Current Trends in ATRA Delivery for Cancer Therapy

 ,
,  ,
, 
 , , and
, , and 
Abstract
1. Introduction
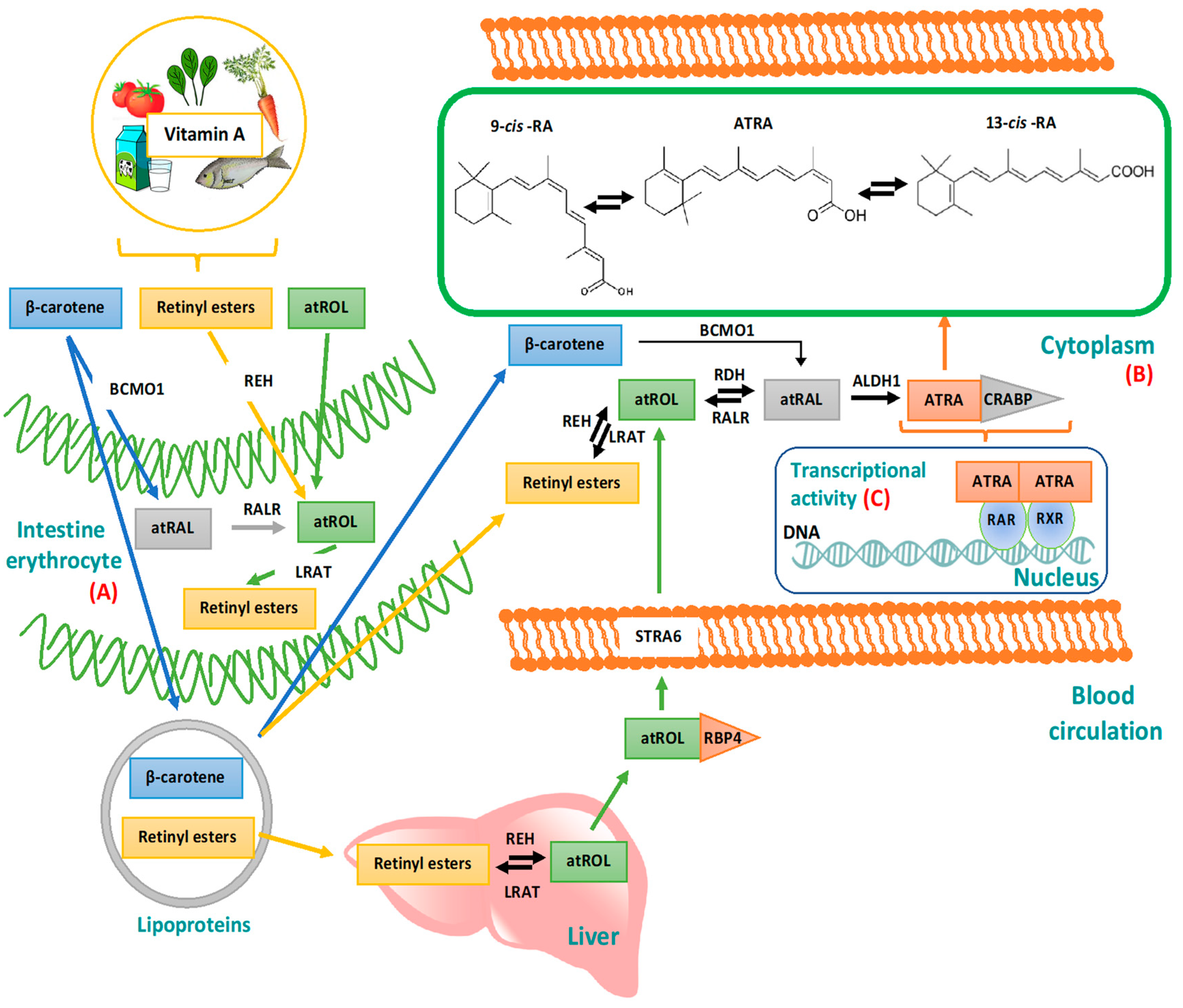
2. From Vitamin A to ATRA
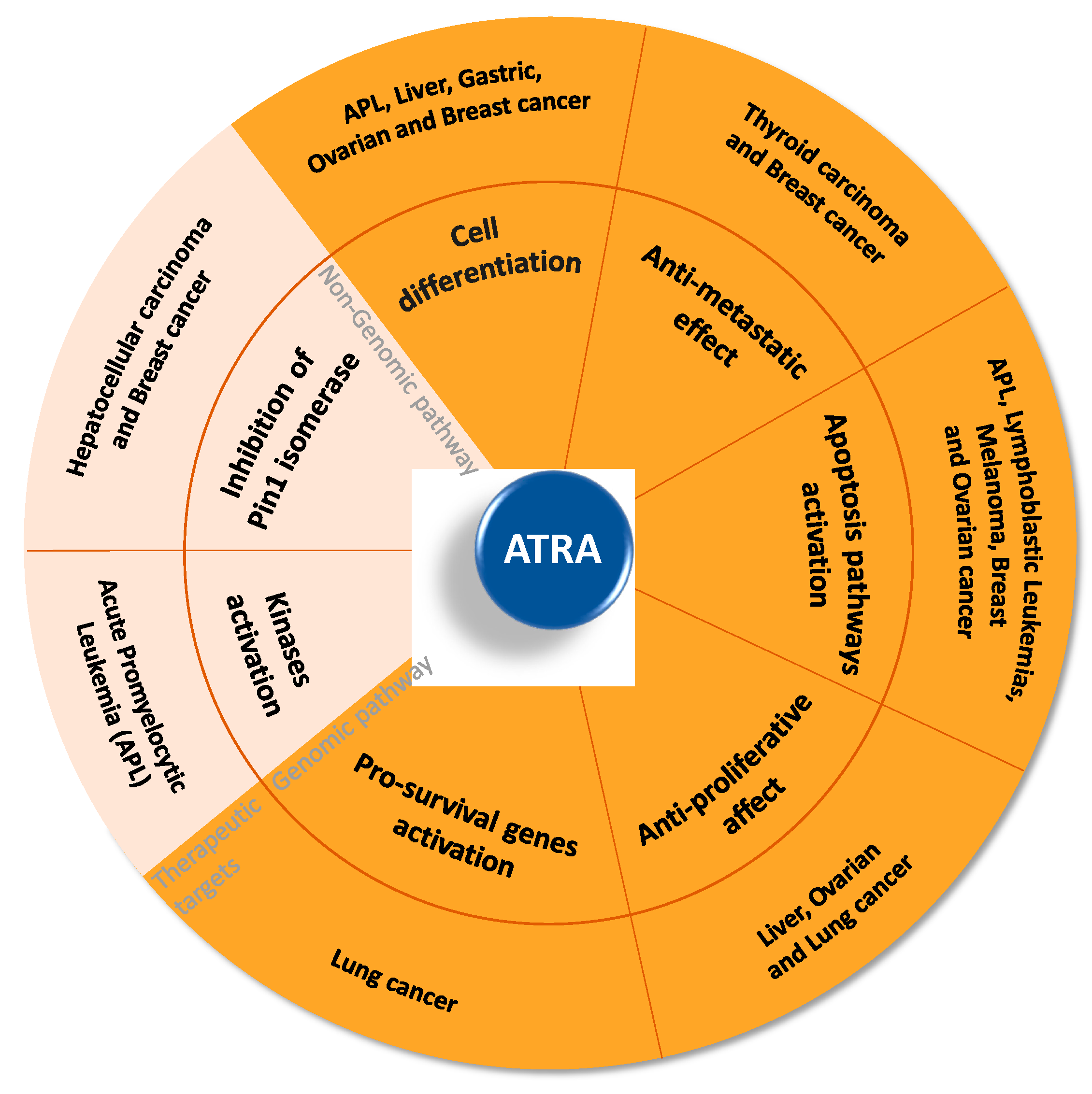
3. ATRA AND CANCER: Successes and Failures
4. ATRA Delivery Strategies: What We Got in the Clinics
5. ATRA Delivery Strategies: Moving Forward
5.1. Oral Administration
5.2. Intravenous Administration
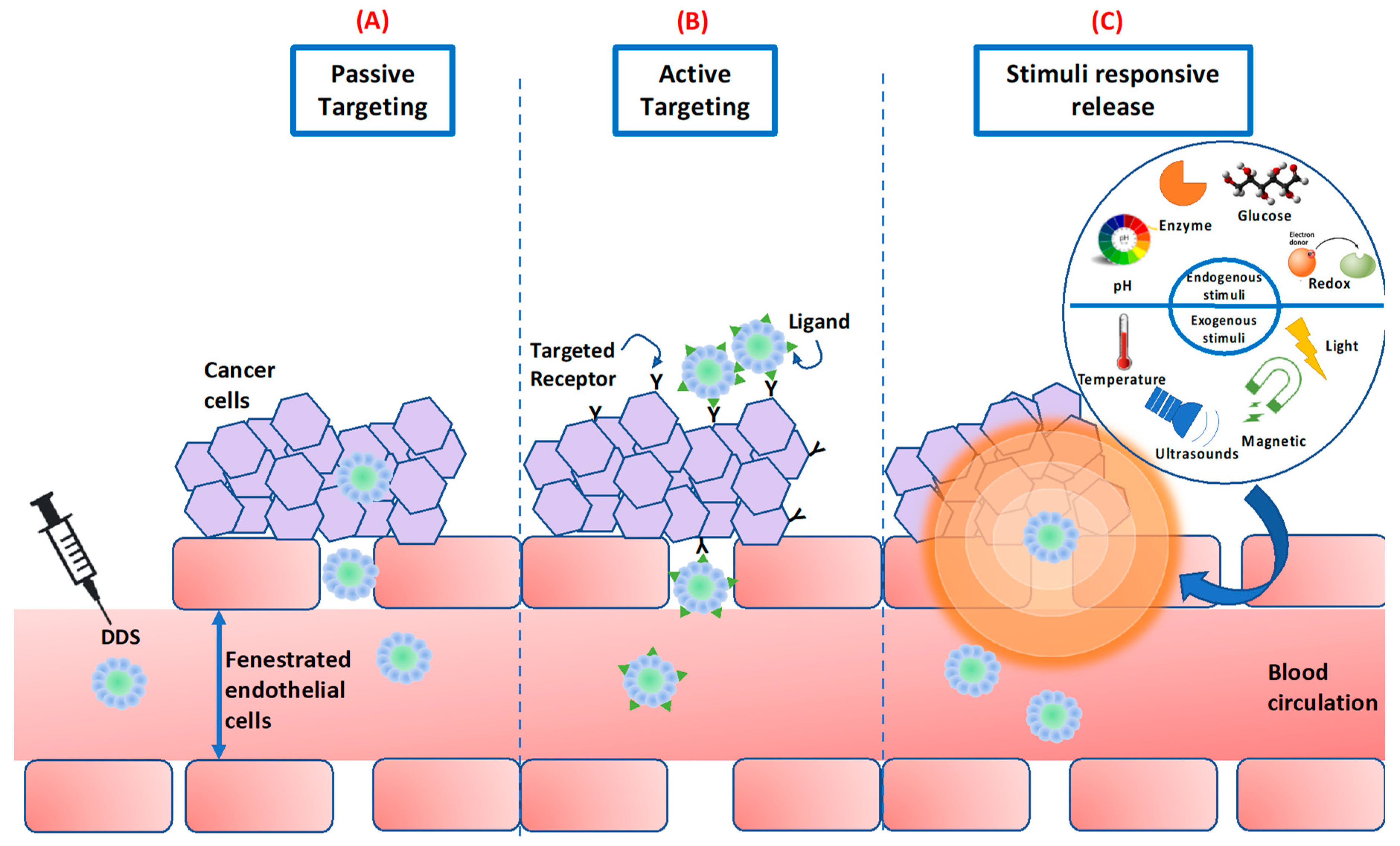
5.2.1. Stealth Strategy
5.2.2. Passive Tumor Accumulation—Enhanced Permeability Retention Effect
5.2.3. Active Targeting—Surface Functionalization with Specific Targeting Ligands
5.2.4. Cellular Uptake and Endosomal Escape
Positive Charge Surface
Proton-Sponge Effect
5.2.5. Stimuli-Responsiveness
5.3. Inhalable Administration
5.4. Alternative Administration

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor | ATRA Delivery System | Model | Pros | Cons | Ref |
|---|---|---|---|---|---|
| Acute Promyelocytic Leukemia | ATRA-loaded microemulsion O/W | Porcine intestinal membrane | Oral delivery of ATRA to enhance drug bioavailability and intestinal absorption | Only in vitro studies | [104] |
| DOX-loaded LMWH–ATRA nanoparticles (DHR nanoparticles) negatively charged | Cell lines: HL-60 and MCF-7; mouse model | Lower risk of bleeding and thrombocytopenia/selective uptaking endocytosis mediated | [137] | ||
| ATRA-loaded in Cholesteryl Butyrate Solid Lipid Nanoparticles | Cell lines: HL-60, Jurkat, and THP1 | High encapsulation efficiency over and enhanced anticancer activity when compared to the free ATRA | Only in vitro studies | [96] | |
| Breast cancer | ATRA-loaded Pluronic F127 micelles | Cell lines: 4T1, MDA-MB-231, EMT6, and BT474; Mouse model | Biocompatibility, high ATRA loading content and synergistic effects with Cisplatin | No biodistribution studies | [98] |
| Human serum albumin (HSA)-based nanoparticles for the co-delivery of ATRA and Paclitaxel (PTX) | Cell line: 4T1 | Increase of individual drug’s efficacy both in vitro and in vivo, inhibition of the migration and invasion of cancer cells in vivo (reduction of cancer cell MMPs activity and of EMT process) | [115] | ||
| Hyaluronic acid (HA) nanoparticle with an inner hydrophobic core containing ATRA and the anticancer drug Gambogic acid (GA) | Cell lines: MCF-7 and KB31 | HA receptor-mediated endocytosis improves the internalization into the tumor cells | [146] | ||
| Nanoparticles co-delivery strategy of an ATRA and DOX based-therapy | Cell line: MDA-MB-231 | Selective uptaking | [127] | ||
| Nanoparticles encapsulating ICG dye with coumarin-containing ATRA (AC), modified with the targeted ligand cyclic (Arg-Gly-Asp-D-Phe-Lys) (cRGD) peptide on the surface | Cell lines: MCF-7 and MDA-MB-231 | Combination of photodynamic therapy (PDT), photothermal therapy (PTT), and chemotherapy | Only in vitro studies | [184] | |
| Amphiphilic zein-chondroitin sulfate (ChS)-based copolymeric micelles containing ATRA/Etoposide | Cell line: MCF-7; mouse model | Enhancing internalization in vitro and reducing tumor volume, decreasing proliferation, and promoting necrosis in vivo | No PK and biodistribution studies | [134] | |
| Gastric cancer | CD44/CD133 antibodyconjugated ATRA-loaded nanoparticles | Cell lines: MKN-45 and NCI-N87 | Specific target of cancer stem cells by using membrane markers | Difficulty reaching the targeted site in vivo | [142] |
| ATRA/Sorafenib/miR-542-3p co-delivery in PEGylated Gelucire-based Solid Lipid Nanoparticles | Cell line: MGC-803; mouse model | Enhanced anti-tumor efficacy of drug co-loading | No biodistribution studies | [121] | |
| Glioblastoma | ATRA-loaded poly(diol citrate) wafers | Cell line: U87MG | Long-term treatment in vitro and reduced ATRA isomerization and degradation | Duration of release in vivo is not known | [197] |
| 3D bioprinted hydrogel mesh loaded with ATRA | Cell line: U87MG | Controlled release and immobilization of DDS close to tumor site | Biocompatibility of the construct in the brain in vivo | [195] | |
| CARD-B6 NPs loaded with ATRA, DOX and CA4 | Cell line: U87MG; Mouse model | Controlled release by using different peptide tools and a tractable DDS by MRI | [178] | ||
| Liver cancer | Poly(amidoamine) (PAMAM) dendrimers | Cell line: HepG2 | pH-responsive DDS and enhanced cellular uptake | Only in vitro studies | [167] |
| Lung cancer | ATRA/Genestein-loaded hybrid lipid nanocore-protein shell | Cell line: A549; Mouse model | Stable inhalable dry powder | [191] | |
| ATRA/Paclitaxel-PEG-b-PBLA micelles (pH and redox dual-responsive) | Cell line: A549; Mouse model | Prolonged circulation time, reduced nonspecific protein adsorption effective delivery to the tumor site and within the tumor cells, controlled drug release, and negligible systemic toxicity | No biodistribution studies | [168] | |
| DOTAP liposomes loaded with ATRA | Mouse model | Higher half-life, Cmax and a lower CL of ATRA loaded liposomes compared to the mice treated with free ATRA. | Strong immune response | [157] | |
| ATRA-loaded niosomes | Inhalable DDS to enhance drug localization in the targeted site | Only in vitro studies | [190] | ||
| Lymphoma | ATRA nanoparticles constituted by a fusion protein scaffold comprising apolipoprotein A1 (APOA1) and a single chain variable antibody fragment (scFv) against CD20 | Lymphoma | Targeted therapy thanks to selective uptake | Only in vitro studies | [150] |
| Melanoma | Polymeric micelles of hyaluronic acid – ATRA for the co-delivery of Paclitaxel and ATRA | Cell line: B16F10; Rat model | Redox-responsive drug release and higher CD44-dependent cellular uptake in vitro, and prolonged circulation time | Antitumor efficacy of the constuct is not known in vivo | [171] |
| CD20-antibody conjugated PLGA nanoparticles | Cell lines: A375 and WM266-4 | Better targeting and stronger inhibitory effects against melanoma-initiating cells (CD20+) with respect to CD20- cells | Only in vitro studies | [140] | |
| Lipid-coated Hollow Mesoporous Silica Nanoparticles-ATRA/Doxorubicin/IL-2 | Mouse model | Excellent encapsulation capacity, satisfactory stability, favorable biodistribution and low systemic toxicity | [99] | ||
| Ovarian cancer | Polymer-oil nanostructued carrier (PONC) | Cell line: SKOV-3 | Controlled and sustained release profile, biological stability and increased cellular uptake by efficient drug permeation | Only in vitro studies | [152] |
| Pancreatic ductal adenocarcinoma | PEGylated polyethylenimine-coated gold nanoparticles for the co-delivery of ATRA and siRNAHSP47 | Cell lines: Pancreatic cancer primary cells; Mouse model | pH-responsive DDS, stability in the systemic circulation, negligible system toxicity, and effective accumulation in the tumor site | Quick clearance of the DDS | [173] |
| Polyamidoamine (PAMAM) dendrimer-coated magnetic iron nanoparticles (DcMNPs) | Cell lines: ductal pancreatic cells and pancreatic stellate cells (PSCs) | Magnetic nanoparticles can be targeted to tumor site in a magnetic field and they successfully taken up by pancreatic cancer and PSC cells | Only in vitro studies | [176] | |
