1. Introduction
The human skin serves as a barrier between the body and the environment. Therefore, it is prone to microbial, thermal, mechanical, and chemical threats, which can cause acute or chronic wounds. Triterpenes have been shown to improve wound healing recovery by inducing cell migration, cell proliferation, and collagen deposition [
1]. Outer birch bark extracts have a high content of triterpenes, which are known for various pharmacological properties such as anti-inflammatory, antimicrobial, antiviral, anticancer activity, and wound healing effects [
2,
3,
4,
5,
6,
7,
8,
9,
10]. A well characterized commercially used triterpene dry extract from the outer bark of birch (TE) contains about 80% (
w/w) betulin as the main component and is obtained by accelerated solvent extraction with n-heptane [
11]. Other disclosed triterpenes of the dry extract include lupeol (LU), erythrodiol (ER), betulinic acid (BA), and oleanolic acid (OA) as shown in
Table 1 [
12]. However, the low solubility of these triterpenes in polar and nonpolar solvents may lead to a poor bioavailability, which might limit their therapeutic application [
8,
13].
A study conducted by Ebeling et al. showed the molecular mechanism of the effects of birch bark extract on wound healing properties in human primary keratinocytes and porcine ex vivo wound healing models. They showed that TE and betulin mainly accelerated reepithelialization in a porcine ex vivo wound healing model after wound treatment with TE-oleogel (10% TE, 90% sunflower oil). Beyond that, they found out that TE led to upregulation of proinflammatory mediators such as COX-2 and IL-6, which play a key role in wound healing und epidermal barrier repair [
6,
14]. Additionally, the effects of TE-based formulations have been already investigated in vivo on different types of wounds including dystrophic epidermolysis bullosa where treatments promote wound healing and was found out to be safe and well tolerated [
15,
16,
17]. It is good to note that TE together with sunflower oil (SO) supports wound healing better than in combination with other oils as examined by Steinbrenner et al. [
8]. At the moment, there are a few available topical formulations containing these triterpenes, including water-in-oil foams [
9], cosmetic water-in-oil creams, and an oleogel consisting of TE and sunflower oil that received European marketing authorization in January 2016 [
18].
The electrospinning technique is a simple and versatile process for preparing fibers having a diameter from few micrometers down to several nanometers [
19,
20]. The resulting nanofibers have special features, such as high surface area to volume ratio, and can form mats/fleeces with high porosity which makes them attractive materials for wound dressing [
21,
22]. Beyond that, electrospun fibrous scaffolds mimic the structure of the native extracellular matrix (ECM) and hence facilitate cell proliferation, improve gaseous exchange, removal of exudate, and act as a physical barrier against entry of microorganisms during wound healing and regeneration of damaged tissues [
21,
23,
24,
25,
26].
Polymeric nanofibers made from biodegradable and biocompatible synthetic or natural polymers have been utilized to develop drug delivery systems to treat various ailments. One of the potential areas to use them is medicated wound dressing [
21,
23]. PVA is a biocompatible and biodegradable hydrophilic polymer with good chemical and mechanical properties, and has been approved by the U. S. Food and Drug Administration (FDA) for different biomedical and pharmaceutical applications [
27,
28]. For instance, PVA has been used to create hydrogels for wound dressing [
29] or electrospun together with active substances, such as silver nanoparticles [
30], and curcumin [
31], to produce wound dressings. Several wound dressings in the market containing active agents such as hydrocolloids or silver dressings often used on wound healing have shown negative effects such as allergic contact dermatitis [
32], and use of dressings loaded with antibiotics has risk of developing antimicrobial resistance [
33]. Our interest in this study is to develop a dressing loaded with birch bark extract with a sustained drug release that is suitable for human use on wound healing with effective therapy even after long-term usage. To satisfy this requirement, we ensured that all the components used in this study are biocompatible and biodegradable when applied to the human body, such that an invasive and/or traumatic intervention for dressing removal, like in the case of deep wounds, is unnecessary. Furthermore, sustained drug release could lead to less frequent dressing changes, which are painful, especially in treatment of chronic wounds, and thereby resulting in better patient compliance. In addition to that, our concept allows one to avoid organic solvents and uses water as the only solvent throughout the whole processing of the wound dressing. This guarantees not only that the solvent is nontoxic and affordable, but also that it is eco-friendly and can thus be regarded as green electrospinning. However, encapsulation of birch bark dry extract into nanofibers may present a challenge. For instance, earlier studies showed that due to their unique structure, the particle sizes of the dry extract cannot be grinded by common techniques to reach sufficiently small particles even when high energy dispersion techniques, e.g., sonication or high-pressure homogenization, were used with aqueous and organic suspensions of the material [
34]. This was solved through processing of oil in water (O/W) colloidal dispersions with birch bark dry extract as the active substance, phospholipids as stabilizer, sunflower oil, and the water phase through a step-wise homogenization process. Hydrogenated soybean phosphatidylcholine (HSPC) types of phospholipids containing mainly esterified stearic and palmitic acid enhanced the producibility of colloidal dispersions with particle sizes < 1 µm and enabled miscibility and stabilization of the lipophilic components in the hydrophilic PVA matrix prior to electrospinning.
