zaller, N.D.; Green, T.C.; Rich, J.D. Buprenorphine and buprenorphine/naloxone diversion, misuse, and illicit use: An international review. Curr. Drug Abuse Rev. 2011, 4, 28–41. [Google Scholar] [CrossRef]
- Rosenthal, R.; Goradia, V. Advances in the delivery of buprenorphine for opioid dependence. Drug Des. Devel. Ther. 2017, 11, 2493–2505. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.C.; Roman-Urrestarazu, A.; Brayne, C. Responses among substance abuse treatment providers to the opioid epidemic in the USA: Variations in buprenorphine and methadone treatment by geography, operational, and payment characteristics, 2007–2016. PLoS ONE 2020, 15, e0229787. [Google Scholar] [CrossRef] [Green Version]
- Dunlap, B.; Cifu, A.S. Clinical Management of Opioid Use Disorder. JAMA 2016, 316, 338. [Google Scholar] [CrossRef]
- Picard, N.; Cresteil, T.; Djebli, N.; Marquet, P. In vitro metabolism study of buprenorphine: Evidence for new metabolic pathways. Drug Metab. Dispos. 2005, 33, 689–695. [Google Scholar] [CrossRef]
- Hanke, N.; Frechen, S.; Moj, D.; Britz, H.; Eissing, T.; Wendl, T.; Lehr, T. PBPK Models for CYP3A4 and P-gp DDI Prediction: A Modeling Network of Rifampicin, Itraconazole, Clarithromycin, Midazolam, Alfentanil, and Digoxin. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 647–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kress, H.G. Clinical update on the pharmacology, efficacy and safety of transdermal buprenorphine. Eur. J. Pain 2009, 13, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Fihlman, M.; Hemmilä, T.; Hagelberg, N.M.; Backman, J.T.; Laitila, J.; Laine, K.; Neuvonen, P.J.; Olkkola, K.T.; Saari, T.I. Voriconazole greatly increases the exposure to oral buprenorphine. Eur. J. Clin. Pharmacol. 2018, 74, 1615–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fihlman, M.; Hemmilä, T.; Hagelberg, N.M.; Kuusniemi, K.; Backman, J.T.; Laitila, J.; Laine, K.; Neuvonen, P.J.; Olkkola, K.T.; Saari, T.I. Voriconazole more likely than posaconazole increases plasma exposure to sublingual buprenorphine causing a risk of a clinically important interaction. Eur. J. Clin. Pharmacol. 2016, 72, 1363–1371. [Google Scholar] [CrossRef] [Green Version]
- Hagelberg, N.M.; Fihlman, M.; Hemmilä, T.; Backman, J.T.; Laitila, J.; Neuvonen, P.J.; Laine, K.; Olkkola, K.T.; Saari, T.I. Rifampicin decreases exposure to sublingual buprenorphine in healthy subjects. Pharmacol. Res. Perspect. 2016, 4, e00271. [Google Scholar] [CrossRef]
- Kovar, L.; Selzer, D.; Britz, H.; Benowitz, N.; St. Helen, G.; Kohl, Y.; Bals, R.; Lehr, T. Comprehensive Parent–Metabolite PBPK/PD Modeling Insights into Nicotine Replacement Therapy Strategies. Clin. Pharmacokinet. 2020. [Google Scholar] [CrossRef] [Green Version]
- Johnson, T.N.; Rostami-Hodjegan, A. Resurgence in the use of physiologically based pharmacokinetic models in pediatric clinical pharmacology: Parallel shift in incorporating the knowledge of biological elements and increased applicability to drug development and clinical practice. Pediatric Anesth. 2011, 21, 291–301. [Google Scholar] [CrossRef]
- Jones, H.M.; Dickins, M.; Youdim, K.; Gosset, J.R.; Attkins, N.J.; Hay, T.L.; Gurrell, I.K.; Logan, Y.R.; Bungay, P.J.; Jones, B.C.; et al. Application of PBPK modelling in drug discovery and development at Pfizer. Xenobiotica 2012, 42, 94–106. [Google Scholar] [CrossRef]