| Thyroid cancer | ATRA/Sorafenib-loaded (PEG–PLGA) polymeric micelles | Cell line: FTC-133; Mouse model | Prolonged circulation time, effective delivery to the tumor site and within the tumor cells, controlled drug release, and negligible system toxicity | [119] |
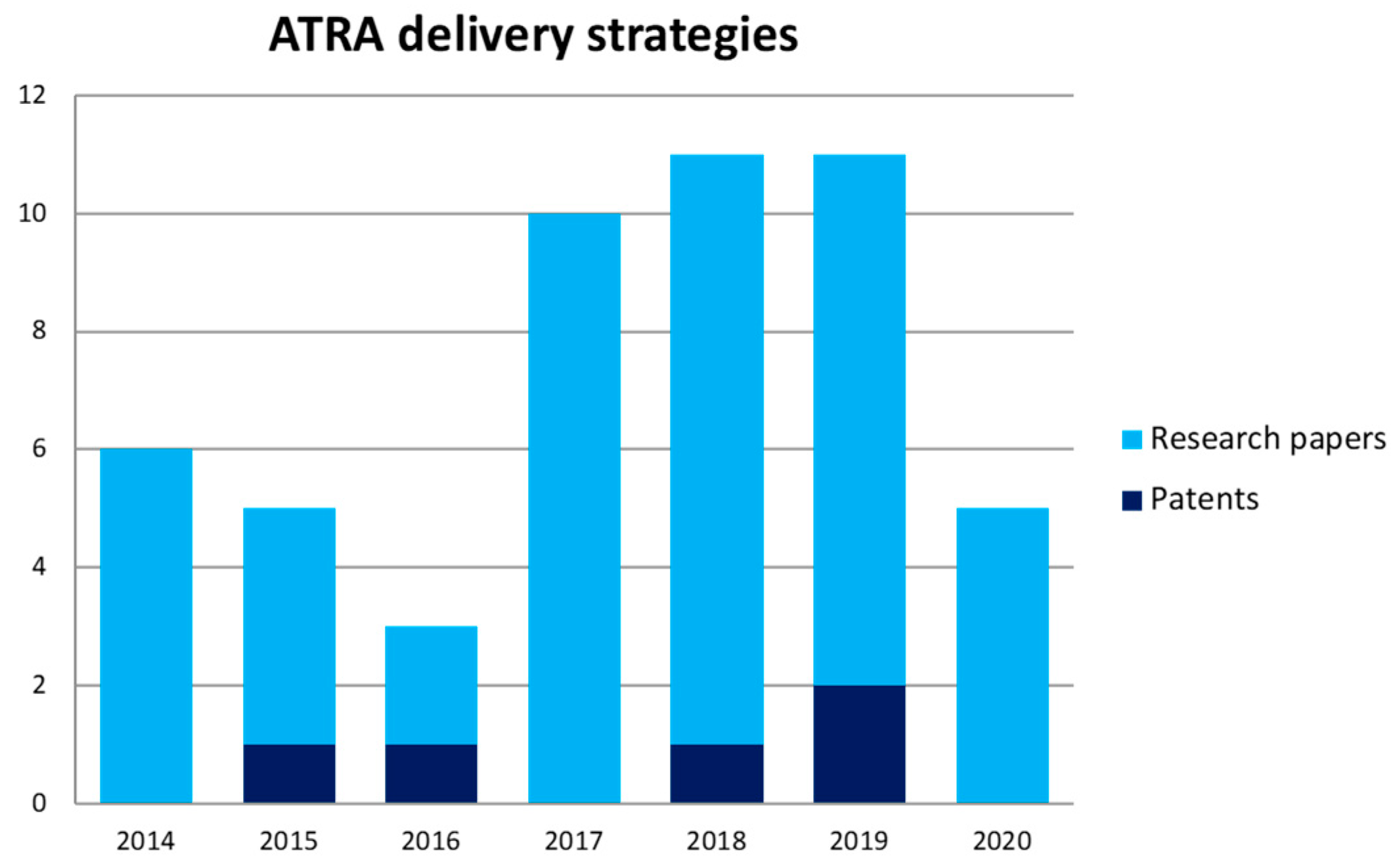
5.5. Patents
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- McCollum, E.V.; Davis, M. The necessity of certain lipins in the diet during growth. J. Biol. Chem. 1913, 15, 167–175. [Google Scholar] [CrossRef]
- Kam, R.K.T.; Deng, Y.; Chen, Y.; Zhao, H. Retinoic acid synthesis and functions in early embryonic development. Cell Biosci. 2012, 2, 11. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, D.N.; Clugston, R.D.; Blaner, W.S. Vitamin A metabolism: An update. Nutrients 2011, 3, 63–103. [Google Scholar] [CrossRef] [PubMed]
- Gropper, S.; Smith, J.; Groff, J. Advanced Nutrition and Human Metabolism, 5th ed.; Cengage Learning: Boston, MA, USA, 2009; pp. 470–488. [Google Scholar]
- Hayashi, K.; Yokozaki, H.; Naka, K.; Yasui, W.; Lotan, R.; Tahara, E. Overexpression of retinoic acid receptor β induces growth arrest and apoptosis in oral cancer cell lines. Jpn. J. Cancer Res. 2001, 92, 42–50. [Google Scholar] [CrossRef]
- Choi, Y.; Kim, S.Y.; Kim, S.H.; Yang, J.; Park, K.; Byun, Y. Inhibition of tumor growth by biodegradable microspheres containing all-trans-retinoic acid in a human head-and-neck cancer xenograft. Int. J. Cancer 2003, 107, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-F.; Bao, B.-Y.; Chang, C. Modulation of the retinoic acid-induced cell apoptosis and differentiation by the human TR4 orphan nuclear receptor. Biochem. Biophys. Res. Commun. 2004, 323, 876–883. [Google Scholar] [CrossRef]
- Park, E.Y.; Dillard, A.; Williams, E.A.; Wilder, E.T.; Pepper, M.R.; Lane, M.A. Retinol inhibits the growth of all-trans-retinoic acid–sensitive and all-trans-retinoic acid–resistant colon cancer cells through a retinoic acid receptor–independent mechanism. Cancer Res. 2005, 65, 9923–9933. [Google Scholar] [CrossRef][Green Version]
- Chlapek, P.; Slavikova, V.; Mazanek, P.; Sterba, J.; Veselska, R. Why differentiation therapy sometimes fails: Molecular mechanisms of resistance to Retinoids. Int. J. Mol. Sci. 2018, 19, 132. [Google Scholar] [CrossRef]
- Masetti, R.; Biagi, C.; Zama, D.; Vendemini, F.; Martoni, A.; Morello, W.; Gasperini, P.; Pession, A. Retinoids in pediatric onco-hematology: The model of acute promyelocytic leukemia and neuroblastoma. Adv. Ther. 2012, 29, 747–762. [Google Scholar] [CrossRef]
- Szuts, E.Z.; Harosi, F.I. Solubility of retinoids in water. Arch. Biochem. Biophys. 1991, 287, 297–304. [Google Scholar] [CrossRef]
- Muindi, J.; Frankel, S.; Miller, W.; Jakubowski, A.; Scheinberg, D.; Young, C.; Dimitrovsky, E. Continuous treatment with all-trans RA progressively decreases plasma drug concentrations: Implications for relapse and resistance in acute promyelocytic leukemia. Blood 1992, 79, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Reboul, E. Mechanisms of Carotenoid Intestinal Absorption: Where Do We Stand? Nutrients 2019, 11, 838. [Google Scholar] [CrossRef] [PubMed]
- Duester, G. Families of retinoid dehydrogenases regulating vitamin A function. Eur. J. Biochem. 2000, 267, 4315–4324. [Google Scholar] [CrossRef] [PubMed]
- Blomhoff, R.; Blomhoff, H.K. Overview of retinoid metabolism and function. J. Neurobiol. 2006, 66, 606–630. [Google Scholar] [CrossRef]
- Napoli, J.L. Cellular retinoid binding-proteins, CRBP, CRABP, FABP5: Effects on retinoid metabolism, function and related diseases. Pharm. Ther. 2017, 173, 19–33. [Google Scholar] [CrossRef]
- Schenk, T.; Stengel, S.; Zelent, A. Unlocking the potential of retinoic acid in anticancer therapy. Br. J. Cancer 2014, 111, 2039–2045. [Google Scholar] [CrossRef]
- Heyman, R.A.; Mangelsdorf, D.J.; Dyck, J.A.; Stein, R.B.; Eichele, G.; Evans, R.M.; Thaller, C. 9-cis retinoic acid is a high affinity ligand for the retinoid X receptor. Cell 1992, 68, 397–406. [Google Scholar] [CrossRef]
- Di Masi, A.; Leboffe, L.; De Marinis, E.; Pagano, F.; Cicconi, L.; Rochette-Egly, C.; Lo-Coco, F.; Ascenzi, P.; Nervi, C. Retinoic acid receptors: From molecular mechanisms to cancer therapy. Mol. Asp. Med. 2015, 41, 1–115. [Google Scholar] [CrossRef]
- Glass, C.K.; Rosenfeld, M.G. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000, 14, 121–141. [Google Scholar]
- Gudas, L.J.; Wagner, J.A. Retinoids regulate stem cell differentiation. J. Cell. Physiol. 2011, 226, 322–330. [Google Scholar] [CrossRef]
- Costantini, L.; Molinari, R.; Farinon, B.; Merendino, N. Retinoic Acids in the Treatment of Most Lethal Solid Cancers. J. Clin. Med. 2020, 9, 360. [Google Scholar] [CrossRef] [PubMed]
- Al Tanoury, Z.; Piskunov, A.; Rochette-Egly, C. Vitamin a and retinoid signaling: Genomic and nongenomic effects thematic review series: Fat-soluble vitamins: Vitamin a. J. Lipid Res. 2013, 54, 1761–1775. [Google Scholar] [CrossRef]
- Breitman, T.; Selonick, S.E.; Collins, S.J. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc. Natl. Acad. Sci. USA 1980, 77, 2936–2940. [Google Scholar] [CrossRef] [PubMed]
- Grimwade, D.; Ivey, A.; Huntly, B.J. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood Am. Soc. Hematol. 2016, 127, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, M.; Zangrilli, D.; Pandolfi, P.P.; Longo, L.; Mencarelli, A.; Giacomucci, a.; Rocchi, M.; Biondi, A.; Rambaldi, A.; Coco, F.L. Translocation breakpoint of acute promyelocytic leukemia lies within the retinoic acid receptor alpha locus. Proc. Natl. Acad. Sci. USA 1991, 88, 1977–1981. [Google Scholar] [CrossRef]
- Kakizuka, A.; Miller, W., Jr.; Umesono, K.; Warrell, R.P., Jr.; Frankel, S.R.; Murty, V.V.; Dmitrovsky, E.; Evans, R.M. Chromosomal translocation t (1.5: 17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell 1991, 66, 663. [Google Scholar] [CrossRef]
- de Thé, H. Differentiation therapy revisited. Nat. Rev. Cancer 2018, 18, 117. [Google Scholar] [CrossRef]
- Taga, T.; Tomizawa, D.; Takahashi, H.; Adachi, S. Acute myeloid leukemia in children: Current status and future directions. Pediatrics Int. 2016, 58, 71–80. [Google Scholar] [CrossRef]
- Iland, H.J.; Collins, M.; Bradstock, K.; Supple, S.G.; Catalano, A.; Hertzberg, M.; Browett, P.; Grigg, A.; Firkin, F.; Campbell, L.J. Use of arsenic trioxide in remission induction and consolidation therapy for acute promyelocytic leukaemia in the Australasian Leukaemia and Lymphoma Group (ALLG) APML4 study: A non-randomised phase 2 trial. Lancet Haematol. 2015, 2, e357–e366. [Google Scholar] [CrossRef]
- Sanz, M.A.; Fenaux, P.; Tallman, M.S.; Estey, E.H.; Löwenberg, B.; Naoe, T.; Lengfelder, E.; Döhner, H.; Burnett, A.K.; Chen, S.-J. Management of acute promyelocytic leukemia: Updated recommendations from an expert panel of the European LeukemiaNet. Blood 2019, 133, 1630–1643. [Google Scholar] [CrossRef]