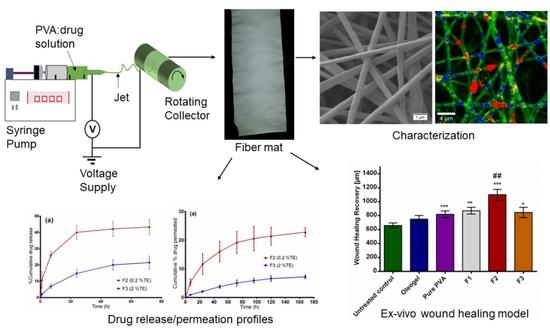
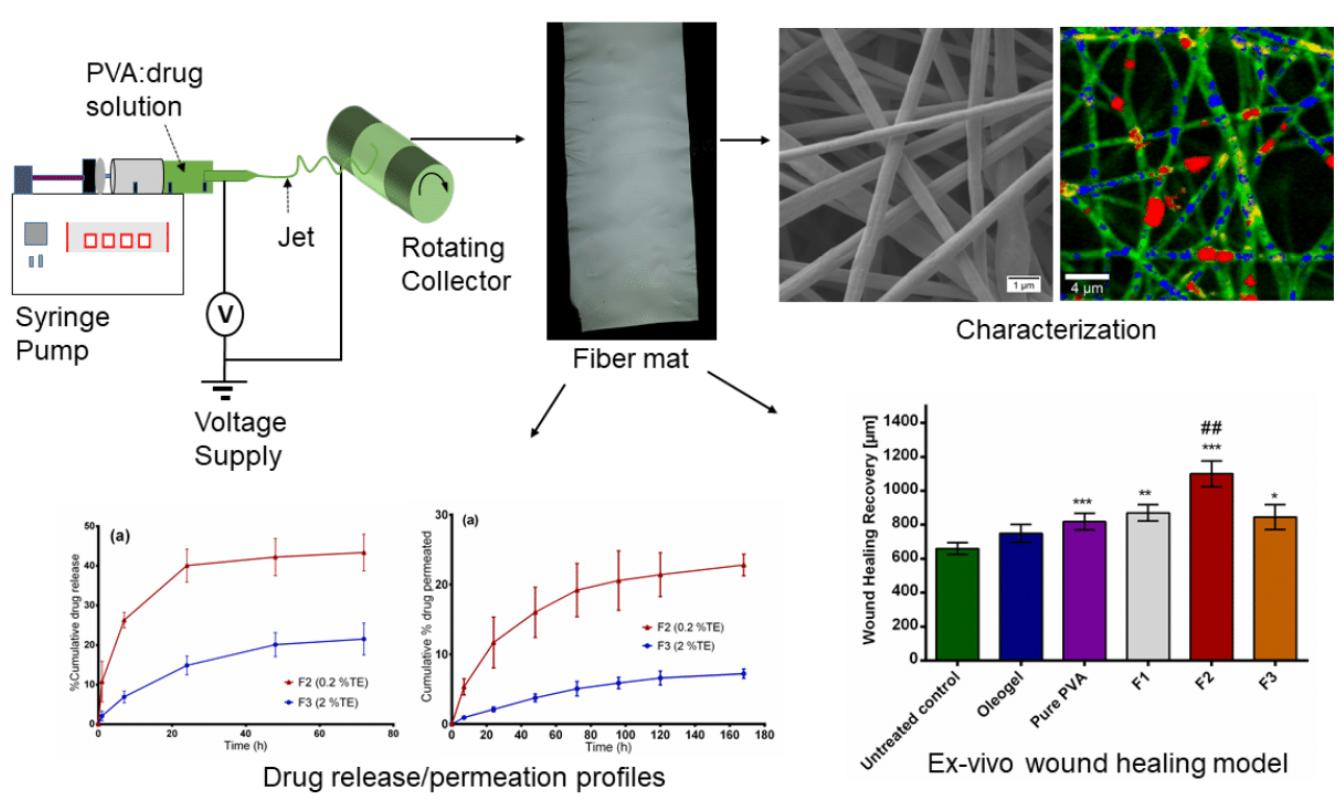
The purpose of this study was to (1) develop wound dressing materials with incorporated colloidal dispersions of TE and to (2) study their efficiency related to wound healing performance and (3) drug release. The surface properties of electrospun fiber mats were characterized by using SEM, thermal analysis with differential scanning calorimetry (DSC) and confocal Raman spectral imaging for visualization of all components within the fiber mats. We also conducted ex vivo permeation and in vitro release studies using Franz diffusion cell. The efficacy of the developed wound dressings was evaluated through an ex vivo wound healing assay. Here, low and highly TE-loaded dressings were tested and compared to an oleogel similar to the authorized product. To the best of our knowledge, there are no reports focusing on the development of this kind of wound dressing; therefore, this will be the first polymer-based wound dressing containing birch bark dry extract aimed for future potential use in wound therapy.
2. Materials and Methods
2.1. Materials
PVA with a molecular weight of 146–186 kDa was purchased from Sigma Aldrich (Steinheim, Germany), hydrogenated phospholipids from soybean lecithin (Phospholipon 90 H) were supplied by Lipoid GmbH (Ludwigshafen, Germany). SO was purchased from Caesar & Loretz GmbH (Hilden, Germany). Birch bark extract was obtained from Amryt AG (Niefern-Öschelbronn, Germany). Reverse osmosis water (ELGA Labwater, Celle, Germany) was used for the preparation of all solutions. Parafilm
® was from Bemis Company Inc., (Oshkosh, WI, USA). Whatman Nuclepore polycarbonate membrane filters were purchased from Sigma-Aldrich (Steinheim, Germany). Pig ears for permeation studies were obtained from the Department of Experimental Medicine at the University of Tuebingen [
35] and for wound healing studies from a slaughter house in Schleswig-Holstein after slaughtering the pigs for human consumption.
2.2. Preparing the Colloidal Dispersions
Three different dispersions were prepared with varying concentrations of TE as shown in
Table 2. Dispersions 1 and 2 (D1 and D2) were necessary to investigate the impact of high or low TE concentration on wound healing, whereas dispersion 3 (D3) was used as a placebo. It is important to mention that it was only practically possible to produce dispersions consisting of high TE (5%) loading by use of high concentrations of PL90H (8%). That is why 8% PL90H was used in preparation of dispersion consisting 5% TE. Dispersion containing 2.5% PL90H was the optimized colloidal dispersion from our previous study. Briefly, during preparation of each formulation, a predispersion was prepared by first dispersing Phospholipon 90H (PL90H) and TE in water at 70 °C for 30 min under magnetic stirring at 400 rpm using Heidolph MR 3001 K magnetic hotplate stirrer (Schwabach, Germany). Then, this mixture was homogenized for 5 min using a rotor-stator system (Ultra Turrax T25, IKA, Staufen, Germany), at 9500 rpm. Thereafter, the formed dispersion was added into the SOand homogenized for 3 min. Finally, the predispersion was homogenized using a high-pressure homogenizer (Emulsiflex C-3, Avestin, Mannheim, Germany) for eight cycles at a pressure of 100 MPa and their particle sizes measured with a Zetasizer Nano-ZS (Malvern Instruments, Herrenberg, Germany).
2.3. Preparation of Electrospinning Solutions
The 12 wt.% PVA solution was prepared by dissolving PVA in water at 90 °C under magnetic stirring for 5 h. The solution was allowed to cool to room temperature and used the following day. Subsequently, for colloidal dispersion-PVA electrospinning experiments, the above aqueous dispersions (D1–D3) were blended with a PVA polymer solution in the ratio of 60:40 using a magnetic stirrer for 2 h at 40 °C to form a homogeneous solution. Samples were then cooled down to room temperature prior to electrospinning.
2.4. Electrospinning of Nanofibers
The electrospinning setup purchased from Nanolab Instruments Sdn. Bhd., Subang Jaya, Malaysia consisted of a syringe pump holding a 5 mL plastic syringe with a blunt-end needle (18-gauge), a grounded rotating collector, and a high-voltage power supply. Electrospun fibers were prepared from solutions and dispersions as listed in
Table 3. Pure polymer solution serves as a reference mainly for the physicochemical characterization. F1 represents the placebo formulation. F2 and F3 were chosen to investigate the effect of high and low TE concentration. The syringe was filled with the solution and electrospun at a target collector distance of 10 cm and an applied voltage of 15 kV. Solutions were pumped through the syringe at a flow rate of 0.5 mL/h and the rotating speed of the collector was fixed at 1000 rpm. All electrospinning studies were carried out at ambient temperature (24 °C) and a relative humidity of 45%.
