Miktoarm Star Polymers: Branched Architectures in Drug Delivery



Abstract
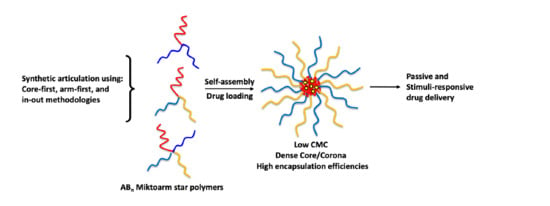
:
1. Introduction
2. Synthetic Approaches to Miktoarm Star Polymers
2.1. Chlorosilane Based Synthesis
2.2. Core-First Synthesis
2.3. Arm-First Synthesis
2.4. In-Out Synthesis
2.5. Miktoarm Polymer Characterization
3. Amphiphilic Miktoarm Star Polymers: Self-Assembly
3.1. Micelle Characteristics: CMC and Stability
3.2. Micelle Drug Loading and Release
3.3. Non-Spherical Micelles
3.4. Polymersomes
4. Drug Delivery
4.1. pH-Responsive Drug Delivery
4.2. Temperature-Responsive Drug Delivery
4.3. Redox-Responsive Drug Delivery
4.4. Light- and Dual-Responsive Drug Delivery
4.5. Polyplex Delivery
5. Conclusions and Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins: Basic science and product development. J. Pharm. Pharmacol. 2010, 62, 1607–1621. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.; DeGiovanni, P.-J.; Piel, B.; Rai, P. Cancer nanomedicine: A review of recent success in drug delivery. Clin. Transl. Med. 2017, 6, 44. [Google Scholar] [CrossRef] [Green Version]
- Croy, S.R.; Kwon, G.S. Polymeric Micelles for Drug Delivery. Curr. Pharm. Des. 2006, 12, 4669–4684. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, E.; Yang, J.; Cao, Z. Strategies to improve micelle stability for drug delivery. Nano Res. 2018, 11, 4985–4998. [Google Scholar] [CrossRef] [PubMed]
- Haag, R.; Kratz, F. Polymer Therapeutics: Concepts and Applications. Angew. Chem. 2006, 45, 1198–1215. [Google Scholar] [CrossRef] [PubMed]
- Blanazs, A.; Armes, S.P.; Ryan, A.J. Self-Assembled Block Copolymer Aggregates: From Micelles to Vesicles and their Biological Applications. Macromol. Rapid Commun. 2009, 30, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Shi, Y.; Kim, J.Y.; Park, K.; Cheng, J.-X. Overcoming the barriers in micellar drug delivery: Loading efficiency, in vivo stability, and micelle–cell interaction. Expert Opin. Drug Deliv. 2010, 7, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Kaditi, E.; Mountrichas, G.; Pispas, S.; Demetzos, C. Block Copolymers for Drug Delivery Nano Systems (DDnSs). Curr. Med. Chem. 2012, 19, 5088–5100. [Google Scholar] [CrossRef]
- Gaucher, G.; Dufresne, M.-H.; Sant, V.P.; Kang, N.; Maysinger, D.; Leroux, J.-C. Block copolymer micelles: Preparation, characterization and application in drug delivery. J. Control. Release 2005, 109, 169–188. [Google Scholar] [CrossRef]
- Mirza, A.Z.; Siddiqui, F.A. Nanomedicine and drug delivery: A mini review. Int. Nano Lett. 2014, 4, 94. [Google Scholar] [CrossRef] [Green Version]
- Farokhzad, O.C.; Langer, R. Nanomedicine: Developing smarter therapeutic and diagnostic modalities. Advanced Drug Deliv. Rev. 2006, 58, 1456–1459. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Hunter, A.C.; Murray, J.C. Long-Circulating and Target-Specific Nanoparticles: Theory to Practice. Pharmacol. Rev. 2001, 53, 283. [Google Scholar] [PubMed]
- Allen, C.; Maysinger, D.; Eisenberg, A. Nano-engineering block copolymer aggregates for drug delivery. Colloids Surf. B Biointerfaces 1999, 16, 3–27. [Google Scholar] [CrossRef]
- Yoshida, E. Control of Micellar Size and Critical Micelle Concentration for “Nonamphiphilic” Poly(vinyl phenol)-block-Polystyrene Diblock Copolymers. Polym. J. 2003, 35, 965–971. [Google Scholar] [CrossRef]
- Lo, C.-L.; Lin, S.-J.; Tsai, H.-C.; Chan, W.-H.; Tsai, C.-H.; Cheng, C.-H.D.; Hsiue, G.-H. Mixed micelle systems formed from critical micelle concentration and temperature-sensitive diblock copolymers for doxorubicin delivery. Biomaterials 2009, 30, 3961–3970. [Google Scholar] [CrossRef]
- Kosa, S.A.; Al-Harbi, L.M.; Baloch, M.K.; Ullah, I.; El-Mossalamy, E.H. Impact of Block Length and Temperature over Self-Assembling Behavior of Block Copolymers. Int. J. Polym. Sci. 2016, 2016, 6732790. [Google Scholar] [CrossRef]
- Kulthe, S.S.; Choudhari, Y.M.; Inamdar, N.N.; Mourya, V. Polymeric micelles: Authoritative aspects for drug delivery. Des. Monomers Polym. 2012, 15, 465–521. [Google Scholar] [CrossRef]