- Bolis, M.; Garattini, E.; Paroni, G.; Zanetti, A.; Kurosaki, M.; Castrignano, T.; Garattini, S.K.; Biancardi, F.; Barzago, M.; Gianni’, M. Network-guided modeling allows tumor-type independent prediction of sensitivity to all-trans-retinoic acid. Ann. Oncol. 2017, 28, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Garattini, E.; Bolis, M.; Garattini, S.K.; Fratelli, M.; Centritto, F.; Paroni, G.; Zanetti, A.; Pagani, A.; Fisher, J.N.; Zambelli, A. Retinoids and breast cancer: From basic studies to the clinic and back again. Cancer Treat. Rev. 2014, 40, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Gudas, L.J. Emerging roles for retinoids in regeneration and differentiation in normal and disease states. Biochim. Biophys. Acta (Bba)-Mol. Cell Biol. Lipids 2012, 1821, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Lan, L.; Cui, D.; Luo, Y.; Shi, B.; Deng, L.; Zhang, G.; Wang, H. Inhibitory effects of retinoic acid on invasiveness of human thyroid carcinoma cell lines in vitro. J. Endocrinol. Investig. 2009, 32, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zang, C.; Fenner, M.; Possinger, K.; Elstner, E. PPARγ ligands and ATRA inhibit the invasion of human breast cancer cells in vitro. Breast Cancer Res. Treat. 2003, 79, 63–74. [Google Scholar] [CrossRef]
- Lokman, N.A.; Ho, R.; Gunasegaran, K.; Bonner, W.M.; Oehler, M.K.; Ricciardelli, C. Anti-tumour effects of all-trans retinoid acid on serous ovarian cancer. J. Exp. Clin. Cancer Res. 2019, 38, 10. [Google Scholar] [CrossRef]
- Huang, J.; Zhou, Y.-F.; Xu, J.; Liang, P.; Liu, Z.-G.; Wang, J.; Zhang, D.; Dong, Q.-M.; Shen, W.-M.; Zhuang, S.-L. Unveiling the growth mechanism of SiO2/Ag hybrid nanospheres and using for Surface Enhanced Raman Scattering detection. Appl. Surf. Sci. 2019, 463, 115–120. [Google Scholar] [CrossRef]
- Bama, E.S.; Grace, V.B.; Sundaram, V.; Jesubatham, P.D. Synergistic effect of co-treatment with all-trans retinoic acid and 9-cis retinoic acid on human lung cancer cell line at molecular level. 3 Biotech. 2019, 9, 159. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target 2020, 5, 1–35. [Google Scholar] [CrossRef]
- Spira, A.I.; Carducci, M.A. Differentiation therapy. Curr. Opin. Pharm. 2003, 3, 338–343. [Google Scholar] [CrossRef]
- Bjerkvig, R.; Tysnes, B.B.; Aboody, K.S.; Najbauer, J.; Terzis, A. The origin of the cancer stem cell: Current controversies and new insights. Nat. Rev. Cancer 2005, 5, 899–904. [Google Scholar] [CrossRef]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Dong, J.; Haiech, J.; Kilhoffer, M.-C.; Zeniou, M. Cancer stem cell quiescence and plasticity as major challenges in cancer therapy. Stem Cells Int. 2016, 2016, 1740936. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Huang, Y.-H.; Chen, J.-L. Understanding and targeting cancer stem cells: Therapeutic implications and challenges. Acta Pharmacol. Sin. 2013, 34, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Prabavathy, D.; Swarnalatha, Y.; Ramadoss, N. Lung cancer stem cells—origin, characteristics and therapy. Stem Cell Investig. 2018, 5. [Google Scholar] [CrossRef]
- Fu, Y.; Du, P.; Zhao, J.; Hu, C.E.; Qin, Y.; Huang, G. Gastric cancer stem cells: Mechanisms and therapeutic approaches. Yonsei Med. J. 2018, 59, 1150–1158. [Google Scholar] [CrossRef]
- Li, N.; Zhu, Y. Targeting liver cancer stem cells for the treatment of hepatocellular carcinoma. Ther. Adv. Gastroenterol. 2019, 12, 1756284818821560. [Google Scholar] [CrossRef]
- Szotek, P.P.; Pieretti-Vanmarcke, R.; Masiakos, P.T.; Dinulescu, D.M.; Connolly, D.; Foster, R.; Dombkowski, D.; Preffer, F.; MacLaughlin, D.T.; Donahoe, P.K. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc. Natl. Acad. Sci. USA 2006, 103, 11154–11159. [Google Scholar] [CrossRef]
- Giovannelli, P.; Di Donato, M.; Galasso, G.; Di Zazzo, E.; Medici, N.; Bilancio, A.; Migliaccio, A.; Castoria, G. Breast cancer stem cells: The role of sex steroid receptors. World J. Stem Cells 2019, 11, 594. [Google Scholar] [CrossRef]
- Croker, A.K.; Allan, A.L. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDH hi CD44+ human breast cancer cells. Breast Cancer Res. Treat. 2012, 133, 75–87. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhu, J.; Zhao, X.; Yang, K.; Lu, L.; Zhang, F.; Shen, W.; Zhang, R. All-trans retinoic acid ameliorates myocardial ischemia/reperfusion injury by reducing cardiomyocyte apoptosis. PLoS ONE 2015, 10, e0133414. [Google Scholar] [CrossRef] [PubMed]
- Modarai, S.R.; Gupta, A.; Opdenaker, L.M.; Kowash, R.; Masters, G.; Viswanathan, V.; Zhang, T.; Fields, J.Z.; Boman, B.M. The anti-cancer effect of retinoic acid signaling in CRC occurs via decreased growth of ALDH+ colon cancer stem cells and increased differentiation of stem cells. Oncotarget 2018, 9, 34658. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Liu, C. All-trans retinoic acid therapy induces asymmetric division of glioma stem cells from the U87MG cell line. Oncol. Lett. 2019, 18, 3646–3654. [Google Scholar] [CrossRef]
- Moog-Lutz, C.; Cavé-Riant, F.; Guibal, F.C.; Breau, M.A.; Di Gioia, Y.; Couraud, P.O.; Cayre, Y.E.; Bourdoulous, S.; Lutz, P.G. JAML, a novel protein with characteristics of a junctional adhesion molecule, is induced during differentiation of myeloid leukemia cells. Blood 2003, 102, 3371–3378. [Google Scholar] [CrossRef] [PubMed]
- Ara, C.; DEVIRGILIIS, L.C.; Massimi, M. Influence of retinoic acid on adhesion complexes in human hepatoma cells: A clue to its antiproliferative effects. Cell Commun. Adhes. 2004, 11, 13–23. [Google Scholar] [CrossRef][Green Version]
- Kominsky, S.L.; Argani, P.; Korz, D.; Evron, E.; Raman, V.; Garrett, E.; Rein, A.; Sauter, G.; Kallioniemi, O.-P.; Sukumar, S. Loss of the tight junction protein claudin-7 correlates with histological grade in both ductal carcinoma in situ and invasive ductal carcinoma of the breast. Oncogene 2003, 22, 2021–2033. [Google Scholar] [CrossRef]
- Martin, T.A.; Mansel, R.E.; Jiang, W.G. Loss of occludin leads to the progression of human breast cancer. Int. J. Mol. Med. 2010, 26, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Leech, A.O.; Cruz, R.G.; Hill, A.D.; Hopkins, A.M. Paradigms lost—an emerging role for over-expression of tight junction adhesion proteins in cancer pathogenesis. Ann. Transl. Med. 2015, 3, 184–199. [Google Scholar]
- Solimando, A.G.; Da Vià, M.C.; Leone, P.; Borrelli, P.; Croci, G.A.; Tabares, P.; Brandl, A.; Di Lernia, G.; Bianchi, F.P.; Tafuri, S. Halting the vicious cycle within the multiple myeloma ecosystem: Blocking JAM-A on bone marrow endothelial cells restores the angiogenic homeostasis and suppresses tumor progression. Haematologica 2020, 105. [Google Scholar] [CrossRef]
- Nguyen, E.; Gausdal, G.; Varennes, J.; Pendino, F.; Lanotte, M.; Døskeland, S.O.; Ségal-Bendirdjian, E. Activation of both protein kinase A (PKA) type I and PKA type II isozymes is required for retinoid-induced maturation of acute promyelocytic leukemia cells. Mol. Pharmacol. 2013, 83, 1057–1065. [Google Scholar] [CrossRef]
- Weng, X.-q.; Sheng, Y.; Ge, D.-z.; Wu, J.; Shi, L.; Cai, X. RAF-1/MEK/ERK pathway regulates ATRA-induced differentiation in acute promyelocytic leukemia cells through C/EBPβ, C/EBPε and PU. 1. Leuk. Res. 2016, 45, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Kozono, S.; Kats, L.; Nechama, M.; Li, W.; Guarnerio, J.; Luo, M.; You, M.-H.; Yao, Y.; Kondo, A. Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nat. Med. 2015, 21, 457. [Google Scholar] [CrossRef]