2.5. Characterization of Nanofiber Morphology
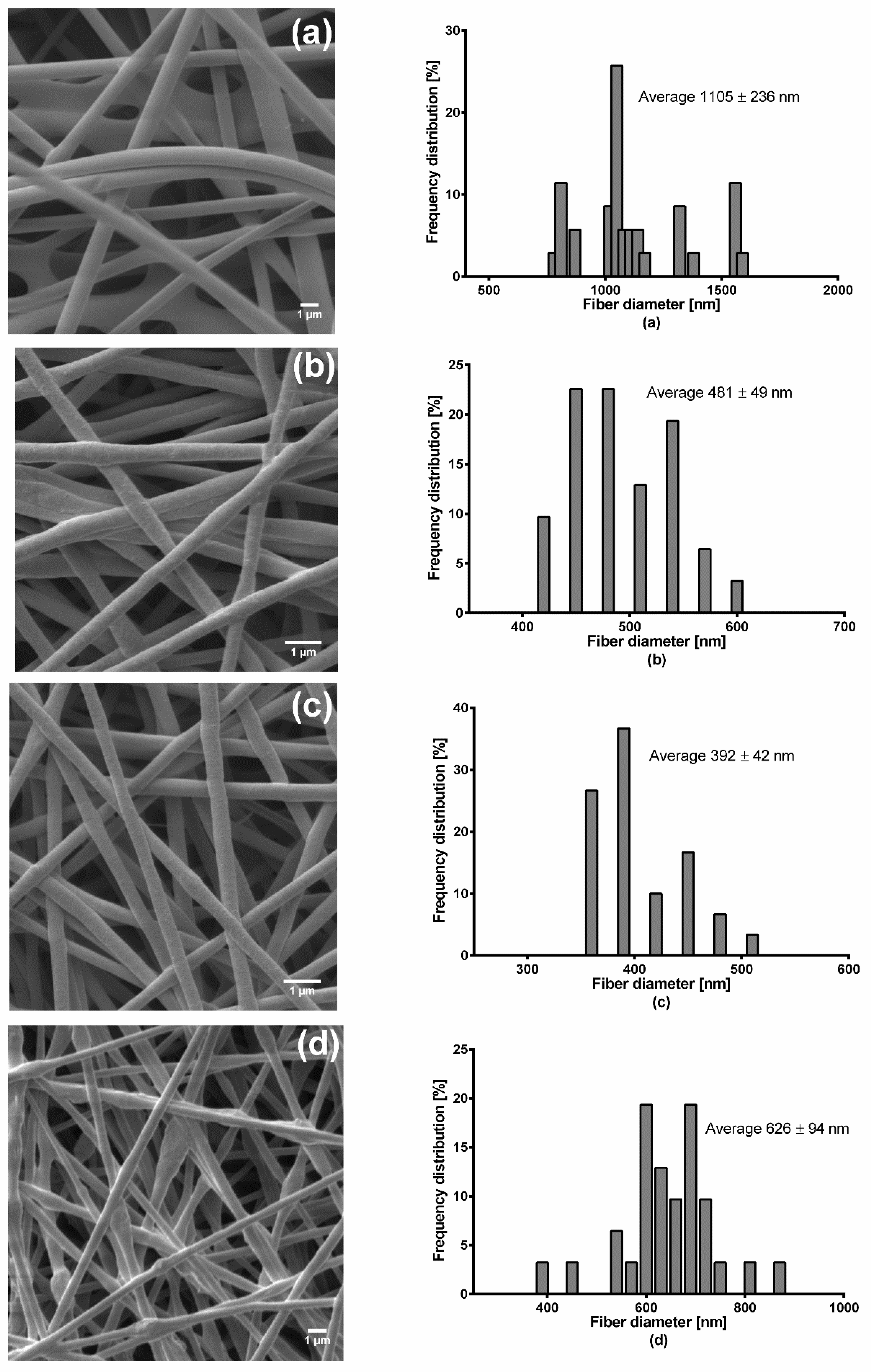
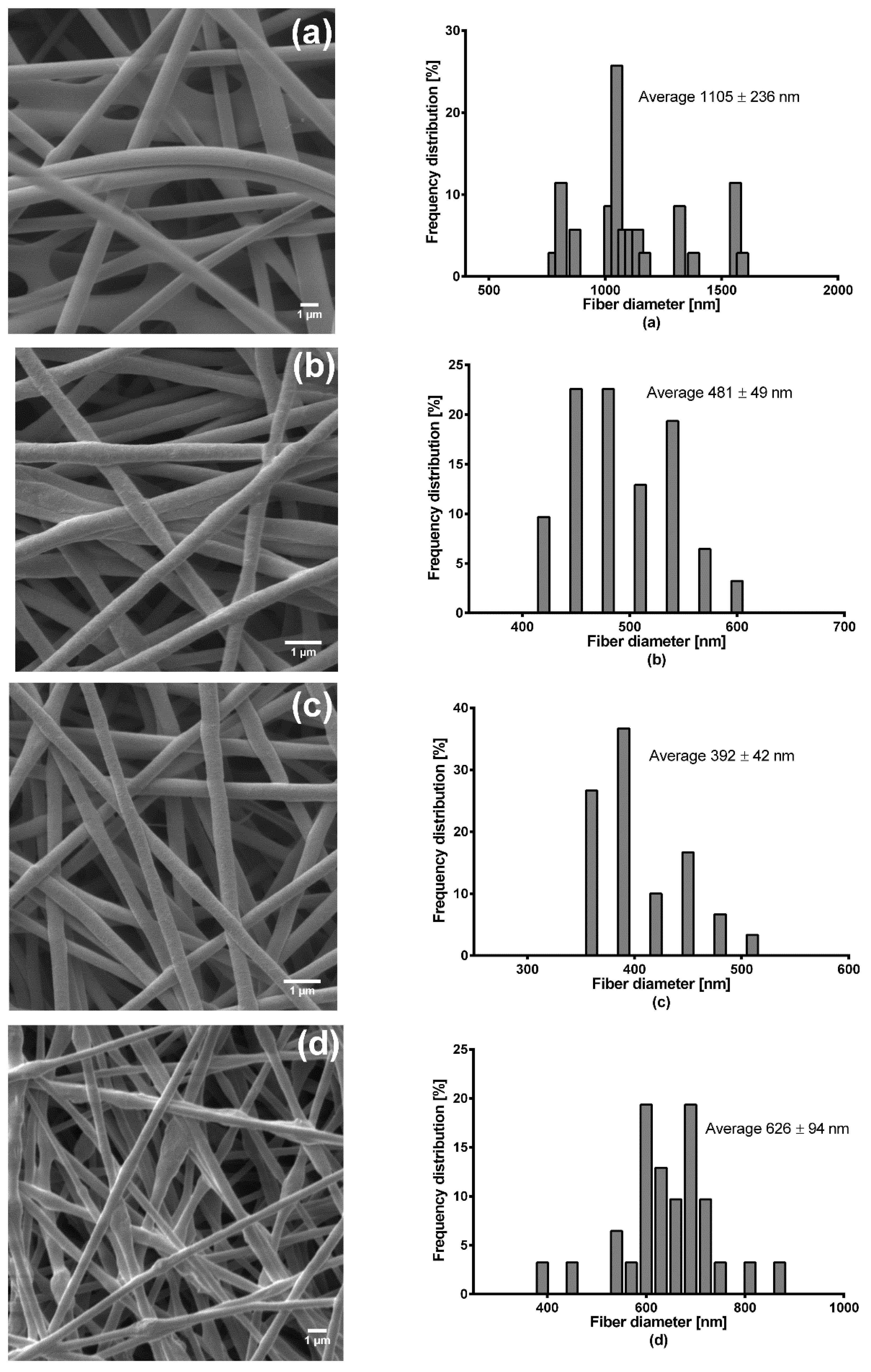
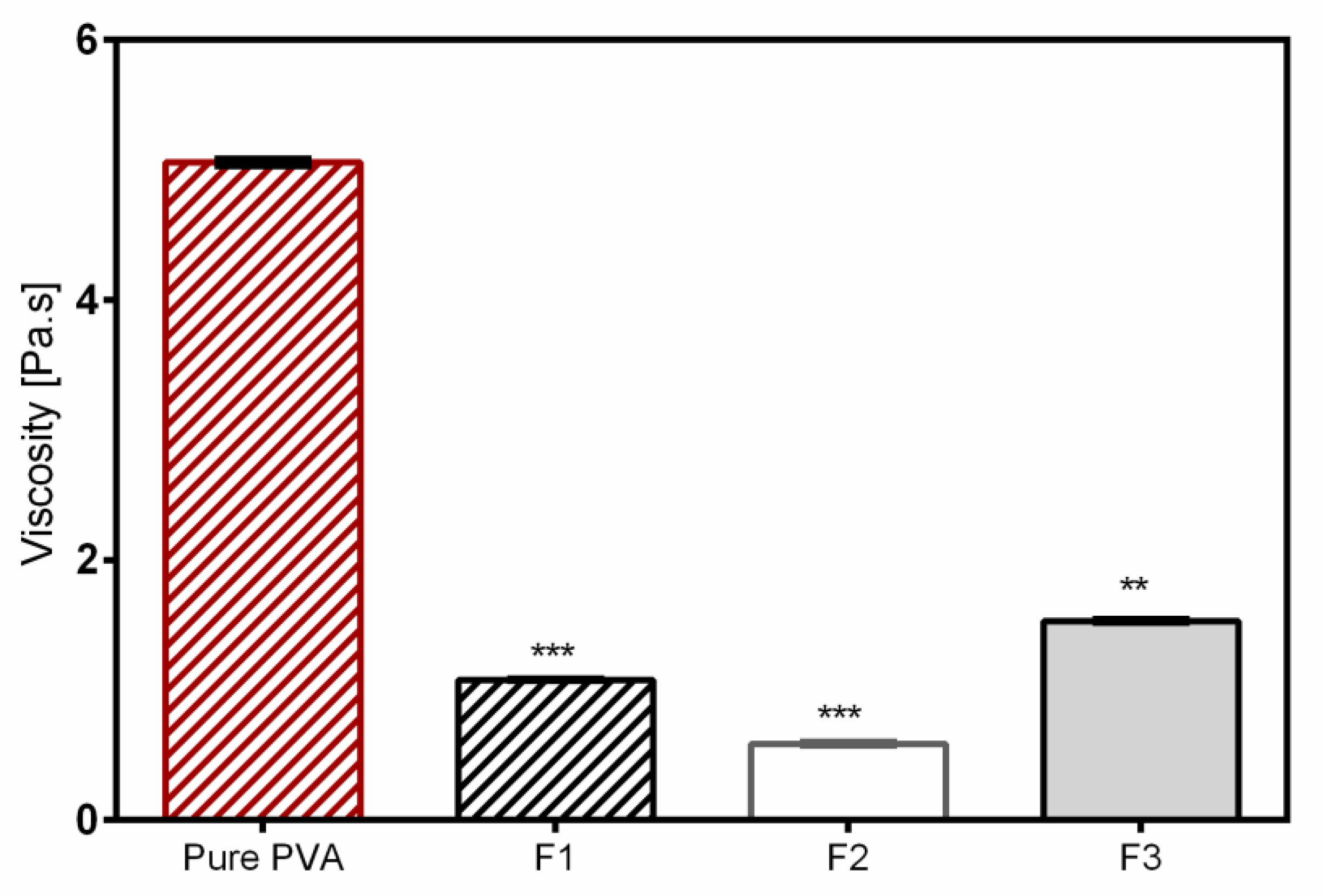
Fiber morphology was analyzed through SEM, Zeiss DSM 940 A, (Carl Zeiss GmbH, Oberkochen, Germany). Fiber mats of 0.5 cm × 0.5 cm sizes were placed of a conductive double-sided tape and sputter coated with gold using Biorad E 5100 Sputter Coater (Bio-Rad GmbH, Munich, Germany) at 2.1 kV and 20 mA for 240 s and imaged using the microscope. Thereafter, the average fiber diameters were determined from the SEM images by randomly selecting a minimum of 30 segments and their diameters calculated using ImageJ software (National Institute of Health, Bethesda, MD USA). For fiber diameter distribution, numbers of fiber diameters were converted into their percentage total values and plotted against grouped fiber diameter. The thicknesses of the fiber mats were measured with a digital micrometer using both magnetic induction and eddy current methods (Dualscope FMP20, Helmut Fischer GmbH, Sindelfingen Germany) taking the average of 15 measurements at randomly selected places.