- Hadjichristidis, N. Synthesis of miktoarm star (μ-star) polymers. J. Polym. Sci. Part A Polym. Chem. 1999, 37, 857–871. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Iatrou, H.; Pitsikalis, M.; Mays, J. Macromolecular architectures by living and controlled/living polymerizations. Prog. Polym. Sci. 2006, 31, 1068–1132. [Google Scholar] [CrossRef]
- Sharma, A.; Kakkar, A. Designing Dendrimer and Miktoarm Polymer Based Multi-Tasking Nanocarriers for Efficient Medical Therapy. Molecules 2015, 20, 16987–17015. [Google Scholar] [CrossRef]
- Khanna, K.; Varshney, S.; Kakkar, A. Miktoarm star polymers: Advances in synthesis, self-assembly, and applications. Polym. Chem. 2010, 1, 1171–1185. [Google Scholar] [CrossRef]
- Pispas, S.; Hadjichristidis, N.; Potemkin, I.; Khokhlov, A. Effect of Architecture on the Micellization Properties of Block Copolymers: A2B Miktoarm Stars vs AB Diblocks. Macromolecules 2000, 33, 1741–1746. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Pitsikalis, M.; Iatrou, H.; Driva, P.; Sakellariou, G.; Chatzichristidi, M. 6.03-Polymers with Star-Related Structures: Synthesis, Properties, and Applications. In Polymer Science: A Comprehensive Reference; Matyjaszewski, K., Möller, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Soliman, G.M.; Sharma, R.; Choi, A.O.; Varshney, S.K.; Winnik, F.M.; Kakkar, A.K.; Maysinger, D. Tailoring the efficacy of nimodipine drug delivery using nanocarriers based on A2B miktoarm star polymers. Biomaterials 2010, 31, 8382–8392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wais, U.; Liu, J.; He, T.; Zhang, H. Micellar and Emulsion-Assisted Drug Delivery: Comparison of Miktoarm Star Polymers and Block Copolymers. In Miktoarm Star Polymers: From Basics of Branched Architecture to Synthesis, Self-assembly and Applications; The Royal Society of Chemistry: Cambridge, UK, 2017; Volume 5. [Google Scholar]
- Aghajanzadeh, M.; Zamani, M.; Rostamizadeh, K.; Sharafi, A.; Danafar, H. The role of miktoarm star copolymers in drug delivery systems. J. Macromol. Sci. Part A 2018, 55, 559–571. [Google Scholar] [CrossRef]
- Li, Y.-Y.; Zhang, X.-Z.; Cheng, H.; Kim, G.-C.; Cheng, S.-X.; Zhuo, R.-X. Novel Stimuli-Responsive Micelle Self-Assembled from Y-Shaped P(UA-Y-NIPAAm) Copolymer for Drug Delivery. Biomacromolecules 2006, 7, 2956–2960. [Google Scholar] [CrossRef]
- Van Butsele, K.V.; Fustin, C.A.; Gohy, J.F.; Jérôme, R.; Jérôme, C. Self-Assembly and pH-Responsiveness of ABC Miktoarm Star Terpolymers. Langmuir 2009, 25, 107–111. [Google Scholar] [CrossRef]
- Cajot, S.; Van Butsele, K.; Paillard, A.; Passirani, C.; Garcion, E.; Benoit, J.P.; Varshney, S.K.; Jérôme, C. Smart nanocarriers for pH-triggered targeting and release of hydrophobic drugs. Acta Biomater. 2012, 8, 4215–4223. [Google Scholar] [CrossRef]
- Van Butsele, K.V.; Stoffelbach, F.; Jérôme, R.; Jérôme, C. Synthesis of Novel Amphiphilic and pH-Sensitive ABC Miktoarm Star Terpolymers. Macromolecules 2006, 39, 5652–5656. [Google Scholar] [CrossRef]
- Wei, H.; Zhang, X.; Cheng, C.; Cheng, S.-X.; Zhuo, R.-X. Self-assembled, thermosensitive micelles of a star block copolymer based on PMMA and PNIPAAm for controlled drug delivery. Biomaterials 2007, 28, 99–107. [Google Scholar] [CrossRef]
- Zhang, H.-H.; Huang, Z.-Q.; Sun, B.-W.; Guo, J.-X.; Wang, J.-L.; Chen, Y.-Q. Y-shaped poly(ethylene glycol) and poly(trimethylene carbonate) amphiphilic copolymer: Synthesis and for drug delivery. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 8131–8140. [Google Scholar] [CrossRef]
- Gou, P.-F.; Zhu, W.-P.; Xu, N.; Shen, Z.-Q. Synthesis, self-assembly and drug-loading capacity of well-defined drug-conjugated amphiphilic A2B2 type miktoarm star copolymers based on poly(ε-caprolactone) and poly(ethylene glycol). J. Polym. Sci. Part A Polym. Chem. 2009, 47, 6962–6976. [Google Scholar] [CrossRef]
- Yin, H.; Kang, S.-W.; Bae, Y.H. Polymersome Formation from AB2 Type 3-Miktoarm Star Copolymers. Macromolecules 2009, 42, 7456–7464. [Google Scholar] [CrossRef]