- Liu, H.-L.; Fan, C.-H.; Ting, C.-Y.; Yeh, C.-K. Combining microbubbles and ultrasound for drug delivery to brain tumors: Current progress and overview. Theranostics 2014, 4, 432. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.Z.; Lu, K.P. The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nat. Rev. Cancer 2016, 16, 463. [Google Scholar] [CrossRef] [PubMed]
- Franciosa, G.; Diluvio, G.; Del Gaudio, F.; Giuli, M.; Palermo, R.; Grazioli, P.; Campese, A.; Talora, C.; Bellavia, D.; D’Amati, G. Prolyl-isomerase Pin1 controls Notch3 protein expression and regulates T-ALL progression. Oncogene 2016, 35, 4741–4751. [Google Scholar] [CrossRef]
- Liao, X.-H.; Zhang, A.L.; Zheng, M.; Li, M.-Q.; Chen, C.P.; Xu, H.; Chu, Q.-S.; Yang, D.; Lu, W.; Tsai, T.-F. Chemical or genetic Pin1 inhibition exerts potent anticancer activity against hepatocellular carcinoma by blocking multiple cancer-driving pathways. Sci. Rep. 2017, 7, 1–11. [Google Scholar]
- Zhang, M.; Sun, H.; Zhao, S.; Wang, Y.; Pu, H.; Wang, Y.; Zhang, Q. Expression of PD-L1 and prognosis in breast cancer: A meta-analysis. Oncotarget 2017, 8, 31347. [Google Scholar] [CrossRef]
- Yang, D.; Luo, W.; Wang, J.; Zheng, M.; Liao, X.-H.; Zhang, N.; Lu, W.; Wang, L.; Chen, A.-Z.; Wu, W.-G. A novel controlled release formulation of the Pin1 inhibitor ATRA to improve liver cancer therapy by simultaneously blocking multiple cancer pathways. J. Control. Release 2018, 269, 405–422. [Google Scholar] [CrossRef]
- Campaner, E.; Rustighi, A.; Zannini, A.; Cristiani, A.; Piazza, S.; Ciani, Y.; Kalid, O.; Golan, G.; Baloglu, E.; Shacham, S. A covalent PIN1 inhibitor selectively targets cancer cells by a dual mechanism of action. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Kozono, S.; Lin, Y.-M.; Seo, H.-S.; Pinch, B.; Lian, X.; Qiu, C.; Herbert, M.K.; Chen, C.-H.; Tan, L.; Gao, Z.J. Arsenic targets Pin1 and cooperates with retinoic acid to inhibit cancer-driving pathways and tumor-initiating cells. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Huang, S.; Chen, Y.; Liang, Z.-M.; Li, N.-N.; Liu, Y.; Zhu, Y.; Liao, D.; Zhou, X.Z.; Lu, K.P.; Yao, Y. Targeting Pin1 by all-trans retinoic acid (ATRA) overcomes tamoxifen resistance in breast cancer via multifactorial mechanisms. Front. Cell Dev. Biol. 2019, 7, 322. [Google Scholar] [CrossRef] [PubMed]
- Freemantle, S.J.; Spinella, M.J.; Dmitrovsky, E. Retinoids in cancer therapy and chemoprevention: Promise meets resistance. Oncogene 2003, 22, 7305–7315. [Google Scholar] [CrossRef]
- Connolly, R.M.; Nguyen, N.K.; Sukumar, S. Molecular pathways: Current role and future directions of the retinoic acid pathway in cancer prevention and treatment. Clin. Cancer Res. 2013, 19, 1651–1659. [Google Scholar] [CrossRef] [PubMed]
- Duong, V.; Rochette-Egly, C. The molecular physiology of nuclear retinoic acid receptors. From health to disease. Biochim. Et Biophys. Acta (Bba)-Mol. Basis Dis. 2011, 1812, 1023–1031. [Google Scholar] [CrossRef]
- Imaizumi, M.; Suzuki, H.; Yoshinari, M.; Sato, A.; Saito, T.; Sugawara, A.; Tsuchiya, S.; Hatae, Y.; Fujimoto, T.; Kakizuka, A. Mutations in the E-domain of RARα portion of the PML/RARα chimeric gene may confer clinical resistance to all-trans retinoic acid in acute promyelocytic leukemia. Blood Am. Soc. Hematol. 1998, 92, 374–382. [Google Scholar]
- Ohnuma-Ishikawa, K.; Morio, T.; Yamada, T.; Sugawara, Y.; Ono, M.; Nagasawa, M.; Yasuda, A.; Morimoto, C.; Ohnuma, K.; Dang, N.H. Knockdown of XAB2 Enhances All-Trans Retinoic Acid–Induced Cellular Differentiation in All-Trans Retinoic Acid–Sensitive and–Resistant Cancer Cells. Cancer Res. 2007, 67, 1019–1029. [Google Scholar] [CrossRef]
- Cheung, B.B. Combination therapies improve the anticancer activities of retinoids in neuroblastoma. World J. Clin. Oncol. 2015, 6, 212. [Google Scholar] [CrossRef]
- Adamson, P.C.; Widemann, B.C.; Reaman, G.H.; Seibel, N.L.; Murphy, R.F.; Gillespie, A.F.; Balis, F.M. A phase I trial and pharmacokinetic study of 9-cis-retinoic acid (ALRT1057) in pediatric patients with refractory cancer: A joint Pediatric Oncology Branch, National Cancer Institute, and Children’s Cancer Group study. Clin. Cancer Res. 2001, 7, 3034–3039. [Google Scholar]
- Sutton, L.M.; Warmuth, M.A.; Petros, W.P.; Winer, E.P. Pharmacokinetics and clinical impact of all-trans retinoic acid in metastatic breast cancer: A phase II trial. Cancer Chemother. Pharmacol. 1997, 40, 335–341. [Google Scholar] [CrossRef]
- Schultze, E.; Collares, T.; Lucas, C.G.; Seixas, F.K. Synergistic and additive effects of ATRA in combination with different anti-tumor compounds. Chem.-Biol. Interact. 2018, 285, 69–75. [Google Scholar] [CrossRef]
- Ho, B.N.; Pfeffer, C.M.; Singh, A.T. Update on nanotechnology-based drug delivery systems in cancer treatment. Anticancer Res. 2017, 37, 5975–5981. [Google Scholar] [PubMed]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An updated review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Rinaldi, F.; Hanieh, P.N.; Del Favero, E.; Rondelli, V.; Brocca, P.; Pereira, M.C.; Andreev, O.A.; Reshetnyak, Y.K.; Marianecci, C.; Carafa, M. Decoration of nanovesicles with pH (low) insertion peptide (pHLIP) for targeted delivery. Nanoscale Res. Lett. 2018, 13, 391. [Google Scholar] [CrossRef] [PubMed]
- Ozpolat, B.; Lopez-Berestein, G.; Adamson, P.; Fu, C.; Williams, A.H. Pharmacokinetics of intravenously administered liposomal all-trans-retinoic acid (ATRA) and orally administered ATRA in healthy volunteers. J. Pharm. Pharm. Sci. 2003, 6, 292–301. [Google Scholar]
- Tallman, M.S. Therapy of acute promyelocytic leukemia: All-trans retinoic acid and beyond. Leukemia 1998, 12. [Google Scholar]
- Douer, D.; Estey, E.; Santillana, S.; Bennett, J.M.; Lopez-Bernstein, G.; Boehm, K.; Williams, T. Treatment of newly diagnosed and relapsed acute promyelocytic leukemia with intravenous liposomal all-trans retinoic acid. Blood Am. Soc. Hematol. 2001, 97, 73–80. [Google Scholar]
- Tsimberidou, A.-M.; Tirado-Gomez, M.; Andreeff, M.; O’Brien, S.; Kantarjian, H.; Keating, M.; Lopez-Berestein, G.; Estey, E. Single-agent liposomal all-trans retinoic acid can cure some patients with untreated acute promyelocytic leukemia: An update of The University of Texas MD Anderson Cancer Center Series. Leuk. Lymphoma 2006, 47, 1062–1068. [Google Scholar] [CrossRef]
- Tallman, M.S.; Nabhan, C.; Feusner, J.H.; Rowe, J.M. Acute promyelocytic leukemia: Evolving therapeutic strategies. BloodJ. Am. Soc. Hematol. 2002, 99, 759–767. [Google Scholar] [CrossRef]
- Estey, E.H.; Giles, F.J.; Kantarjian, H.; O’Brien, S.; Cortes, J.; Freireich, E.J.; Lopez-Berestein, G.; Keating, M. Molecular remissions induced by liposomal-encapsulated all-trans retinoic acid in newly diagnosed acute promyelocytic leukemia. BloodJ. Am. Soc. Hematol. 1999, 94, 2230–2235. [Google Scholar] [CrossRef]
- Grignani, F.; Fagioli, M.; Alcalay, M.; Longo, L.; Pandolfi, P.P.; Donti, E.; Biondi, A.; Lo Coco, F.; Grignani, F.; Pelicci, P.G. Acute promyelocytic leukemia: From genetics to treatment. 1994, 83, 10–25. [Google Scholar]