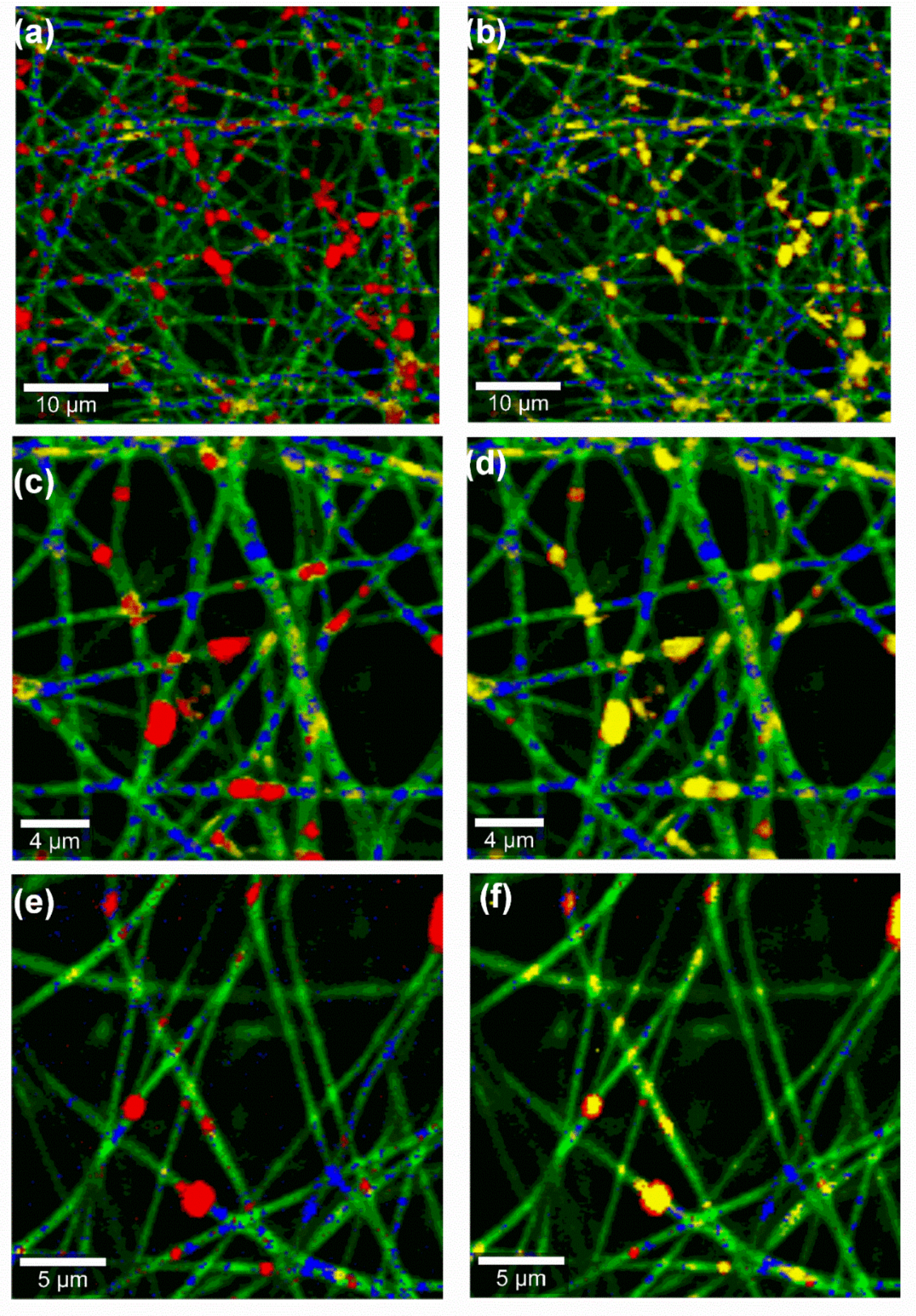
2.6. Confocal Raman Spectral Imaging
Raman images of wound dressings were acquired using an alpha 500R Raman microscope (WiTec GmbH, Ulm, Germany) equipped with a DV401-BV charge coupled device (CCD) detector, connected via an optical fiber to a UHTS 300 spectrometer and a 532 nm laser excitation source. A 100× air objective (EC Epiplan-Neofluor, Carl Zeiss, Oberkochen, Germany) with a numerical aperture of 0.9 was used to view the sample while the laser intensity was adjusted to 35 mW. The spectral range covered the fingerprint region between 710 and 1820 cm
−1 and region between 2700 and 3580 cm
−1. Spectra of all single components (PL90H, SO, PVA and TE) were first collected and used to monitor the presence of each component in the wound dressing. Subsequently, nanofiber mats were presented to the microscope on glass slides (VWR-International, Darmstadt, Germany) without the use of a coverslip. Image scans of wound dressings using 25 µm × 25 µm area of the surface were taken at an integration time of 0.01 s. Henceforth, color-coded images were obtained by initial cosmic ray removal and spectral background subtraction using the WiTec Project data analysis software 4.1 (WITec GmbH, Ulm, Germany, 2016). By assigning the spectrum of each component to a color, the distribution of all components within the formulation can be indicated, resulting in a color-coded image of the scanned area [
36].
2.7. Differential Scanning Calorimetry (DSC)
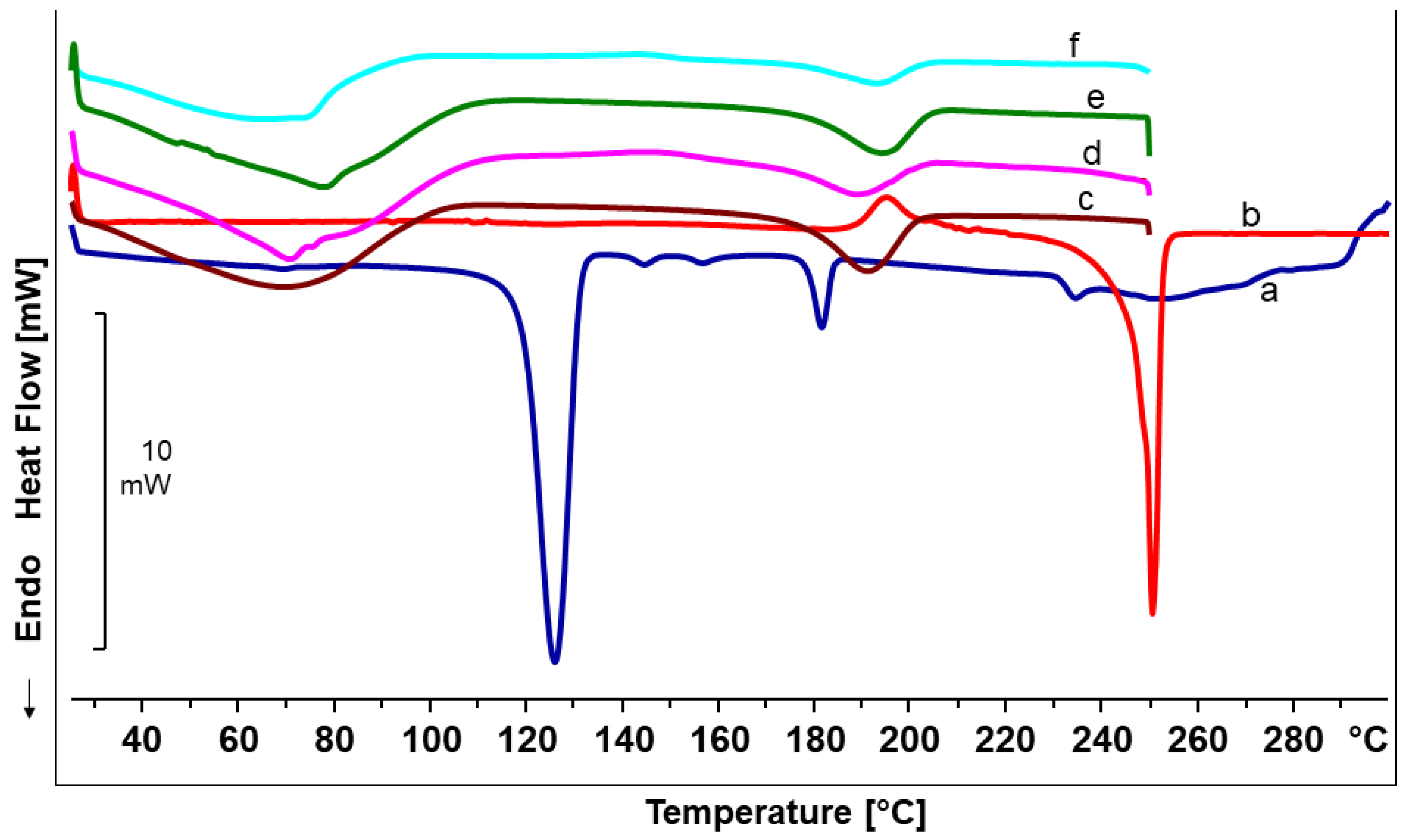
Differential scanning calorimetry (DSC) studies of the produced nanofiber mats were carried out using a DSC 820 differential scanning calorimeter (Mettler-Toledo GmbH, Gießen, Germany). The DSC was calibrated for temperature and enthalpy using indium as a standard. Samples of 4–7 mg were sealed in an aluminium pan with one pinhole in the lid. The furnace was purged with nitrogen at a flow rate of 80 mL/min. The DSC scans were obtained by heating in the range of 25,250 °C at a rate of 10 °C/min.