- Wojtyniak, J.-G.; Britz, H.; Selzer, D.; Schwab, M.; Lehr, T. Data Digitizing: Accurate and Precise Data Extraction for Quantitative Systems Pharmacology and Physiologically-Based Pharmacokinetic Modeling. CPT Pharmacomet. Syst. Pharmacol. Pharmacol. 2020. accepted for publication. [Google Scholar] [CrossRef]
- Maharaj, A.R.; Barrett, J.S.; Edginton, A.N. A workflow example of PBPK modeling to support pediatric research and development: Case study with lorazepam. AAPS J. 2013, 15, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Maharaj, A.R.; Edginton, A.N. Physiologically Based Pharmacokinetic Modeling and Simulation in Pediatric Drug Development. CPT Pharmacomet. Syst. Pharmacol. 2014, 3, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ince, I.; Solodenko, J.; Frechen, S.; Dallmann, A.; Niederalt, C.; Schlender, J.; Burghaus, R.; Lippert, J.; Willmann, S. Predictive Pediatric Modeling and Simulation Using Ontogeny Information. J. Clin. Pharmacol. 2019, 59, S95–S103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oechsler, S.; Skopp, G. An in vitro approach to estimate putative inhibition of buprenorphine and norbuprenorphine glucuronidation. Int. J. Legal Med. 2010, 124, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Everhart, E.; Cheung, P.; Mendelson, J.; Upton, R.; Jones, R. The mass balance of buprenorphine in humans. Clin. Pharmacol. Ther. 1999, 65, 152. [Google Scholar] [CrossRef]
- Chang, Y.; Moody, D.E. Glucuronidation of buprenorphine and norbuprenorphine by human liver microsomes and UDP-glucuronosyltransferases. Drug Metab. Lett. 2009, 3, 101–107. [Google Scholar] [CrossRef]
- Brown, S.M.; Campbell, S.D.; Crafford, A.; Regina, K.J.; Holtzman, M.J.; Kharasch, E.D. P-glycoprotein is a major determinant of norbuprenorphine brain exposure and antinociception. J. Pharmacol. Exp. Ther. 2012, 343, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Open Systems Pharmacology Suite Community Open Systems Pharmacology Suite Manual, Version 7.4. Available online: https://github.com/Open-Systems-Pharmacology/OSPSuite.Documentation/blob/master/Open%20Systems%20Pharmacology%20Suite.pdf (accessed on 25 March 2020).
- PK-Sim® Ontogeny Database, Version 7.3. Available online: https://github.com/Open-Systems-Pharmacology/OSPSuite.Documentation/blob/master/PK-Sim Ontogeny Database Version 7.3.pdf (accessed on 25 March 2020).
- McNamara, P.J.; Alcorn, J. Protein binding predictions in infants. AAPS PharmSci 2002, 4, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, Y.; Ishii, S.; Arizono, H.; Nishimura, S.; Tsuruda, K.; Saito, N.; Nemoto, H.; Jin, Y.; Esumi, Y. Pharmacokinetics of Buprenorphine Hydrochloride (BN HCl) (1): Absorption, Distribution, Metabolism and Excretion after Percutaneous (TSN-09: BN HCl Containing Tape Application) or Subcutaneous Administration of BN HCl in Rats. Drug Metab. Pharmacokinet. 2001, 16, 569–583. [Google Scholar] [CrossRef] [Green Version]
- Tod, M.; Jullien, V.; Pons, G. Facilitation of Drug Evaluation in Children by Population Methods and Modelling†. Clin. Pharmacokinet. 2008, 47, 231–243. [Google Scholar] [CrossRef]
- Kajosaari, L.I.; Laitila, J.; Neuvonen, P.J.; Backman, J.T. Metabolism of repaglinide by CYP2C8 and CYP3A4 in vitro: Effect of fibrates and rifampicin. Basic Clin. Pharmacol. Toxicol. 2005, 97, 249–256. [Google Scholar] [CrossRef]