- Goldberg, J.S.; Vargas, M.; Rosmarin, A.S.; Milowsky, M.I.; Papanicoloau, N.; Gudas, L.J.; Shelton, G.; Feit, K.; Petrylak, D.; Nanus, D.M. Phase I trial of interferon α2b and liposome-encapsulated all-trans retinoic acid in the treatment of patients with advanced renal cell carcinoma. Cancer 2002, 95, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Boorjian, S.A.; Milowsky, M.I.; Kaplan, J.; Albert, M.; Cobham, M.V.; Coll, D.M.; Mongan, N.P.; Shelton, G.; Petrylak, D.; Gudas, L.J. Phase 1/2 clinical trial of interferon α2b and weekly liposome-encapsulated all-trans retinoic acid in patients with advanced renal cell carcinoma. J. Immunother. 2007, 30, 655–662. [Google Scholar] [CrossRef]
- Bernstein, Z.P.; Rios, A.; Scadden, D.; Groopman, J.; Northfelt, D.; Lang, W.; Fischl, M.; Cohen, P.; Bock, A.; Gill, P. A MULTICENTER, PHASE II/III STUDY of ATRAGEN™(Tretinoin Liposomal) in PATIENTS with AIDS-ASSOCIATED KAPOSI’S SARCOMA. JAIDS J. Acquir. Immune Defic. Syndr. 1998, 17, A24. [Google Scholar] [CrossRef]
- Cristiano, M.C.; Cosco, D.; Celia, C.; Tudose, A.; Mare, R.; Paolino, D.; Fresta, M. Anticancer activity of all-trans retinoic acid-loaded liposomes on human thyroid carcinoma cells. Colloids Surf. B: Biointerfaces 2017, 150, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Silva, E.L.; Lima, F.A.; Carneiro, G.; Ramos, J.P.; Gomes, D.A.; de Souza-Fagundes, E.M.; Miranda Ferreira, L.A. Improved in vitro antileukemic activity of all-trans retinoic acid loaded in cholesteryl butyrate solid lipid nanoparticles. J. Nanosci. Nanotechnol. 2016, 16, 1291–1300. [Google Scholar] [CrossRef]
- Li, C.; Han, X. Co-delivery of Dacarbazine and All-Trans Retinoic Acid (ATRA) Using Lipid Nanoformulations for Synergistic Antitumor Efficacy Against Malignant Melanoma. Nanoscale Res. Lett. 2020, 15, 1–10. [Google Scholar] [CrossRef]
- Zhu, Y.-H.; Ye, N.; Tang, X.-F.; Khan, M.I.; Liu, H.-L.; Shi, N.; Hang, L.-F. Synergistic Effect of Retinoic Acid Polymeric Micelles and Prodrug for the Pharmacodynamic Evaluation of Tumor Suppression. Front. Pharmacol. 2019, 10, 447. [Google Scholar] [CrossRef]
- Kong, M.; Tang, J.; Qiao, Q.; Wu, T.; Qi, Y.; Tan, S.; Gao, X.; Zhang, Z. Biodegradable hollow mesoporous silica nanoparticles for regulating tumor microenvironment and enhancing antitumor efficiency. Theranostics 2017, 7, 3276. [Google Scholar] [CrossRef]
- Wang, Z.; Du, Y.; He, W.; Zhou, W.; Xia, Q.; Li, X. Doxorubicin-Loaded All-Trans Retinoic Acid Dimer Phospholipid Liposomes as Co-Delivery System to Reverse Drug Resistance in Breast Cancer. Nanosci. Nanotechnol. Lett. 2019, 11, 749–759. [Google Scholar] [CrossRef]
- Sabzichi, M.; Mohammadian, J.; Ghorbani, M.; Saghaei, S.; Chavoshi, H.; Ramezani, F.; Hamishehkar, H. Fabrication of all-trans-retinoic acid-loaded biocompatible precirol: A strategy for escaping dose-dependent side effects of doxorubicin. Colloids Surf. B: Biointerfaces 2017, 159, 620–628. [Google Scholar] [CrossRef]
- Jang, E.-J.; Choi, W.R.; Kim, S.-Y.; Hong, S.-S.; Rhee, I.; Lee, S.-J.; Choi, S.W.; Choi, H.-G.; Lim, S.-J. 2-Hydroxyoleic acid-inserted liposomes as a multifunctional carrier of anticancer drugs. Drug Deliv. 2017, 24, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.H.; Shieh, W.J. Aqueous media for effective delivery of tretinoin. J. Incl. Phenom. Macrocycl. Chem. 2002, 44, 133–136. [Google Scholar] [CrossRef]
- Subongkot, T.; Ngawhirunpat, T. Development of a novel microemulsion for oral absorption enhancement of all-trans retinoic acid. Int. J. Nanomed. 2017, 12, 5585. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, F.; Hanieh, P.N.; Longhi, C.; Carradori, S.; Secci, D.; Zengin, G.; Ammendolia, M.G.; Mattia, E.; Del Favero, E.; Marianecci, C. Neem oil nanoemulsions: Characterisation and antioxidant activity. J. Enzym. Inhib. Med. Chem. 2017, 32, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Campos, J.; Severino, P.; Santini, A.; Silva, A.; Shegokar, R.; Souto, S.; Souto, E. Solid lipid nanoparticles (SLN): Prediction of toxicity, metabolism, fate and physicochemical properties. In Nanopharmaceuticals; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1–15. [Google Scholar]
- Hu, L.; Tang, X.; Cui, F. Solid lipid nanoparticles (SLNs) to improve oral bioavailability of poorly soluble drugs. J. Pharm. Pharmacol. 2004, 56, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Mehta, K.; Sadeghi, T.; McQueen, T.; Lopez-Berestein, G. Liposome encapsulation circumvents the hepatic clearance mechanisms of all-trans-retinoic acid. Leuk. Res. 1994, 18, 587–596. [Google Scholar] [CrossRef]
- Sharma, M. Transdermal and intravenous nano drug delivery systems: Present and future. In Applications of Targeted Nano Drugs and Delivery Systems; Elsevier: Amsterdam, The Netherlands, 2019; pp. 499–550. [Google Scholar]
- Allen, T.M.; Cullis, P.R. Drug delivery systems: Entering the mainstream. Science 2004, 303, 1818–1822. [Google Scholar] [CrossRef]
- Acharya, S.; Sahoo, S.K. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 170–183. [Google Scholar] [CrossRef]
- Kong, N.; Deng, M.; Sun, X.-N.; Chen, Y.-D.; Sui, X.-B. Polydopamine-functionalized CA-(PCL-ran-PLA) nanoparticles for target delivery of docetaxel and chemo-photothermal therapy of breast cancer. Front. Pharmacol. 2018, 9, 125. [Google Scholar] [CrossRef]
- Yan, S.; Zhang, H.; Piao, J.; Chen, Y.; Gao, S.; Lu, C.; Niu, L.; Xia, Y.; Hu, Y.; Ji, R. Studies on the preparation, characterization and intracellular kinetics of JD27-loaded human serum albumin nanoparticles. Procedia Eng. 2015, 102, 590–601. [Google Scholar] [CrossRef]
- Foote, M. Using nanotechnology to improve the characteristics of antineoplastic drugs: Improved characteristics of nab-paclitaxel compared with solvent-based paclitaxel. Biotechnol. Annu. Rev. 2007, 13, 345–357. [Google Scholar] [PubMed]
- Huang, H.; Shi, H.; Liu, J.; Min, Y.; Wang, Y.; Wang, A.Z.; Wang, J.; Liu, Y. Co-delivery of all-trans-retinoic acid enhances the anti-metastasis effect of albumin-bound paclitaxel nanoparticles. Chem. Commun. 2017, 53, 212–215. [Google Scholar] [CrossRef]
- Kobayashi, H.; Turkbey, B.; Watanabe, R.; Choyke, P.L. Cancer drug delivery: Considerations in the rational design of nanosized bioconjugates. Bioconjugate Chem. 2014, 25, 2093–2100. [Google Scholar] [CrossRef]
- Mu, L.; Feng, S. A novel controlled release formulation for the anticancer drug paclitaxel (Taxol®): PLGA nanoparticles containing vitamin E TPGS. J. Control. Release 2003, 86, 33–48. [Google Scholar] [CrossRef]
- Pisano, C.; Cecere, S.C.; Di Napoli, M.; Cavaliere, C.; Tambaro, R.; Facchini, G.; Scaffa, C.; Losito, S.; Pizzolorusso, A.; Pignata, S. Clinical trials with pegylated liposomal Doxorubicin in the treatment of ovarian cancer. J. Drug Deliv. 2013, 2013, 898146. [Google Scholar] [CrossRef]
- Li, S.; Dong, S.; Xu, W.; Jiang, Y.; Li, Z. Polymer Nanoformulation of Sorafenib and All-Trans Retinoic Acid for Synergistic Inhibition of Thyroid Cancer. Front. Pharmacol. 2020, 10, 1676. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Xu, W.; Wei, W.; Wen, D.; Lu, Z.; Yang, S.; Chen, T.; Wang, Y.; Wang, Y.; Ji, Q. Clinicopathological and survival outcomes of well-differentiated thyroid carcinoma undergoing dedifferentiation: A retrospective study from FUSCC. Int. J. Endocrinol. 2018, 2018, 1–11. [Google Scholar] [CrossRef]