2.8. Skin Permeation and In Vitro Drug Release Studies
Both permeation and release studies were performed using modified vertical Franz diffusion cells (Gauer Glas, Püttlingen, Germany) with a receptor volume of 12 mL. For in vitro release studies, synthetic polycarbonate membranes with a pore size diameter of 0.4 µm were used to separate donor and receptor compartments. For permeation studies, pig ears were obtained from the Department of Experimental Medicine of the University Hospital Tuebingen. The live animals were kept at the Department of Experimental Medicine and sacrificed during their experiments, with the approval of the ethics committee of the University Hospital Tuebingen. The ears were delivered directly after the death of the animals. The Department of Pharmaceutical Technology is registered for the use of animal products at the District Office of Tuebingen (registration number: DE 08 416 1052 21). Fresh pig ears were first cleaned with isotonic saline using cotton balls and after postauricular skin excision, they were wrapped in aluminium foil and stored at −30 °C until use. During the day of experiment, the skin samples were thawed at room temperature, skin strips of 3 cm width were made and pinned to a block of Styrofoam precovered with aluminium foil. Thereafter, wounded skin samples were prepared according to [
9] by a skin grafting method using a Dermatom (Dermatom GA 630, Aesculap AG & Co. KG, Tüttlingen, Germany). The skin was first “wounded” through removal of the outermost layers of the skin with a thickness of 0.2 mm using Dermatom. Subsequently, the remaining skin was then dermatomed to a thickness of 0.4 mm. From the prepared porcine skin, a specimen was punched to obtain discs of 25 mm in diameter using a circular hole punch (Eduard Gottfried Ferne, Remscheid, Germany) [
37].
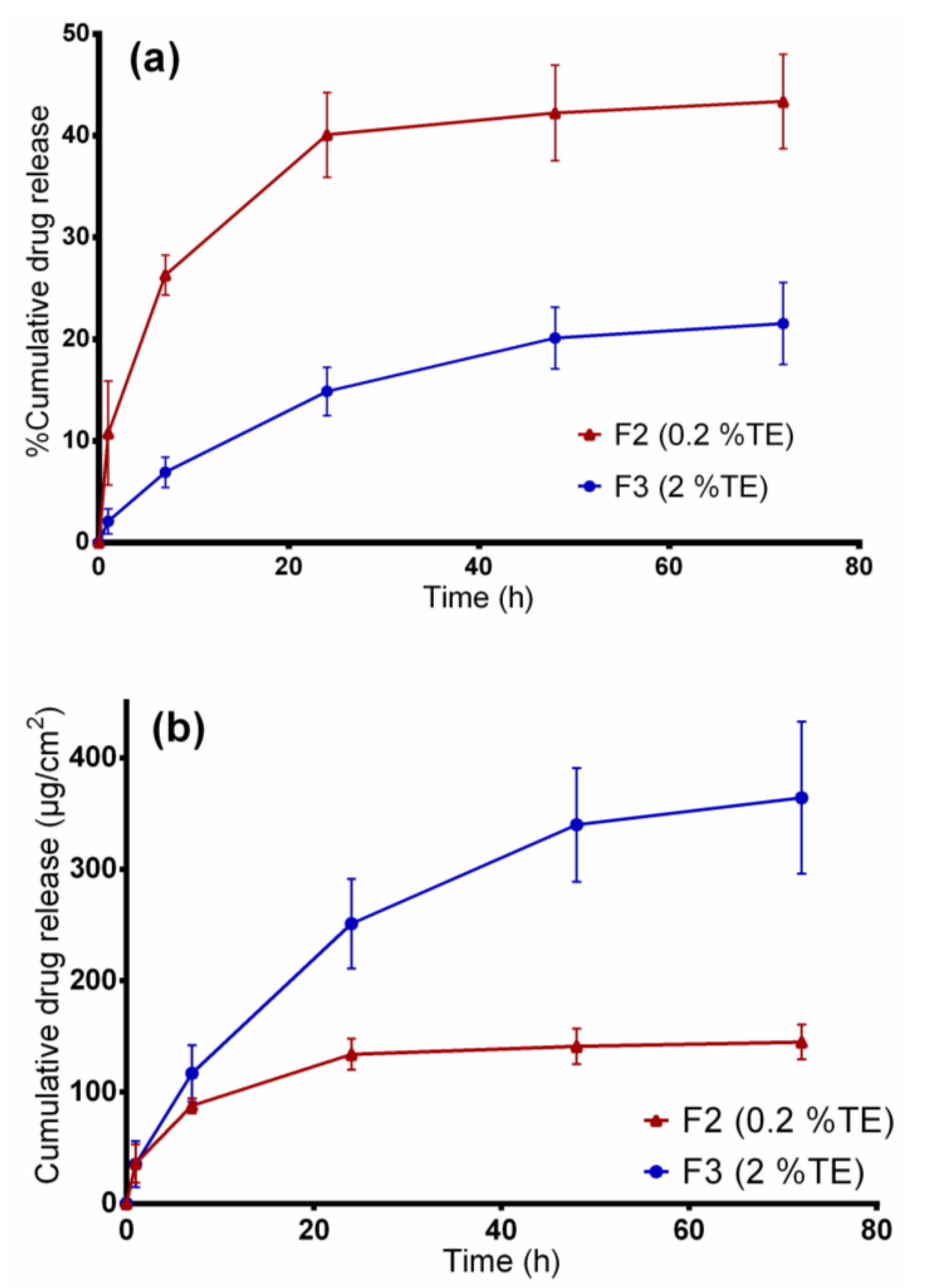
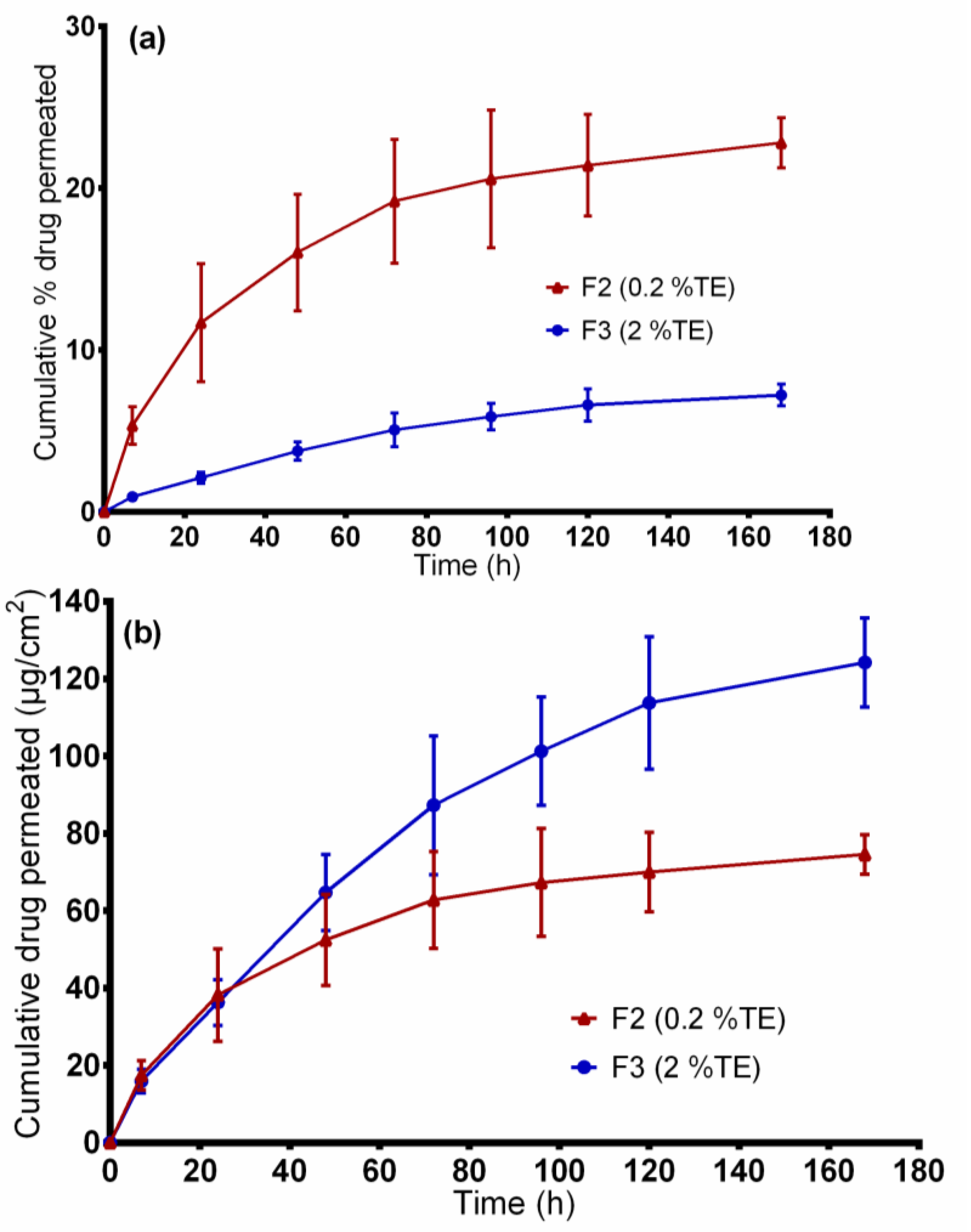
The synthetic membrane/dermal porcine skin was mounted between the compartments of diffusion cells, and the effective diffusion area was 1.77 cm2. For both in vitro release and ex vivo permeation experiments studies, samples of 20 mg fiber mats (F2 and F3), exactly weighed, were used. Consequently, all samples were loaded in the donor compartments and covered with parafilm to