- Rajaonarison, J.; Lacarelle, B.; Catalin, J.; Placidi, M.; Rahmani, R. 3’-azido-3’-deoxythymidine drug interactions. Screening for inhibitors in human liver microsomes. Drug Metab. Dispos. 1992, 20, 578–584. [Google Scholar] [PubMed]
- Chiou, W.J.; de Morais, S.M.; Kikuchi, R.; Voorman, R.L.; Li, X.; Bow, D.A.J. In vitro OATP1B1 and OATP1B3 inhibition is associated with observations of benign clinical unconjugated hyperbilirubinemia. Xenobiotica 2014, 44, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Soars, M.G.; Petullo, D.M.; Eckstein, J.A.; Kasper, S.C.; Wrighton, S.A. An assessment of udp-glucuronosyltransferase induction using primary human hepatocytes. Drug Metab. Dispos. 2004, 32, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Trottier, J.; El Husseini, D.; Perreault, M.; Pâquet, S.; Caron, P.; Bourassa, S.; Verreault, M.; Inaba, T.T.; Poirier, G.G.; Bélanger, A.; et al. The Human UGT1A3 Enzyme Conjugates Norursodeoxycholic Acid into a C 23 -ester Glucuronide in the Liver. J. Biol. Chem. 2010, 285, 1113–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reitman, M.L.; Chu, X.; Cai, X.; Yabut, J.; Venkatasubramanian, R.; Zajic, S.; Stone, J.A.; Ding, Y.; Witter, R.; Gibson, C.; et al. Rifampin’s Acute Inhibitory and Chronic Inductive Drug Interactions: Experimental and Model-Based Approaches to Drug–Drug Interaction Trial Design. Clin. Pharmacol. Ther. 2011, 89, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Greiner, B.; Eichelbaum, M.; Fritz, P.; Kreichgauer, H.-P.; von Richter, O.; Zundler, J.; Kroemer, H.K. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J. Clin. Investig. 1999, 104, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Buckley, D.B.; Wiegand, C.M.; Prentiss, P.L.; Fahmi, O.A. Time-course of cytochrome P450 (CYP450) induction in cultured human hepatocytes: Evaluation of activity and mRNA expression profiles for six inducible CYP450 enzymes [poster no. P186]. In Proceedings of the 10th International ISSX Meeting, Toronto, ON, Canada, 29 September–3 October 2013. [Google Scholar]
- Barrett, D.; Simpson, J.; Rutter, N.; Kurihara-Bergstrom, T.; Shaw, P.; Davis, S. The pharmacokinetics and physiological effects of buprenorphine infusion in premature neonates. Br. J. Clin. Pharmacol. 1993, 36, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.N.; Hawks, R.L. Pharmacokinetics of the combination tablet of buprenorphine and naloxone. Drug Alcohol Depend. 2003, 70, S39–S47. [Google Scholar] [CrossRef]
- Kalluri, H.V.; Zhang, H.; Caritis, S.N.; Venkataramanan, R. A physiologically based pharmacokinetic modelling approach to predict buprenorphine pharmacokinetics following intravenous and sublingual administration. Br. J. Clin. Pharmacol. 2017, 83, 2458–2473. [Google Scholar] [CrossRef]
- Bai, S.A.; Xiang, Q.; Finn, A. Evaluation of the Pharmacokinetics of Single- and Multiple-dose Buprenorphine Buccal Film in Healthy Volunteers. Clin. Ther. 2016, 38, 358–369. [Google Scholar] [CrossRef]