- Li, T.; Zhang, Y.; Meng, Y.-P.; Bo, L.-S.; Ke, W.-B. MiR-542-3p appended sorafenib/all-trans retinoic acid (ATRA)-loaded lipid nanoparticles to enhance the anticancer efficacy in gastric cancers. Pharm. Res. 2017, 34, 2710–2719. [Google Scholar] [CrossRef]
- Marcu, L.G.; Marcu, D. In silico modelling of a cancer stem cell-targeting agent and its effects on tumour control during radiotherapy. Sci. Rep. 2016, 6, 32332. [Google Scholar] [CrossRef]
- Gupta, P.B.; Fillmore, C.M.; Jiang, G.; Shapira, S.D.; Tao, K.; Kuperwasser, C.; Lander, E.S. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 2011, 146, 633–644. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.-M.; Du, J.-Z.; Yao, Y.-D.; Mao, C.-Q.; Dou, S.; Huang, S.-Y.; Zhang, P.-Z.; Leong, K.W.; Song, E.-W.; Wang, J. Simultaneous delivery of siRNA and paclitaxel via a “two-in-one” micelleplex promotes synergistic tumor suppression. Acs. Nano. 2011, 5, 1483–1494. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Lv, P.-P.; Chen, X.-M.; Yue, Z.-G.; Fu, Q.; Liu, S.-Y.; Yue, H.; Ma, G.-H. Codelivery of mTERT siRNA and paclitaxel by chitosan-based nanoparticles promoted synergistic tumor suppression. Biomaterials 2013, 34, 3912–3923. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Liu, Y.; Li, S.-Y.; Shen, S.; Du, X.-J.; Xu, C.-F.; Cao, Z.-T.; Bao, Y.; Zhu, Y.-H.; Li, Y.-P. Co-delivery of all-trans-retinoic acid and doxorubicin for cancer therapy with synergistic inhibition of cancer stem cells. Biomaterials 2015, 37, 405–414. [Google Scholar] [CrossRef]
- Fang, Y.; Xue, J.; Gao, S.; Lu, A.; Yang, D.; Jiang, H.; He, Y.; Shi, K. Cleavable PEGylation: A strategy for overcoming the “PEG dilemma” in efficient drug delivery. Drug Deliv. 2017, 24, 22–32. [Google Scholar] [CrossRef]
- Hatakeyama, H.; Akita, H.; Harashima, H. Polyethyleneglycol?: A classical but innovative material the polyethyleneglycol dilemma?: Advantage and disad-vantage of PEGylation of liposomes for systemic genes and nucleic acids delivery to tumors. Biol. Pharm. Bull. 2013, 36, 892–899. [Google Scholar] [CrossRef]
- Sanchez, L.; Yi, Y.; Yu, Y. Effect of partial PEGylation on particle uptake by macrophages. Nanoscale 2017, 9, 288–297. [Google Scholar] [CrossRef]
- Tagalakis, A.D.; Kenny, G.D.; Bienemann, A.S.; McCarthy, D.; Munye, M.M.; Taylor, H.; Wyatt, M.J.; Lythgoe, M.F.; White, E.A.; Hart, S.L. PEGylation improves the receptor-mediated transfection efficiency of peptide-targeted, self-assembling, anionic nanocomplexes. J. Control. Release 2014, 174, 177–187. [Google Scholar] [CrossRef]
- Smith, S.A.; Selby, L.I.; Johnston, A.P.; Such, G.K. The endosomal escape of nanoparticles: Toward more efficient cellular delivery. Bioconjugate Chem. 2018, 30, 263–272. [Google Scholar] [CrossRef]
- d’Avanzo, N.; Celia, C.; Barone, A.; Carafa, M.; Di Marzio, L.; Santos, H.A.; Fresta, M. Immunogenicity of Polyethylene Glycol Based Nanomedicines: Mechanisms, Clinical Implications and Systematic Approach. Adv. Ther. 2020, 3, 1900170. [Google Scholar] [CrossRef]
- Gaber, M.; Elhasany, K.A.; Sabra, S.; Helmy, M.W.; Fang, J.-Y.; Khattab, S.N.; Bekhit, A.A.; Teleb, M.; Elkodairy, K.A.; Elzoghby, A.O. Co-Administration of Tretinoin Enhances the Anti-Cancer Efficacy of Etoposide via Tumor-Targeted Green Nano-Micelles. Colloids Surfaces B: Biointerfaces 2020, 192, 110997. [Google Scholar] [CrossRef] [PubMed]
- Azzopardi, E.A.; Ferguson, E.L.; Thomas, D.W. The enhanced permeability retention effect: A new paradigm for drug targeting in infection. J. Antimicrob. Chemother. 2013, 68, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H. Tumor-selective delivery of macromolecular drugs via the EPR effect: Background and future prospects. Bioconjugate Chem. 2010, 21, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xiong, H.; Dahmani, F.Z.; Sun, L.; Li, Y.; Yao, L.; Zhou, J.; Yao, J. Combination chemotherapy of doxorubicin, all-trans retinoic acid and low molecular weight heparin based on self-assembled multi-functional polymeric nanoparticles. Nanotechnology 2015, 26, 145101. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv. Drug Deliv. Rev. 2015, 91, 3–6. [Google Scholar] [CrossRef]
- Sabra, S.; Abdelmoneem, M.; Abdelwakil, M.; Mabrouk, M.T.; Anwar, D.; Mohamed, R.; Khattab, S.; Bekhit, A.; Elkhodairy, K.; Freag, M. Self-assembled nanocarriers based on amphiphilic natural polymers for anti-cancer drug delivery applications. Curr. Pharm. Des. 2017, 23, 5213–5229. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Z.; Yang, S.; Chen, H.; Wang, D.; Li, J. All-trans retinoic acid-encapsulated, CD20 antibody-conjugated poly (lactic-co-glycolic acid) nanoparticles effectively target and eliminate melanoma-initiating cells in vitro. Oncotargets Ther. 2018, 11, 6177. [Google Scholar] [CrossRef]
- Gui, K.; Zhang, X.; Chen, F.; Ge, Z.; Zhang, S.; Qi, X.; Sun, J.; Yu, Z. Lipid-polymer nanoparticles with CD133 aptamers for targeted delivery of all-trans retinoic acid to osteosarcoma initiating cells. Biomed. Pharmacother. 2019, 111, 751–764. [Google Scholar] [CrossRef]
- Chen, H.; Lin, J.; Shan, Y.; Zhengmao, L. The promotion of nanoparticle delivery to two populations of gastric cancer stem cells by CD133 and CD44 antibodies. Biomed. Pharmacother. 2019, 115, 108857. [Google Scholar] [CrossRef]
- Xia, P.; Xu, X.Y. DKK3 attenuates the cytotoxic effect of natural killer cells on CD133+ gastric cancer cells. Mol. Carcinog. 2017, 56, 1712–1721. [Google Scholar] [CrossRef]
- Takaishi, S.; Okumura, T.; Tu, S.; Wang, S.S.; Shibata, W.; Vigneshwaran, R.; Gordon, S.A.; Shimada, Y.; Wang, T.C. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 2009, 27, 1006–1020. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shi, S.; Ming, Y.; Wang, L.; Li, C.; Luo, M.; Li, Z.; Li, B.; Chen, J. Specific cancer stem cell-therapy by albumin nanoparticles functionalized with CD44-mediated targeting. J. Nanobiotechnol. 2018, 16, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Li, Y.; Sun, X.; Dahmani, F.Z.; Liu, H.; Zhou, J. Nanoparticle delivery and combination therapy of gambogic acid and all-trans retinoic acid. Int. J. Nanomed. 2014, 9, 3313. [Google Scholar] [CrossRef]
- Shi, S.; Zhou, M.; Li, X.; Hu, M.; Li, C.; Li, M.; Sheng, F.; Li, Z.; Wu, G.; Luo, M. Synergistic active targeting of dually integrin αvβ3/CD44-targeted nanoparticles to B16F10 tumors located at different sites of mouse bodies. J. Control. Release 2016, 235, 1–13. [Google Scholar] [CrossRef]
- Qi, X.; Fan, Y.; He, H.; Wu, Z. Hyaluronic acid-grafted polyamidoamine dendrimers enable long circulation and active tumor targeting simultaneously. Carbohydr. Polym. 2015, 126, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Roesch, A. Melanoma stem cells. JDDG: J. Dtsch. Dermatol. Ges. 2015, 13, 118–124. [Google Scholar] [CrossRef][Green Version]