prevent solvent evaporation. A mixture of ethanol and water, 50:50 (v/v), was used as the receptor medium and the diffusion cells were maintained at 32 °C under constant magnetic stirring at 500 rpm. At predetermined time intervals, samples of 1 mL were taken from the receptor compartment and replaced with the same volume of fresh prewarmed receptor medium to maintain sink condition. The samples removed were analyzed directly using the HPLC-UV method as described below. The experiments were conducted in triplicate.
2.9. Entrapment Efficiency
The amount of betulin entrapped within fiber mats was estimated by dissolving approximately 20 mg of electrospun fiber mat, exactly weighed, in 30 mL ethanol:water (50:50
v/v) solution by means of sonication at 65 °C (Sonorex RK 31H, Bandelin, Berlin). Samples were then filtered using hydrophilic polyethersulfone (PES) syringe filters (Macherey-Nagel, Düren, Germany), and the total amount of drug extracted was quantified by High Performance Liquid Chromatography with Ultraviolet detection (HPLC-UV). The percentage entrapment efficiency (%EE) was calculated by the following Equation (1):
2.10. Betulin Permeation/Release Kinetics Studies
The release/permeation kinetics were estimated by linear regression analysis of the in vitro release and ex vivo permeation data using various mathematical models [
38]. The four models included zero order (Equation (2)), first order (Equation (3)), Korsmeyer–Peppas (Equation (4)), and Higuchi (Equation (5)) and the mathematical model that best fitted the kinetic release profile was selected based on the highest coefficient of determination, R
2. In all equations, M
t represents the amount of betulin released at time t.
Zero-order model, where k
0 is the zero-order release constant:
First order model where K
1 is the first order release rate constant:
Korsmeyer–Peppas model:
where K is the Korsmeyer–Peppas constant, which is connected to the characteristics of the delivery system and the encapsulated drug. The n is the diffusional exponent that shows the drug release mechanism; where
n equal to 0.45 represents a Fickian diffusion mechanism and 0.45 <
n < 1 is referred to as a non-Fickian diffusion mechanism (anomalous transport) in which both Fickian diffusion and Case-II transport occurs [
39].