- Nederberg, F.; Appel, E.; Tan, J.P.K.; Kim, S.H.; Fukushima, K.; Sly, J.; Miller, R.D.; Waymouth, R.M.; Yang, Y.Y.; Hedrick, J.L. Simple Approach to Stabilized Micelles Employing Miktoarm Terpolymers and Stereocomplexes with Application in Paclitaxel Delivery. Biomacromolecules 2009, 10, 1460–1468. [Google Scholar] [CrossRef]
- Gou, P.-F.; Zhu, W.-P.; Shen, Z.-Q. Synthesis, Self-Assembly, and Drug-Loading Capacity of Well-Defined Cyclodextrin-Centered Drug-Conjugated Amphiphilic A14B7 Miktoarm Star Copolymers Based on Poly(ε-caprolactone) and Poly(ethylene glycol). Biomacromolecules 2010, 11, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cheng, J.; Wang, Q.; Zhong, Z.; Zhuo, R. Miktoarm Copolymers Bearing One Poly(ethylene glycol) Chain and Several Poly(ε-caprolactone) Chains on a Hyperbranched Polyglycerol Core. Macromolecules 2010, 43, 6671–6677. [Google Scholar] [CrossRef]
- Li, L.-Y.; He, W.-D.; Li, J.; Zhang, B.-Y.; Pan, T.-T.; Sun, X.-L.; Ding, Z.-L. Shell-Cross-Linked Micelles from PNIPAM-b-(PLL)2 Y-Shaped Miktoarm Star Copolymer as Drug Carriers. Biomacromolecules 2010, 11, 1882–1890. [Google Scholar] [CrossRef] [PubMed]
- Khanna, K.; Varshney, S.; Kakkar, A. Designing Miktoarm Polymers Using a Combination of “Click” Reactions in Sequence with Ring-Opening Polymerization. Macromolecules 2010, 43, 5688–5698. [Google Scholar] [CrossRef]
- Maglio, G.; Nicodemi, F.; Conte, C.; Palumbo, R.; Tirino, P.; Panza, E.; Ianaro, A.; Ungaro, F.; Quaglia, F. Nanocapsules Based on Linear and Y-Shaped 3-Miktoarm Star-Block PEO-PCL Copolymers as Sustained Delivery System for Hydrophilic Molecules. Biomacromolecules 2011, 12, 4221–4229. [Google Scholar] [CrossRef]
- Sharma, A.; Khatchadourian, A.; Khanna, K.; Sharma, R.; Kakkar, A.; Maysinger, D. Multivalent niacin nanoconjugates for delivery to cytoplasmic lipid droplets. Biomaterials 2011, 32, 1419–1429. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Soliman, G.M.; Al-Hajaj, N.; Sharma, R.; Maysinger, D.; Kakkar, A. Design and Evaluation of Multifunctional Nanocarriers for Selective Delivery of Coenzyme Q10 to Mitochondria. Biomacromolecules 2012, 13, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Kang, H.C.; Huh, K.M.; Bae, Y.H. Biocompatible, pH-sensitive AB2 miktoarm polymer-based polymersomes: Preparation, characterization, and acidic pH-activated nanostructural transformation. J. Mater. Chem. 2012, 22, 19168–19178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.; Kang, H.C.; Huh, K.M.; Bae, Y.H. Effects of cholesterol incorporation on the physicochemical, colloidal, and biological characteristics of pH-sensitive AB2 miktoarm polymer-based polymersomes. Colloids Surf. B 2014, 116, 128–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Zhang, Y.-F.; Liu, S.-Y. Drug and plasmid DNA co-delivery nanocarriers based on abctype polypeptide hybrid miktoarm star copolymers. Chin. J. Polym. Sci. 2013, 31, 924–937. [Google Scholar] [CrossRef]
- Blasco, E.; Schmidt, B.V.K.J.; Barner-Kowollik, C.; Piñol, M.; Oriol, L. Dual thermo- and photo-responsive micelles based on miktoarm star polymers. Polym. Chem. 2013, 4, 4506–4514. [Google Scholar] [CrossRef]
- Soliman, G.M.; Sharma, A.; Cui, Y.; Sharma, R.; Kakkar, A.; Maysinger, D. Miktoarm Star Micelles Containing Curcumin Reduce Cell Viability of Sensitized Glioblastoma. J. Nanomed Biother Discov. 2014, 4, 10. [Google Scholar] [CrossRef]
- Chu, Y.; Yu, H.; Zhang, Y.; Zhang, G.; Ma, Y.; Zhuo, R.; Jiang, X. Synthesis and characterization of biodegradable amphiphilic ABC Y-shaped miktoarm terpolymer by click chemistry for drug delivery. J. Polym. Sci. Part A Polym. Chem. 2014, 52, 3346–3355. [Google Scholar] [CrossRef]
- Soliman, G.M.; Redon, R.; Sharma, A.; Mejía, D.; Maysinger, D.; Kakkar, A. Miktoarm Star Polymer Based Multifunctional Traceable Nanocarriers for Efficient Delivery of Poorly Water Soluble Pharmacological Agents. Macromol. Biosci. 2014, 14, 1312–1324. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Nie, S.; Xiong, D.; Guo, X.; Wang, J.; Zhang, L. pH-responsive micelles based on (PCL)2(PDEA-b-PPEGMA)2 miktoarm polymer: Controlled synthesis, characterization, and application as anticancer drug carrier. Nanoscale Res. Lett. 2014, 9, 243. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Nie, S.; Zhong, Q.; Yang, Y.; Cai, C.; Wang, J.; Zhang, L. Amphiphilic miktoarm star copolymer (PCL)3-(PDEAEMA-b-PPEGMA)3 as pH-sensitive micelles in the delivery of anticancer drug. J. Mater. Chem. B 2014, 2, 4008–4020. [Google Scholar] [CrossRef]