- Bartlett, A.J.; Lloyd-Jones, J.G.; Rance, M.J.; Flockhart, I.R.; Dockray, G.J.; Bennett, M.R.D.; Moore, R.A. The radioimmunoassay of buprenorphine. Eur. J. Clin. Pharmacol. 1980, 18, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Bullingham, R.E.; McQuay, H.J.; Moore, A.; Bennett, M.R. Buprenorphine kinetics. Clin. Pharmacol. Ther. 1980, 28, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Bullingham, R.E.; McQuay, H.J.; Porter, E.J.; Allen, M.C.; Moore, R.A. Sublingual buprenorphine used postoperatively: Ten hour plasma drug concentration analysis. Br. J. Clin. Pharmacol. 1982, 13, 665–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, D.S.; Jones, R.T.; Welm, S.; Upton, R.A.; Lin, E.; Mendelson, J. Buprenorphine and naloxone co-administration in opiate-dependent patients stabilized on sublingual buprenorphine. Drug Alcohol Depend. 2000, 61, 85–94. [Google Scholar] [CrossRef]
- Huestis, M.A.; Cone, E.J.; Pirnay, S.O.; Umbricht, A.; Preston, K.L. Intravenous buprenorphine and norbuprenorphine pharmacokinetics in humans. Drug Alcohol Depend. 2013, 131, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Mendelson, J.; Upton, R.A.; Everhart, E.T.; Jacob, P., III; Jones, R.T. Bioavailability of Sublingual Buprenorphine. J. Clin. Pharmacol. 1997, 37, 31–37. [Google Scholar] [CrossRef]
- Olkkola, K.; Maunuksela, E.; Korpela, R. Pharmacokinetics of intravenous buprenorphine in children. Br. J. Clin. Pharmacol. 1989, 28, 202–204. [Google Scholar] [CrossRef] [Green Version]
- Cone, E.J.; Gorodetzky, C.W.; Yousefnejad, D.; Buchwald, W.F.; Johnson, R.E. The metabolism and excretion of buprenorphine in humans. Drug Metab. Dispos. 1984, 12, 577–581. [Google Scholar]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Yeola, G.S.; Darandale, S.; Khire, A.; Vavia, P.R. Fabrication and statistical optimization of a polysaccharide-based sublingual film of buprenorphine hydrochloride for breakthrough pain management: In vitro and in vivo performance. Drug Deliv. Transl. Res. 2014, 4, 116–125. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Heel, R.C.; Brogden, R.N.; Speight, T.M.; Avery, G.S. Buprenorphine: A review of its pharmacological properties and therapeutic efficacy. Drugs 1979, 17, 81–110. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, W. General approach for the calculation of tissue to plasma partition coefficients. Toxicol. In Vitro 2008, 22, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Obach, R.S. Nonspecific binding to microsomes: Impact on scale-up of in vitro intrinsic clearance to hepatic clearance as assessed through examination of warfarin, imipramine, and propranolol. Drug Metab. Dispos. 1997, 25, 1359–1369. [Google Scholar] [PubMed]
- Mahmood, I.; Tegenge, M.A. A Comparative Study Between Allometric Scaling and Physiologically Based Pharmacokinetic Modeling for the Prediction of Drug Clearance From Neonates to Adolescents. J. Clin. Pharmacol. 2019, 59, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Kapil, R.P.; Cipriano, A.; Friedman, K.; Michels, G.; Shet, M.S.; Colucci, S.V.; Apseloff, G.; Kitzmiller, J.; Harris, S.C. Once-Weekly Transdermal Buprenorphine Application Results in Sustained and Consistent Steady-State Plasma Levels. J. Pain Symptom Manag. 2013, 46, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Duan, P.; Fisher, J.W.; Yoshida, K.; Zhang, L.; Burckart, G.J.; Wang, J. Physiologically Based Pharmacokinetic Prediction of Linezolid and Emtricitabine in Neonates and Infants. Clin. Pharmacokinet. 2017, 56, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Edginton, A.N.; Schmitt, W.; Voith, B.; Willmann, S. A Mechanistic Approach for the Scaling of Clearance in Children. Clin. Pharmacokinet. 2006, 45, 683–704. [Google Scholar] [CrossRef]