- Stauffer, R.G.; Mohammad, M.; Singh, A.T. Novel nanoscale delivery particles encapsulated with anticancer drugs, all-trans retinoic acid or curcumin, enhance apoptosis in lymphoma cells predominantly expressing CD20 antigen. Anticancer Res. 2015, 35, 6425–6429. [Google Scholar]
- Solimando, A.G.; Ribatti, D.; Vacca, A.; Einsele, H. Targeting B-cell non Hodgkin lymphoma: New and old tricks. Leuk. Res. 2016, 42, 93–104. [Google Scholar] [CrossRef]
- Narvekar, M.; Xue, H.Y.; Tran, N.T.; Mikhael, M.; Wong, H.L. A new nanostructured carrier design including oil to enhance the pharmaceutical properties of retinoid therapy and its therapeutic effects on chemo-resistant ovarian cancer. Eur. J. Pharm. Biopharm. 2014, 88, 226–237. [Google Scholar] [CrossRef]
- Gao, J.; Chen, H.; Song, H.; Su, X.; Niu, F.; Li, W.; Li, B.; Dai, J.; Wang, H.; Guo, Y. Antibody-targeted immunoliposomes for cancer treatment. Mini Rev. Med. Chem. 2013, 13, 2026–2035. [Google Scholar] [CrossRef]
- Krasnici, S.; Werner, A.; Eichhorn, M.E.; Schmitt-Sody, M.; Pahernik, S.A.; Sauer, B.; Schulze, B.; Teifel, M.; Michaelis, U.; Naujoks, K. Effect of the surface charge of liposomes on their uptake by angiogenic tumor vessels. Int. J. Cancer 2003, 105, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, S.; Suzuki, S.; Yamashita, F.; Hashida, M. Induction of apoptosis in A549 human lung cancer cells by all-trans retinoic acid incorporated in DOTAP/cholesterol liposomes. J. Control. Release 2006, 110, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Kawakami, S.; Chansri, N.; Yamashita, F.; Hashida, M. Inhibition of pulmonary metastasis in mice by all-trans retinoic acid incorporated in cationic liposomes. J. Control. Release 2006, 116, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Grace, V.B.; Viswanathan, S. Pharmacokinetics and therapeutic efficiency of a novel cationic liposome nano-formulated all trans retinoic acid in lung cancer mice model. J. Drug Deliv. Sci. Technol. 2017, 39, 223–236. [Google Scholar] [CrossRef]
- Magesh, V.; Singh, J.P.V.; Selvendiran, K.; Ekambaram, G.; Sakthisekaran, D. Antitumour activity of crocetin in accordance to tumor incidence, antioxidant status, drug metabolizing enzymes and histopathological studies. Mol. Cell. Biochem. 2006, 287, 127–135. [Google Scholar] [CrossRef]
- Rahman, S.A.; Abdelmalak, N.S.; Badawi, A.; Elbayoumy, T.; Sabry, N.; El Ramly, A. Tretinoin-loaded liposomal formulations: From lab to comparative clinical study in acne patients. Drug Deliv. 2016, 23, 1184–1193. [Google Scholar] [CrossRef]
- Sahay, G.; Querbes, W.; Alabi, C.; Eltoukhy, A.; Sarkar, S.; Zurenko, C.; Karagiannis, E.; Love, K.; Chen, D.; Zoncu, R. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat. Biotechnol. 2013, 31, 653–658. [Google Scholar] [CrossRef]
- Shapero, K.; Fenaroli, F.; Lynch, I.; Cottell, D.C.; Salvati, A.; Dawson, K.A. Time and space resolved uptake study of silica nanoparticles by human cells. Mol. Biosyst. 2011, 7, 371–378. [Google Scholar] [CrossRef]
- Vermeulen, L.M.; De Smedt, S.C.; Remaut, K.; Braeckmans, K. The proton sponge hypothesis: Fable or fact? Eur. J. Pharm. Biopharm. 2018, 129, 184–190. [Google Scholar] [CrossRef]
- Mu, L.-M.; Liu, L.; Liu, R.; Duan, J.-L.; Ma, S.; Li, X.-Q.; Cui, Y.-N.; Su, Z.-B.; Zhang, X.; Hu, J.-X. Development of functional dendrisomes based on a single molecule of polyesterbenzylether dendrimer and their application in cancer stem cell therapy. Npg Asia Mater. 2019, 11, 1–16. [Google Scholar] [CrossRef]
- Mutalabisin, M.F.; Chatterjee, B.; Jaffri, J.M. pH Responsive Polymers in Drug Delivery. Res. J. Pharm. Technol. 2018, 11, 5115–5122. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhang, L.; Yang, T.; Wu, H. Stimuli-responsive polymeric micelles for drug delivery and cancer therapy. Int. J. Nanomed. 2018, 13, 2921. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhang, J.; Hao, W.; Wang, T.; Liu, J.; Xie, Y.; Xu, S.; Liu, H. Copolymer micelles function as pH-responsive nanocarriers to enhance the cytotoxicity of a HER2 aptamer in HER2-positive breast cancer cells. Int. J. Nanomed. 2018, 13, 537. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Lv, X.; Liu, C.; Qi, L.; Song, X.; Yu, A. Enhancement of all-trans retinoic acid-induced differentiation by pH-sensitive nanoparticles for solid tumor cells. Macromol. Biosci. 2014, 14, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Peng, L.; Chu, J.; Zhang, M.; Sun, L.; Zhong, B.; Wu, Q. pH and redox dual-responsive copolymer micelles with surface charge reversal for co-delivery of all-trans-retinoic acid and paclitaxel for cancer combination chemotherapy. Int. J. Nanomed. 2018, 13, 6499. [Google Scholar] [CrossRef] [PubMed]
- Taghizadeh, B.; Taranejoo, S.; Monemian, S.A.; Salehi Moghaddam, Z.; Daliri, K.; Derakhshankhah, H.; Derakhshani, Z. Classification of stimuli–responsive polymers as anticancer drug delivery systems. Drug Deliv. 2015, 22, 145–155. [Google Scholar] [CrossRef]
- Sun, J.; Liu, Y.; Chen, Y.; Zhao, W.; Zhai, Q.; Rathod, S.; Huang, Y.; Tang, S.; Kwon, Y.T.; Fernandez, C. Doxorubicin delivered by a redox-responsive dasatinib-containing polymeric prodrug carrier for combination therapy. J. Control. Release 2017, 258, 43–55. [Google Scholar] [CrossRef]
- Han, L.; Hu, L.; Liu, F.; Wang, X.; Huang, X.; Liu, B.; Feng, F.; Liu, W.; Qu, W. Redox-sensitive micelles for targeted intracellular delivery and combination chemotherapy of paclitaxel and all-trans-retinoid acid. Asian J. Pharm. Sci. 2019, 14, 531–542. [Google Scholar] [CrossRef]
- Karimi, N.; Mansouri, K.; Soleiman-Beigi, M.; Fattahi, A. All-Trans Retinoic Acid Grafted Poly Beta-Amino Ester Nanoparticles: A Novel Anti-angiogenic Drug Delivery System. Adv. Pharm. Bull. 2020, 10, 221A–232A. [Google Scholar] [CrossRef]
- Han, X.; Li, Y.; Xu, Y.; Zhao, X.; Zhang, Y.; Yang, X.; Wang, Y.; Zhao, R.; Anderson, G.J.; Zhao, Y. Reversal of pancreatic desmoplasia by re-educating stellate cells with a tumour microenvironment-activated nanosystem. Nat. Commun. 2018, 9, 1–18. [Google Scholar] [CrossRef]
- Ji, T.; Lang, J.; Wang, J.; Cai, R.; Zhang, Y.; Qi, F.; Zhang, L.; Zhao, X.; Wu, W.; Hao, J. Designing liposomes to suppress extracellular matrix expression to enhance drug penetration and pancreatic tumor therapy. Acs. Nano. 2017, 11, 8668–8678. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Lee, J.S.; Zhang, M. Magnetic nanoparticles in MR imaging and drug delivery. Adv. Drug Deliv. Rev. 2008, 60, 1252–1265. [Google Scholar] [CrossRef] [PubMed]
- Yalçın, S.; Erkan, M.; Ünsoy, G.; Parsian, M.; Kleeff, J.; Gündüz, U. Effect of gemcitabine and retinoic acid loaded PAMAM dendrimer-coated magnetic nanoparticles on pancreatic cancer and stellate cell lines. Biomed. Pharmacother. 2014, 68, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Rasheed, T.; Nabeel, F.; Hayat, U.; Bilal, M.; Iqbal, H. Endogenous and exogenous stimuli-responsive drug delivery systems for programmed site-specific release. Molecules 2019, 24, 1117. [Google Scholar] [CrossRef]
- Lu, Z.; Li, Y.; Shi, Y.; Li, Y.; Xiao, Z.; Zhang, X. Traceable nanoparticles with spatiotemporally controlled release ability for synergistic glioblastoma multiforme treatment. Adv. Funct. Mater. 2017, 27, 1703967. [Google Scholar] [CrossRef]