- Sui, B.; Xu, H.; Jin, J.; Gou, J.; Liu, J.; Tang, X.; Zhang, Y.; Xu, J.; Zhang, H.; Jin, X. Self-Assembled Micelles Composed of Doxorubicin Conjugated Y-Shaped PEG-Poly(glutamic acid)2 Copolymers via Hydrazone Linkers. Molecules 2014, 19, 11915–11932. [Google Scholar] [CrossRef] [Green Version]
- Blasco, E.; Schmidt, B.V.K.J.; Barner-Kowollik, C.; Piñol, M.; Oriol, L. A Novel Photoresponsive Azobenzene-Containing Miktoarm Star Polymer: Self-Assembly and Photoresponse Properties. Macromolecules 2014, 47, 3693–3700. [Google Scholar] [CrossRef]
- Yoon, K.; Kang, H.C.; Li, L.; Cho, H.; Park, M.-K.; Lee, E.; Bae, Y.H.; Huh, K.M. Amphiphilic poly(ethylene glycol)-poly(ε-caprolactone) AB2 miktoarm copolymers for self-assembled nanocarrier systems: Synthesis, characterization, and effects of morphology on antitumor activity. Polym. Chem. 2015, 6, 531–542. [Google Scholar] [CrossRef]
- Moquin, A.; Sharma, A.; Cui, Y.; Lau, A.; Maysinger, D.; Kakkar, A. Asymmetric AB3 Miktoarm Star Polymers: Synthesis, Self-Assembly, and Study of Micelle Stability Using AF4 for Efficient Drug Delivery. Macromol. Biosci. 2015, 15, 1744–1754. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, R.; Ghaemy, M. pH-responsive ABC type miktoarm star terpolymers: Synthesis via combination of click reaction and SET-LRP, characterization, self-assembly, and controlled drug release. Polymer 2015, 66, 179–191. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, M.; Luo, X.; Zhang, H.; Liu, C.; Li, H.; Li, X. Tuning multiple arms for camptothecin and folate conjugations on star-shaped copolymers to enhance glutathione-mediated intracellular drug delivery. Polym. Chem. 2015, 6, 2192–2203. [Google Scholar] [CrossRef]
- Zhou, Q.-H.; Lin, J.; Li, L.-D.; Shang, L. Biodegradable micelles self-assembled from miktoarm star block copolymers for MTX delivery. Colloid Polym. Sci. 2015, 293, 2291–2300. [Google Scholar] [CrossRef]
- Zhu, M.-M.; Song, F.; Nie, W.-C.; Wang, X.-L.; Wang, Y.-Z. A facile chemoenzymatic synthesis of amphiphilic miktoarm star copolymers from a sugar core and their potential for anticancer drug delivery. Polymer 2016, 93, 159–166. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhang, Y.X.; Wu, Z.F.; Peng, X.Y.; Su, T.; Cao, J.; He, B.; Li, S. Biodegradable poly(ethylene glycol)–poly(ε-carprolactone) polymeric micelles with different tailored topological amphiphilies for doxorubicin (DOX) drug delivery. RSC Adv. 2016, 6, 58160–58172. [Google Scholar] [CrossRef]
- Huang, J.; Liang, H.; Cheng, D.; Lu, J. Polypeptide–poly(ethylene glycol) miktoarm star copolymers with a fluorescently labeled core: Synthesis, delivery and imaging of siRNA. Polym. Chem. 2016, 7, 1792–1802. [Google Scholar] [CrossRef]
- Huang, L.-M.; Li, L.-D.; Shang, L.; Zhou, Q.-H.; Lin, J. Preparation of pH-sensitive micelles from miktoarm star block copolymers by ATRP and their application as drug nanocarriers. React. Funct. Polym. 2016, 107, 28–34. [Google Scholar] [CrossRef]
- Mielańczyk, A.; Odrobińska, J.; Grządka, S.; Mielańczyk, Ł.; Neugebauer, D. Miktoarm star copolymers from D-(−)-salicin core aggregated into dandelion-like structures as anticancer drug delivery systems: Synthesis, self-assembly and drug release. Int. J. Pharm. 2016, 515, 515–526. [Google Scholar] [CrossRef]
- Xu, W.; Steinschulte, A.A.; Plamper, F.A.; Korolovych, V.F.; Tsukruk, V.V. Hierarchical Assembly of Star Polymer Polymersomes into Responsive Multicompartmental Microcapsules. Chem. Mater. 2016, 28, 975–985. [Google Scholar] [CrossRef]
- Zhu, J.; Liu, Y.; Xiao, L.; Zhou, P. Temperature-Sensitive (BA)(AC)2 Miktoarm Star Diblock Copolymer Based on PMMA, PPEGMA, and PNIPAm. Macromol. Chem. Phys. 2016, 217, 773–782. [Google Scholar] [CrossRef]