- Edginton, A.N.; Willmann, S. Physiology-based versus allometric scaling of clearance in children; an eliminating process based comparison. Paediatric Perinat. Drug Ther. 2006, 7, 146–153. [Google Scholar] [CrossRef]
- Kearns, G.L.; Abdel-Rahman, S.M.; Alander, S.W.; Blowey, D.L.; Leeder, J.S.; Kauffman, R.E. Developmental pharmacology--drug disposition, action, and therapy in infants and children. N. Engl. J. Med. 2003, 349, 1157–1167. [Google Scholar] [CrossRef]
- Kapil, R.P.; Cipriano, A.; Michels, G.H.; Perrino, P.; O’Keefe, S.A.; Shet, M.S.; Colucci, S.V.; Noveck, R.J.; Harris, S.C. Effect of Ketoconazole on the Pharmacokinetic Profile of Buprenorphine following Administration of a Once-Weekly Buprenorphine Transdermal System. Clin. Drug Investig. 2012, 32, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.N.; Jamei, M.; Rowland-Yeo, K. How Does In Vivo Biliary Elimination of Drugs Change with Age? Evidence from In Vitro and Clinical Data Using a Systems Pharmacology Approach. Drug Metab. Dispos. 2016, 44, 1090–1098. [Google Scholar] [CrossRef]
- Zhang, H.; Kalluri, H.V.; Bastian, J.R.; Chen, H.; Alshabi, A.; Caritis, S.N.; Venkataramanan, R. Gestational changes in buprenorphine exposure: A physiologically-based pharmacokinetic analysis. Br. J. Clin. Pharmacol. 2018, 84, 2075–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, B.; Liu, S.; Xue, Y.; He, X.; Man, V.H.; Xie, X.-Q.; Wang, J. Prediction of Drug–Drug Interactions Between Opioids and Overdosed Benzodiazepines Using Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulation. Drugs R D 2019, 19, 297–305. [Google Scholar] [CrossRef] [Green Version]
- Jensen, M.L.; Foster, D.; Upton, R.; Grant, C.; Martinez, A.; Somogyi, A. Comparison of cerebral pharmacokinetics of buprenorphine and norbuprenorphine in an in vivo sheep model. Xenobiotica 2007, 37, 441–457. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, M.; Kotaki, H.; Nishitateno, K.; Sawada, Y.; Iga, T. Kinetics of respiratory depression in rats induced by buprenorphine and its metabolite, norbuprenorphine. J. Pharmacol. Exp. Ther. 1997, 281, 428–433. [Google Scholar] [PubMed]
- Strang, J.; Knight, A.; Baillie, S.; Reed, K.; Bogdanowicz, K.; Bell, J. Norbuprenorphine and respiratory depression: Exploratory analyses with new lyophilized buprenorphine and sublingual buprenorphine. Int. J. Clin. Pharmacol. Ther. 2018, 56, 81–85. [Google Scholar] [CrossRef]
- Hans, G.; Robert, D. Transdermal buprenorphine—A critical appraisal of its role in pain management. J. Pain Res. 2009, 2, 117–134. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Study | Dose [mg] | Administration | n | Female [%] | Age [Years] | Weight [kg] | Blood Sample a | Norbuprenorphine Measurements | Dataset | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| Adults | ||||||||||
| Bai et al. 2016 | 0.3 | iv (2 min) | 25 | 24 | 35.5 (20–53) | 76.1 (62.6–93.0) | arterial | no | e | [54] |
| Bartlett et al. 1980 | 0.3 | iv (1 min) b | 1 | - | - | - | arterial | no | e | [55] |
| Bullingham et al. 1980 (1) | 0.3 | iv (1 min) | 24 | 42 | 64.5 ± 1.6 | 67.7 ± 2.4 | arterial | no | e | [56] |
| Bullingham et al. 1980 (2) | 0.3 | iv (1 min, m.d.) | 10 | 40 | 67.5 ± 6.5 | 67.5 ± 2.1 | arterial | no | e | [56] |
| Bullingham et al. 1982 (1) | 0.3 | iv (1 min) | 5 | 60 | 66.8 ± 2.9 | 65.0 ± 4.0 | arterial | no | e | [57] |
| Bullingham et al. 1982 (2) | 0.3 | iv (1 min) | 5 | 60 | 64.2 ± 2.5 | 66.4 ± 2.9 | arterial | no | i | [57] |