- Li, H.; Zeng, D.; Wang, Z.; Fang, L.; Li, F.; Wang, Z. Ultrasound-enhanced delivery of doxorubicin/all-trans retinoic acid-loaded nanodiamonds into tumors. Nanomedicine 2018, 13, 981–996. [Google Scholar] [CrossRef]
- Wang, Q.; Manmi, K.; Liu, K.-K. Cell mechanics in biomedical cavitation. Interface Focus 2015, 5, 20150018. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, P.; Liu, P.; Zhao, Y.; Gao, S.; Tan, K.; Liu, Z. Endothelial adhesion of targeted microbubbles in both small and great vessels using ultrasound radiation force. Mol. Imaging 2012, 11, 58–66. [Google Scholar] [CrossRef]
- Whitlow, J.; Pacelli, S.; Paul, A. Multifunctional nanodiamonds in regenerative medicine: Recent advances and future directions. J. Control. Release 2017, 261, 62–86. [Google Scholar] [CrossRef]
- Morales-Cruz, M.; Delgado, Y.; Castillo, B.; Figueroa, C.M.; Molina, A.M.; Torres, A.; Milián, M.; Griebenow, K. Smart Targeting To Improve Cancer Therapeutics. Drug Des. Dev. Ther. 2019, 13, 3753. [Google Scholar] [CrossRef]
- Jiao, J.; Wu, H.; Chen, F.; Chen, R.; Sun, B.; Wang, M. Delivery of coumarin-containing all-trans retinoic acid derivatives via targeted nanoparticles encapsulating indocyanine green for chemo/photothermal/photodynamic therapy of breast cancer. New J. Chem. 2018, 42, 8805–8814. [Google Scholar] [CrossRef]
- Cai, Y.; Wang, H.-E.; Zhao, X.; Huang, F.; Wang, C.; Deng, Z.; Li, Y.; Cao, G.; Su, B.-L. Walnut-like porous core/shell TiO2 with hybridized phases enabling fast and stable lithium storage. Acs Appl. Mater. Interfaces 2017, 9, 10652–10663. [Google Scholar] [CrossRef] [PubMed]
- Wauthoz, N.; Deleuze, P.; Saumet, A.; Duret, C.; Kiss, R.; Amighi, K. Temozolomide-based dry powder formulations for lung tumor-related inhalation treatment. Pharm. Res. 2011, 28, 762–775. [Google Scholar] [CrossRef]
- Brajtburg, J.; Powderly, W.; Kobayashi, G.; Medoff, G. Amphotericin B: Delivery systems. Antimicrob. Agents Chemother. 1990, 34, 381. [Google Scholar] [CrossRef][Green Version]
- Marianecci, C.; Petralito, S.; Rinaldi, F.; Hanieh, P.N.; Carafa, M. Some recent advances on liposomal and niosomal vesicular carriers. J. Drug Deliv. Sci. Technol. 2016, 32, 256–269. [Google Scholar] [CrossRef]
- Marianecci, C.; Di Marzio, L.; Rinaldi, F.; Celia, C.; Paolino, D.; Alhaique, F.; Esposito, S.; Carafa, M. Niosomes from 80s to present: The state of the art. Adv. Colloid Interface Sci. 2014, 205, 187–206. [Google Scholar] [CrossRef] [PubMed]
- Desai, T.R.; Finlay, W.H. Nebulization of niosomal all-trans-retinoic acid: An inexpensive alternative to conventional liposomes. Int. J. Pharm. 2002, 241, 311–317. [Google Scholar] [CrossRef]
- Kamel, N.M.; Helmy, M.W.; Abdelfattah, E.-Z.; Khattab, S.N.; Ragab, D.; Samaha, M.W.; Fang, J.-Y.; Elzoghby, A.O. Inhalable dual-targeted hybrid lipid nanocore–protein shell composites for combined delivery of genistein and all-trans retinoic acid to lung cancer cells. Acs Biomater. Sci. Eng. 2019, 6, 71–87. [Google Scholar] [CrossRef]
- Karmakar, S.; Banik, N.L.; Ray, S.K. Combination of all-trans retinoic acid and paclitaxel-induced differentiation and apoptosis in human glioblastoma U87MG xenografts in nude mice. Cancer: Interdiscip. Int. J. Am. Cancer Soc. 2008, 112, 596–607. [Google Scholar] [CrossRef]
- Liang, C.; Yang, L.; Guo, S. All-trans retinoic acid inhibits migration, invasion and proliferation, and promotes apoptosis in glioma cells in vitro. Oncol. Lett. 2015, 9, 2833–2838. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Eisenberg, J.; Yang, J. Human blood—brain barrier insulin receptor. J. Neurochem. 1985, 44, 1771–1778. [Google Scholar] [CrossRef] [PubMed]
- Mirani, B.; Pagan, E.; Shojaei, S.; Duchscherer, J.; Toyota, B.D.; Ghavami, S.; Akbari, M. A 3D bioprinted hydrogel mesh loaded with all-trans retinoic acid for treatment of glioblastoma. Eur. J. Pharmacol. 2019, 854, 201–212. [Google Scholar] [CrossRef]
- Hu, J.; Liu, Y.-F.; Wu, C.-F.; Xu, F.; Shen, Z.-X.; Zhu, Y.-M.; Li, J.-M.; Tang, W.; Zhao, W.-L.; Wu, W. Long-term efficacy and safety of all-trans retinoic acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 2009, 106, 3342–3347. [Google Scholar] [CrossRef]
- Jones, T.; Zhang, B.; Major, S.; Webb, A. All-trans retinoic acid eluting poly (diol citrate) wafers for treatment of glioblastoma. J. Biomed. Mater. Res. Part. B: Appl. Biomater. 2020, 108, 619–628. [Google Scholar] [CrossRef]
- Potter, K.A.; Buck, A.C.; Self, W.K.; Capadona, J.R. Stab injury and device implantation within the brain results in inversely multiphasic neuroinflammatory and neurodegenerative responses. J. Neural Eng. 2012, 9, 046020. [Google Scholar] [CrossRef]




| Novelty | Number of the Patent | Priority Date | Publication Date | National | International |
|---|---|---|---|---|---|
| Nano-fibular nanoparticles polymer-ATRA conjugate for sustained dermal delivery | WO2016210087A1; US2018185513A1 | 23rd June 2015 | 29th December 2016 (WO2016210087A1); 5th July 2018 (US2018185513A1) | USA | PCT |
| ATRA-loaded liposomal aerosols for delivery to the lungs | US6334999B1 | 27th August 1999 | 1st January 2002 | USA | - |
| ATRA/TGFβ-loaded (PLGA) polymeric nanoparticles for the treatment of Type 1 Diabetes Mellitus | WO2015109245A1; US2016338984A1; US10105334B2 | 17th January 2014 | 23rd July 2015 (WO2015109245A1); 24th November 2016 (US2016338984A1); 23rd October 2018 (US10105334B2) | USA | PCT |
| ATRA quasicrystal-loaded liposomes for the treatment of solid tumors | CN109364027A | 12nd December 2018 | 22nd February 2019 | China | - |
| ATRA-loaded liposomes for the treatment of solid tumors | WO2018033118A1; CN107753427A; EP3501500A1 | 18th August 2016 | 22nd February 2018 (WO2018033118A1); 6th March 2018 (CN107753427A); 26th June 2019 (EP3501500A1) | China | PCT; European patent |
| ATRA/aPD-L1-loaded (PLGA-PEG) polymeric nanoparticles for the treatment of oral dysplasia and oral squamous carcinoma | CN110623942A | 30th September 2019 | 31st December 2019 | China | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giuli, M.V.; Hanieh, P.N.; Giuliani, E.; Rinaldi, F.; Marianecci, C.; Screpanti, I.; Checquolo, S.; Carafa, M. Current Trends in ATRA Delivery for Cancer Therapy. Pharmaceutics 2020, 12, 707. https://doi.org/10.3390/pharmaceutics12080707
Giuli MV, Hanieh PN, Giuliani E, Rinaldi F, Marianecci C, Screpanti I, Checquolo S, Carafa M. Current Trends in ATRA Delivery for Cancer Therapy. Pharmaceutics. 2020; 12(8):707. https://doi.org/10.3390/pharmaceutics12080707
Chicago/Turabian StyleGiuli, Maria Valeria, Patrizia Nadia Hanieh, Eugenia Giuliani, Federica Rinaldi, Carlotta Marianecci, Isabella Screpanti, Saula Checquolo, and Maria Carafa. 2020. "Current Trends in ATRA Delivery for Cancer Therapy" Pharmaceutics 12, no. 8: 707. https://doi.org/10.3390/pharmaceutics12080707
APA StyleGiuli, M. V., Hanieh, P. N., Giuliani, E., Rinaldi, F., Marianecci, C., Screpanti, I., Checquolo, S., & Carafa, M. (2020). Current Trends in ATRA Delivery for Cancer Therapy. Pharmaceutics, 12(8), 707. https://doi.org/10.3390/pharmaceutics12080707