- Huo, H.; Ma, X.; Dong, Y.; Qu, F. Light/temperature dual-responsive ABC miktoarm star terpolymer micelles for controlled release. Eur. Polym. J. 2017, 87, 331–343. [Google Scholar] [CrossRef]
- Nieto-Orellana, A.; Di Antonio, M.; Conte, C.; Falcone, F.H.; Bosquillon, C.; Childerhouse, N.; Mantovani, G.; Stolnik, S. Effect of polymer topology on non-covalent polymer–protein complexation: Miktoarm versus linear mPEG-poly(glutamic acid) copolymers. Polym. Chem. 2017, 8, 2210–2220. [Google Scholar] [CrossRef]
- Nieto-Orellana, A.; Coghlan, D.; Rothery, M.; Falcone, F.H.; Bosquillon, C.; Childerhouse, N.; Mantovani, G.; Stolnik, S. Dry-powder formulations of non-covalent protein complexes with linear or miktoarm copolymers for pulmonary delivery. Int. J. Pharm. 2018, 540, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Patil, Y.; Bilalis, P.; Polymeropoulos, G.; Almahdali, S.; Hadjichristidis, N.; Rodionov, V. A Novel Poly(vinylidene fluoride)-Based 4-Miktoarm Star Terpolymer: Synthesis and Self-Assembly. Mol. Pharm. 2018, 15, 3005–3009. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, X.; Peng, H.; Zhang, M.; Zhang, X.; Liu, Z.; Ma, L.; Wei, H. Optimization of Amphiphilic Miktoarm Star Copolymers for Anticancer Drug Delivery. ACS Biomater. Sci. Eng. 2018, 4, 2903–2910. [Google Scholar] [CrossRef]
- Aghajanzadeh, M.; Zamani, M.; Rashidzadeh, H.; Rostamizadeh, K.; Sharafi, A.; Danafar, H. Amphiphilic Y shaped miktoarm star copolymer for anticancer hydrophobic and hydrophilic drugs codelivery: Synthesis, characterization, in vitro, and in vivo biocompatibility study. J. Biomed. Mater. Res. Part A 2018, 106, 2817–2826. [Google Scholar] [CrossRef]
- Ramesh, K.; Thangagiri, B.; Mishra, A.K.; Ahn, B.-H.; Gal, Y.-S.; Lim, K.T. AB2-type miktoarm poly(l-lactide)-b-poly(N-acryloylmorpholine) amphiphilic star block copolymers as nanocarriers for drug delivery. React. Funct. Polym. 2018, 132, 112–119. [Google Scholar] [CrossRef]
- Kim, Y.; Uthaman, S.; Nurunnabi, M.; Mallick, S.; Oh, K.S.; Kang, S.-W.; Cho, S.; Kang, H.C.; Lee, Y.-K.; Huh, K.M. Synthesis and characterization of bioreducible cationic biarm polymer for efficient gene delivery. Int. J. Biol. Macromol. 2018, 110, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Saravanakumar, G.; Park, H.; Kim, J.; Park, D.; Pramanick, S.; Kim, D.H.; Kim, W.J. Miktoarm Amphiphilic Block Copolymer with Singlet Oxygen-Labile Stereospecific β-Aminoacrylate Junction: Synthesis, Self-Assembly, and Photodynamically Triggered Drug Release. Biomacromolecules 2018, 19, 2202–2213. [Google Scholar] [CrossRef] [PubMed]
- Chong, Y.K.; Zainol, I.; Ng, C.H.; Ooi, I.H. Miktoarm star polymers nanocarrier: Synthesis, characterisation, and in-vitro drug release study. J. Polym. Res. 2019, 26, 79. [Google Scholar] [CrossRef]
- Nieto-Orellana, A.; Li, H.; Rosiere, R.; Wauthoz, N.; Williams, H.; Monteiro, C.J.; Bosquillon, C.; Childerhouse, N.; Keegan, G.; Coghlan, D.; et al. Targeted PEG-poly(glutamic acid) complexes for inhalation protein delivery to the lung. J. Control. Release 2019, 316, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, S.J.; Kalhapure, R.S.; Jadhav, M.; Rambharose, S.; Mocktar, C.; Govender, T. AB2-type amphiphilic block copolymer containing a pH-cleavable hydrazone linkage for targeted antibiotic delivery. Int. J. Pharm. 2020, 575, 118948. [Google Scholar] [CrossRef]
- Aghajanzadeh, M.; Andalib, S.; Danafar, H.; Rostamizadeh, K.; Sharafi, A. The effect of baicalein-loaded Y-shaped miktoarm copolymer on spatial memory and hippocampal expression of DHCR24, SELADIN and SIRT6 genes in rat model of Alzheimer. Int. J. Pharm. 2020, 586, 119546. [Google Scholar] [CrossRef]
- Tomalia, D.A.; Baker, H.; Dewald, J.; Hall, M.; Kallos, G.; Martin, S.; Roeck, J.; Ryder, J.; Smith, P. Dendritic macromolecules: Synthesis of starburst dendrimers. Macromolecules 1986, 19, 2466–2468. [Google Scholar] [CrossRef]
- Hawker, C.J.; Frechet, J.M.J. Preparation of polymers with controlled molecular architecture. A new convergent approach to dendritic macromolecules. J. Am. Chem. Soc. 1990, 112, 7638–7647. [Google Scholar] [CrossRef]