| Bullingham et al. 1982 (3) | 0.3 | iv (1 min) | 5 | 60 | 66.0 ± 3.2 | 64.8 ± 3.9 | arterial | no | e | [57] |
| Everhart et al. 1999 | 1 | iv (60 min) | 6 | - | - | - | - | yes | i | [35] |
| Hagelberg et al. 2016 (1) | 1 | iv (bolus) | 12 | 42 | - (19–23) | - (57–95) | venous | no | e | [26] |
| Hagelberg et al. 2016 (2) c | 1 | iv (bolus) | 12 | 42 | - (19–23) | - (57–95) | venous | no | e | [26] |
| Harris et al. 2000 | 4 | iv (10 min) | 9 | 11 | 34 (21–42) | - | venous | no | e | [58] |
| Huestis et al. 2013 (1) | 2 | iv (1 min) | 5 | - | 34.6 (32–39) | 74.7 (62.1–82.6) | venous | yes | i | [59] |
| Huestis et al. 2013 (2) | 4 | iv (1 min) | 5 | - | 34.6 (32–39) | 74.7 (62.1–82.6) | venous | yes | e | [59] |
| Huestis et al. 2013 (3) | 8 | iv (1 min) | 5 | - | 34.6 (32–39) | 74.7 (62.1–82.6) | venous | yes | e | [59] |
| Huestis et al. 2013 (4) | 12 | iv (1 min) | 5 | - | 34.6 (32–39) | 74.7 (62.1–82.6) | venous | yes | e | [59] |
| Huestis et al. 2013 (5) | 16 | iv (1 min) | 5 | - | 34.6 (32–39) | 74.7 (62.1–82.6) | venous | yes | i | [59] |
| Kuhlman et al. 1996 | 1.2 | iv (1 min) | 5 | 0 | 34.4 (27–40) | 67.7 (62.6–72.7) | venous | yes | i | [6] |
| Mendelson et al. 1997 | 1 | iv (30 min) | 6 | 17 | 29 (21–38) | - | venous | no | e | [60] |
| Pediatrics | ||||||||||
| Barrett et al. 1993 d | 3 µg/kg + 0.72–2.16 µg/kg/h | iv (30 min + 11–118 h) | 12 | - | 31 weeks (27–34) e | 1.5 (0.9–2.4) | arterial | no | e | [51] |
| Olkkola et al. 1989 | 3 µg/kg | iv (2 min) | 10 | - | 5.9 (4.6–7.5) | 21.4 (18.5–25) | venous | no | e | [61] |
| Parameter | Value | Unit | Source | Literature | Reference | Value | Unit | Source | Literature | Reference | Description |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Buprenorphine | Norbuprenorphine | ||||||||||
| MW | 467.64 | g/mol | lit. | 467.64 | [63] a | 413.55 | g/mol | lit. | 413.55 | [63] b | Molecular weight |
| pKa1 (base) | 12.54 | lit. | 12.54 | [63] a | 10.49 | lit. | 10.49 | [63] b | Acid dissociation constant | ||
| pKa2 (acid) | 7.50 | lit. | 7.50 | [63] a | 9.80 | lit. | 9.80 | [63] b | Acid dissociation constant | ||
| logP | 3.40 | lit. | 3.40 | [64] | 3.19 | lit. | 3.19 | [65] c | Lipophilicity | ||
| fu (adults) | 4.0 | % | lit. | 4.0 | [66] | 21.7 | % | optim. | - | - | Fraction unbound |
| fu (children) | 5.1 | % | calc. | 5.1 | [40,66] | Fraction unbound | |||||
| fu (preterm neonates) | 7.2 | % | calc. | 7.2 | [40,66] | Fraction unbound | |||||
| CYP2C8 Km -> norbup | 5.2 | µmol/L | lit. | 5.2 | [21] e | Michaelis-Menten constant | |||||
| CYP2C8 vmax -> norbup | f1∙176.3 | pmol/min/mg protein | lit. | 176.3 | [21] | Maximum reaction velocity | |||||
| CYP3A4 Km -> norbup | 5.7 | µmol/L | lit. | 5.7 | [21] e | Michaelis-Menten constant | |||||
| CYP3A4 vmax -> norbup | f1∙520.0 | pmol/min/mg protein | lit. | 520.0 | [21] | Maximum reaction velocity | |||||
| CYP3A4 Km -> undef | 5.7 | µmol/L | ass. | - | - | Michaelis-Menten constant | |||||
| CYP3A4 vmax -> undef | f2∙1352.1 | pmol/min/mg protein | calc.d | - | - | Maximum reaction velocity | |||||
| CYP3A7 Km -> undef | 29.1 | µmol/L | calc.d | - | - | Michaelis-Menten constant | |||||
| CYP3A7 vmax -> undef | f2∙632.6 | pmol/min/mg protein | calc.d | - | - | Maximum reaction velocity | |||||
| UGT1A1 Km -> undef | 10.4 | µmol/L | lit. | 10.4 | [34] e | 21.8 | µmol/L | lit. | 21.8 | [34] e | Michaelis-Menten constant |