Higuchi model, where k is Higuchi constant:
2.11. HPLC Analysis
Betulin quantification was performed using an LC-20A prominence HPLC system (Shimadzu, Kyoto, Germany). The mobile phase, a mixture of acetonitrile and water with the addition of 0.1% (
v/v) phosphoric acid, was used as a gradient system for chromatographic separation. For efficient separation, gradient conditions were used according to the isocratic/gradient modes developed by Armbruster et al. [
40]. A Nucleosil 100-5 C18 EC 125/4 column with a precolumn Universal RP EC 4/3 (Macherey-Nagel, Düren, Germany) was kept at 40 °C and a flow rate of 1.2 mL/min was used. A sample volume of 100 µL was injected and the peaks were detected at 210 nm. The retention time of betulin was approximately 7.5 min.
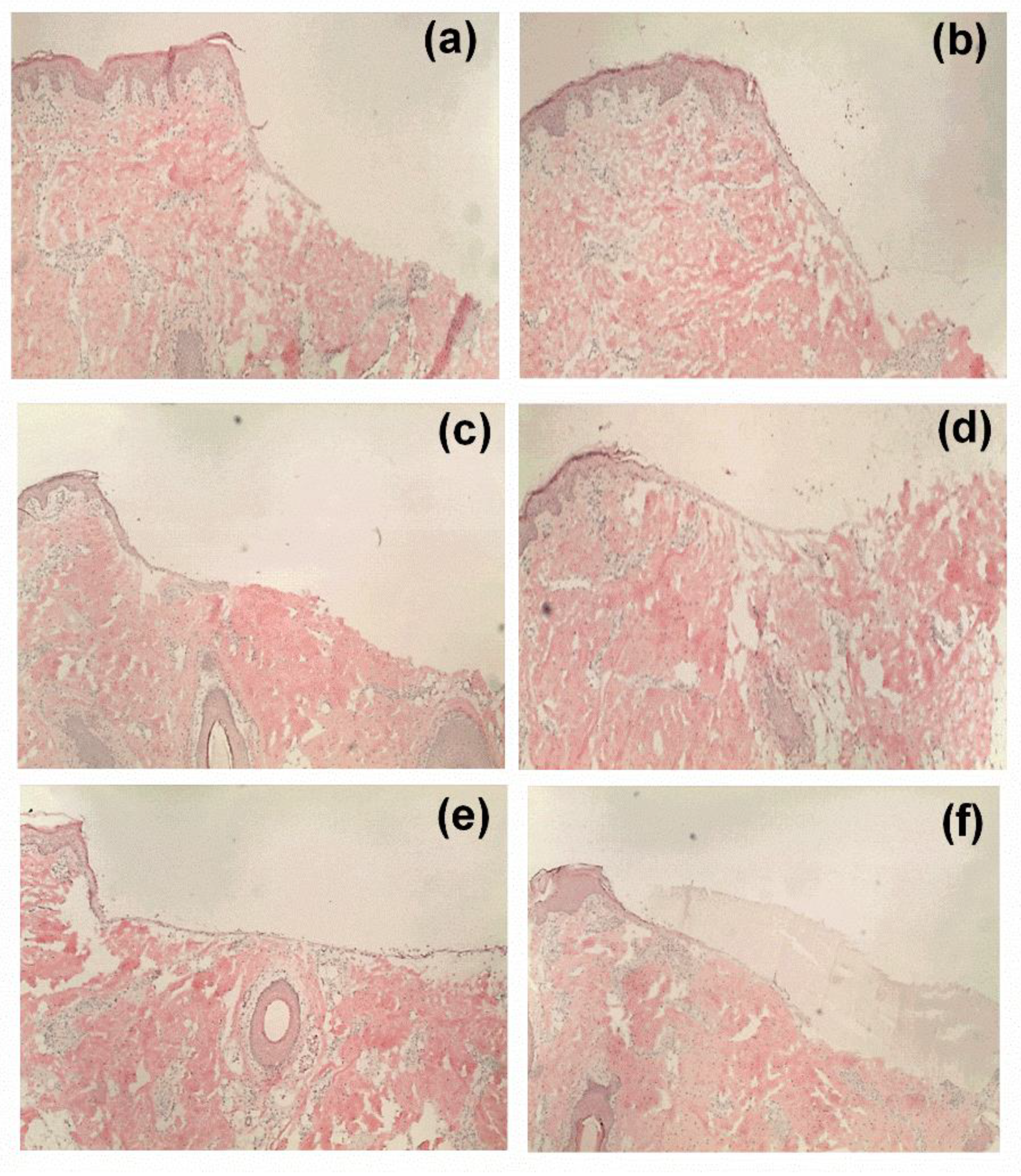
2.12. Ex Vivo Wound Healing Assay
In order to characterize the wound healing efficiency, a porcine ex-vivo wound healing assay was performed as described elsewhere [
8]. Briefly, pig ears were immediately delivered after slaughtering for human consumption to the laboratory, cleaned, and disinfected. Thereafter, 6 mm punch biopsies were taken from the plicae of the ears and fat and subcutis were removed. Consequently, wounds were generated by the removal of the epidermis and upper dermis in a central area of 7.1 mm
2. Then, the so formed ex vivo wound healing model was placed dermis-down on gauze in culture dishes and incubated at the air–liquid interface with Dulbecco’s modified Eagle’s medium supplemented with hydrocortisone, 2% fetal calf serum, penicillin, and streptomycin. The tested sample groups were divided into six groups (
n = 10): (i) untreated control, (ii) TE-oleogel, (iii) pure PVA mat (iv) F1, (v) F2, and (vi) F3 fiber mats. Subsequently, sections of the electrospun fiber mats (4 mm in diameter)/5 µL of the oleogel were immediately applied after wounding, and the models were incubated for 48 h at 37 °C and 5% CO
2. Further steps involved shock freezing, preparations of cryostat sections of the central parts of the wound healing models, and staining with hematoxylin and eosin. Wound healing progress (reepithelialization) was assessed by measuring the distance between the wound margin and the tip of the regenerated epidermis using a Leica DMLS microscope (10×), a Leica MC 170 HD CCD camera, and the Leica LAS V4.9 software (Leica, Wetzlar, Germany, 2017). Means of left and right wound margins were calculated.
2.13. Statistical Analysis
All the experiments unless mentioned otherwise were carried out in triplicate and the results were expressed as mean ± standard deviation (mean ± SD). Statistical analysis of the acquired data was performed by Student’s t-test and analysis of variance (ANOVA). p-value less than 0.05 was considered statistically significant, and where significance has been proven, it is indicated by * p < 0.05, ** p < 0.01, and *** p < 0.001.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}