- Meldal, M.; Tornøe, C.W. Cu-Catalyzed Azide−Alkyne Cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Fetters, L.J. Star-Branched Polymers. 4. Synthesis of 18-Arm Polyisoprenes. Macromolecules 1980, 13, 191–193. [Google Scholar] [CrossRef]
- Roovers, J.; Hadjichristidis, N.; Fetters, L.J. Analysis and dilute solution properties of 12- and 18-arm-star polystyrenes. Macromolecules 1983, 16, 214–220. [Google Scholar] [CrossRef]
- Alward, D.B.; Kinning, D.J.; Thomas, E.L.; Fetters, L.J. Effect of arm number and arm molecular weight on the solid-state morphology of poly(styrene-isoprene) star block copolymers. Macromolecules 1986, 19, 215–224. [Google Scholar] [CrossRef]
- Nguyen, A.B.; Hadjichristidis, N.; Fetters, L.J. Static light scattering study of high-molecular weight 18-arm star block copolymers. Macromolecules 1986, 19, 768–773. [Google Scholar] [CrossRef]
- Pennisi, R.W.; Fetters, L.J. Preparation of asymmetric 3-arm polybutadiene and polystyrene stars. Macromolecules 1988, 21, 1094–1099. [Google Scholar] [CrossRef]
- Bauer, B.J.; Fetters, L.J.; Graessley, W.W.; Hadjichristidis, N.; Quack, G.F. Chain dimensions in dilute polymer solutions: A light-scattering and viscometric study of multiarmed polyisoprene stars in good and.THETA. solvents. Macromolecules 1989, 22, 2337–2347. [Google Scholar] [CrossRef]
- Iatrou, H.; Hadjichristidis, N. Synthesis of a model 3-miktoarm star terpolymer. Macromolecules 1992, 25, 4649–4651. [Google Scholar] [CrossRef]
- Mays, J.W. Synthesis of “simple graft” poly(isoprene-g-styrene) by anionic polymerization. Polym. Bull. 1990, 23, 247–250. [Google Scholar] [CrossRef]
- Iatrou, H.; Hadjichristidis, N. Synthesis and characterization of model 4-miktoarm star co- and quaterpolymers. Macromolecules 1993, 26, 2479–2484. [Google Scholar] [CrossRef]
- Iatrou, H.; Siakali-Kioulafa, E.; Hadjichristidis, N.; Roovers, J.; Mays, J. Hydrodynamic properties of model 3-miktoarm star copolymers. J. Polym. Sci. Part B Polym. Phys. 1995, 33, 1925–1932. [Google Scholar] [CrossRef]
- Pitsikalis, M.; Hadjichristidis, N. Model Mono-, Di-, and Tri-.omega.-Functionalized Three-Arm Star Polybutadienes. Synthesis and Association in Dilute Solutions by Membrane Osmometry and Static Light Scattering. Macromolecules 1995, 28, 3904–3910. [Google Scholar] [CrossRef]
- Allgaier, J.; Young, R.N.; Efstratiadis, V.; Hadjichristidis, N. Synthesis and Characterization of Polyisoprene/Polybutadiene A2B2 Star Copolymers. Macromolecules 1996, 29, 1794–1797. [Google Scholar] [CrossRef]
- Sioula, S.; Tselikas, Y.; Hadjichristidis, N. Synthesis of Model 3-Miktoarm Star Terpolymers of Styrene, Isoprene, and Methyl Methacrylate. Macromolecules 1997, 30, 1518–1520. [Google Scholar] [CrossRef]
- Zioga, A.; Sioula, S.; Hadjichristidis, N. Synthesis and morphology of model 3-miktoarm star terpolymers of styrene, isoprene and 2-vinyl pyridine. Macromol. Symp. 2000, 157, 239–250. [Google Scholar] [CrossRef]
- Bellas, V.; Iatrou, H.; Hadjichristidis, N. Controlled Anionic Polymerization of Hexamethylcyclotrisiloxane. Model Linear and Miktoarm Star Co- and Terpolymers of Dimethylsiloxane with Styrene and Isoprene. Macromolecules 2000, 33, 6993–6997. [Google Scholar] [CrossRef]
- Tsoukatos, T.; Hadjichristidis, N. Synthesis of model polycyclohexylene/polyethylene miktoarm star copolymers with three and four arms. J. Polym. Sci. Part A Polym. Chem. 2002, 40, 2575–2582. [Google Scholar] [CrossRef]
- Mavroudis, A.; Avgeropoulos, A.; Hadjichristidis, N.; Thomas, E.L.; Lohse, D.J. Synthesis and Morphological Behavior of Model Linear and Miktoarm Star Copolymers of 2-Methyl-1,3-Pentadiene and Styrene. Chem. Mater. 2003, 15, 1976–1983. [Google Scholar] [CrossRef]
- Avgeropoulos, A.; Poulos, Y.; Hadjichristidis, N.; Roovers, J. Synthesis of Model 16-Miktoarm (Vergina) Star Copolymers of the A8B8 Type. Macromolecules 1996, 29, 6076–6078. [Google Scholar] [CrossRef]