| UGT1A1 vmax -> undef | f2∙6726.8 | pmol/min/mg protein | lit. | 6726.8 | [34] | f3∙714.6 | pmol/min/mg protein | lit. | 714.6 | [34] | Maximum reaction velocity |
| UGT1A3 Km -> undef | 1.1 | µmol/L | lit. | 1.1 | [34] e | 14.7 | µmol/L | lit. | 14.7 | [34] e | Michaelis-Menten constant |
| UGT1A3 vmax -> undef | f2∙642.6 | pmol/min/mg protein | lit. | 642.6 | [34] | f3∙387.0 | pmol/min/mg protein | lit. | 387.0 | [34] | Maximum reaction velocity |
| UGT2B7 Km -> undef | 1.8 | µmol/L | lit. | 1.8 | [34] e | Michaelis-Menten constant | |||||
| UGT2B7 vmax -> undef | f2∙823.8 | pmol/min/mg protein | lit. | 823.8 | [34] | Maximum reaction velocity | |||||
| P-gp Km | 3.4 | µmol/L | optim. | - | Michaelis-Menten constant | ||||||
| P-gp kcat | 2.14 | 1/min | optim. | - | Transport rate constant | ||||||
| f1 | 2.80 | optim. | - | Factor | |||||||
| f2 | 0.27 | optim. | - | Factor | |||||||
| f3 | 0.43 | optim. | - | Factor | |||||||
| GFR fraction | 1.00 | ass. | - | 1.00 | ass. | - | Filtered drug in the urine | ||||
| UGT1A1 Ki | 14.8 | µmol/L | lit. | 14.8 | [34] e | Conc. for 50% inhibition | |||||
| UGT1A3 Ki | 0.5 | µmol/L | lit. | 0.5 | [34] e | 1.6 | µmol/L | lit. | 1.6 | [34] e | Conc. for 50% inhibition |
| Partition coefficients | Diverse | calc. | Schmitt | [67] | Diverse | calc. | PK-Sim | [38] | Cell to plasma partitioning | ||
| Cellular permeability | 6.91E-03 | cm/min | calc. | PK-Sim | [38] | 8.91E-03 | cm/min | calc. | PK-Sim | [38] | Perm. into the cellular space |
| Victim Drug [Dose] | Perpetrator Drug [Dose] | Pred. AUC Ratio (Buprenorphine) with/without Perpetrator | Obs. AUC Ratio (Buprenorphine) with/without Perpetrator | Pred. AUC Ratio (Norbuprenorphine) with/without Perpetrator | Obs. AUC Ratio (Norbuprenorphine) with/without Perpetrator | Reference |
|---|---|---|---|---|---|---|
| Buprenorphine [1.4 mg, 168 h iv infusion] | Clarithromycin [250 mg, bid, po] | 1.06 | - | 0.82 | - | simulated a |
| Buprenorphine [1.4 mg, 168 h iv infusion] | Itraconazole [100 mg, bid, po] b | 1.11 | - | 0.64 | - | simulated a |
| Buprenorphine [1 mg, iv bolus] | Rifampicin [600 mg, qd, po] | 0.89 | 0.85 | 1.11 | - | [26] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovar, L.; Schräpel, C.; Selzer, D.; Kohl, Y.; Bals, R.; Schwab, M.; Lehr, T. Physiologically-Based Pharmacokinetic (PBPK) Modeling of Buprenorphine in Adults, Children and Preterm Neonates. Pharmaceutics 2020, 12, 578. https://doi.org/10.3390/pharmaceutics12060578
Kovar L, Schräpel C, Selzer D, Kohl Y, Bals R, Schwab M, Lehr T. Physiologically-Based Pharmacokinetic (PBPK) Modeling of Buprenorphine in Adults, Children and Preterm Neonates. Pharmaceutics. 2020; 12(6):578. https://doi.org/10.3390/pharmaceutics12060578
Chicago/Turabian StyleKovar, Lukas, Christina Schräpel, Dominik Selzer, Yvonne Kohl, Robert Bals, Matthias Schwab, and Thorsten Lehr. 2020. "Physiologically-Based Pharmacokinetic (PBPK) Modeling of Buprenorphine in Adults, Children and Preterm Neonates" Pharmaceutics 12, no. 6: 578. https://doi.org/10.3390/pharmaceutics12060578
APA StyleKovar, L., Schräpel, C., Selzer, D., Kohl, Y., Bals, R., Schwab, M., & Lehr, T. (2020). Physiologically-Based Pharmacokinetic (PBPK) Modeling of Buprenorphine in Adults, Children and Preterm Neonates. Pharmaceutics, 12(6), 578. https://doi.org/10.3390/pharmaceutics12060578