- Mavroudis, A.; Avgeropoulos, A.; Hadjichristidis, N.; Thomas, E.L.; Lohse, D.J. Synthesis and Morphological Behavior of Model 6-Miktoarm Star Copolymers, PS(P2MP)5, of Styrene (S) and 2-Methyl-1,3-Pentadiene (P2MP). Chem. Mater. 2006, 18, 2164–2168. [Google Scholar] [CrossRef]
- Tunca, U.; Ozyurek, Z.; Erdogan, T.; Hizal, G. Novel miktofunctional initiator for the preparation of an ABC-type miktoarm star polymer via a combination of controlled polymerization techniques. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 4228–4236. [Google Scholar] [CrossRef]
- Chado, G.R.; Holland, E.N.; Tice, A.K.; Stoykovich, M.P.; Kaar, J.L. Modification of Lipase with Poly(4-acryloylmorpholine) Enhances Solubility and Transesterification Activity in Anhydrous Ionic Liquids. Biomacromolecules 2018, 19, 1324–1332. [Google Scholar] [CrossRef]
- Xu, F.; Li, H.; Luo, Y.-L.; Tang, W. Redox-Responsive Self-Assembly Micelles from Poly(N-acryloylmorpholine-block-2-acryloyloxyethyl ferrocenecarboxylate) Amphiphilic Block Copolymers as Drug Release Carriers. ACS Appl. Mater. Interfaces 2017, 9, 5181–5192. [Google Scholar] [CrossRef] [PubMed]
- Neises, B.; Steglich, W. Simple Method for the Esterification of Carboxylic Acids. Angew. Chem. Int. Ed. 1978, 17, 522–524. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Yuan, Y.-Y.; Wang, Y.-C.; Du, J.-Z.; Wang, J. Synthesis of Amphiphilic ABC 3-Miktoarm Star Terpolymer by Combination of Ring-Opening Polymerization and “Click” Chemistry. Macromolecules 2008, 41, 8620–8625. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, H.; Hu, J.; Li, C.; Liu, S. Synthesis and Aggregation Behavior of Multi-Responsive Double Hydrophilic ABC Miktoarm Star Terpolymer. Macromol. Rapid Commun. 2009, 30, 941–947. [Google Scholar] [CrossRef]
- Ishizu, K.; Kuwahara, K. Synthesis of heteroarm star copolymers by anionic copolymerization of binary macromonomers. Polymer 1994, 35, 4907–4913. [Google Scholar] [CrossRef]
- Du, J.; Chen, Y. PCL Star Polymer, PCL-PS Heteroarm Star Polymer by ATRP, and Core-Carboxylated PS Star Polymer Thereof. Macromolecules 2004, 37, 3588–3594. [Google Scholar] [CrossRef]
- Gao, H.; Tsarevsky, N.V.; Matyjaszewski, K. Synthesis of Degradable Miktoarm Star Copolymers via Atom Transfer Radical Polymerization. Macromolecules 2005, 38, 5995–6004. [Google Scholar] [CrossRef]
- Zhang, D.; Fourie-O’Donohue, A.; Dragovich, P.S.; Pillow, T.H.; Sadowsky, J.D.; Kozak, K.R.; Cass, R.T.; Liu, L.; Deng, Y.; Liu, Y.; et al. Catalytic cleavage of disulfide bonds in small molecules and linkers of antibody- drug conjugates. Drug Metab. Dispos. 2019. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, G.; Li, R.; Guan, M.; Wang, X.; Zou, T.; Zhang, Y.; Wang, C.; Shu, C.; Hong, H.; et al. Biodegradable, Hydrogen Peroxide, and Glutathione Dual Responsive Nanoparticles for Potential Programmable Paclitaxel Release. J. Am. Chem. Soc. 2018, 140, 7373–7376. [Google Scholar] [CrossRef]
- Luo, C.; Sun, J.; Liu, D.; Sun, B.; Miao, L.; Musetti, S.; Li, J.; Han, X.; Du, Y.; Li, L.; et al. Self-Assembled Redox Dual-Responsive Prodrug-Nanosystem Formed by Single Thioether-Bridged Paclitaxel-Fatty Acid Conjugate for Cancer Chemotherapy. Nano Lett. 2016, 16, 5401–5408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiltshire, J.T.; Qiao, G.G. Selectively Degradable Core Cross-Linked Star Polymers. Macromolecules 2006, 39, 9018–9027. [Google Scholar] [CrossRef]
- Bates, M.W.; Barbon, S.M.; Levi, A.E.; Lewis, R.M.; Beech, H.K.; Vonk, K.M.; Zhang, C.; Fredrickson, G.H.; Hawker, C.J.; Bates, C.M. Synthesis and Self-Assembly of ABn Miktoarm Star Polymers. ACS Macro Lett. 2020, 9, 396–403. [Google Scholar] [CrossRef]
- Li, H.; Yang, D.; Gao, Y.; Li, H.; Xu, J. Dual responsive macroemulsion stabilized by Y-shaped amphiphilic AB2 miktoarm star copolymers. RSC Adv. 2015, 5, 96377–96386. [Google Scholar] [CrossRef]
- Englert, C.; Brendel, J.C.; Majdanski, T.C.; Yildirim, T.; Schubert, S.; Gottschaldt, M.; Windhab, N.; Schubert, U.S. Pharmapolymers in the 21st century: Synthetic polymers in drug delivery applications. Prog. Polym. Sci. 2018, 87, 107–164. [Google Scholar] [CrossRef]
- Brown, R.A.; Masters, A.J.; Price, C.; Yuan, X.F. 6-Chain Segregation in Block Copolymers. In Comprehensive Polymer Science and Supplements; Allen, G., Bevington, J.C., Eds.; Pergamon: Amsterdam, The Netherlands, 1989. [Google Scholar]
- Ryan, A.J.; Mai, S.-M.; Fairclough, J.P.A.; Hamley, I.W. Structures of amphiphilic block copolymers in their liquid and solid states. In Amphiphilic Block Copolymers; Alexandridis, P., Lindman, B., Eds.; Elsevier Science, B.V.: Amsterdam, The Netherlands, 2000; pp. 151–167. [Google Scholar]
- Discher, D.E.; Ahmed, F. Polymersomes. Annu. Rev. Biomed. Eng. 2006, 8, 323–341. [Google Scholar] [CrossRef]
- Kedracki, D.; Abraham, J.N.; Prado, E.; Nardin, C. Self-Assembly of Biohybrid Polymers. In Macromolecular Self-Assembly; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 193–229. [Google Scholar]
- Kakkar, A.; Traverso, G.; Farokhzad, O.C.; Weissleder, R.; Langer, R. Evolution of macromolecular complexity in drug delivery systems. Nat. Rev. Chem. 2017, 1, 63. [Google Scholar] [CrossRef]
- Oliver, R.C.; Lipfert, J.; Fox, D.A.; Lo, R.H.; Doniach, S.; Columbus, L. Dependence of Micelle Size and Shape on Detergent Alkyl Chain Length and Head Group. PLoS ONE 2013, 8, e62488. [Google Scholar] [CrossRef]
- Breyton, C.; Gabel, F.; Abla, M.; Pierre, Y.; Lebaupain, F.; Durand, G.; Popot, J.-L.; Ebel, C.; Pucci, B. Micellar and biochemical properties of (hemi)fluorinated surfactants are controlled by the size of the polar head. Biophys. J. 2009, 97, 1077–1086. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Zhang, X.-Z.; Zhou, Y.; Cheng, S.-X.; Zhuo, R.-X. Self-assembled thermoresponsive micelles of poly(N-isopropylacrylamide-b-methyl methacrylate). Biomaterials 2006, 27, 2028–2034. [Google Scholar] [CrossRef]
- Gou, J.; Feng, S.; Xu, H.; Fang, G.; Chao, Y.; Zhang, Y.; Xu, H.; Tang, X. Decreased Core Crystallinity Facilitated Drug Loading in Polymeric Micelles without Affecting Their Biological Performances. Biomacromolecules 2015, 16, 2920–2929. [Google Scholar] [CrossRef] [PubMed]
- Aliabadi, H.M.; Lavasanifar, A. Polymeric micelles for drug delivery. Expert Opin. Drug Del. 2006, 3, 139–162. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, T.; Chakraborty, I.; Ghosh, S. The methods of determination of critical micellar concentrations of the amphiphilic systems in aqueous medium. Arab. J. Chem. 2011, 4, 265–270. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Forrest, M.L.; Kwon, G.S.; Zhihong, X.X. Polymeric Micelles in Water-Insoluble Drug Delivery. In Water-Insoluble Drug Formulation, 3rd ed.; Liu, R., Ed.; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Huang, X.; Xiao, Y.; Lang, M. Synthesis and self-assembly behavior of six-armed block copolymers with pH- and thermo-responsive properties. Macromol. Res. 2011, 19, 113–121. [Google Scholar] [CrossRef]
- Chen, L.-J.; Lin, S.-Y.; Huang, C.-C. Effect of Hydrophobic Chain Length of Surfactants on Enthalpy−Entropy Compensation of Micellization. J. Phys. Chem. B 1998, 102, 4350–4356. [Google Scholar] [CrossRef]
- Zhao, S.; Yang, H.; Zuo, C.; Sun, L.; Ma, L.; Wei, H. pH-sensitive drug release of star-shaped micelles with OEG brush corona. RSC Adv. 2016, 6, 111217–111225. [Google Scholar] [CrossRef]
- Soppimath, K.S.; Tan, D.C.-W.; Yang, Y.-Y. pH-Triggered Thermally Responsive Polymer Core–Shell Nanoparticles for Drug Delivery. Adv. Mater. 2005, 17, 318–323. [Google Scholar] [CrossRef]
- Li, H.; Diao, M.; Zhang, S.; Wang, K.; Xue, C. Novel polymeric micelles of AB2 type alpha-methoxy-poly(ethylene glycol)-b-Poly(gamma-benzyl-L-glutamate), copolymers as tamoxifen carriers. J. Nanosci. Nanotechnol. 2009, 9, 4805–4811. [Google Scholar] [CrossRef]
- Zhu, Y.-J.; Chen, F. pH-Responsive Drug-Delivery Systems. Chem. Asian J. 2015, 10, 284–305. [Google Scholar] [CrossRef]
- Karimi, M.; Zangabad, F.S.; Ghasemi, A.; Amiri, M.; Bahrami, M.; Malekzad, H.; Asl, H.G.; Mahdieh, Z.; Bozorgomid, M.; Ghasemi, A.; et al. Temperature-Responsive Smart Nanocarriers for Delivery Of Therapeutic Agents: Applications and Recent Advances. ACS Appl. Mater. Interfaces 2016, 8, 21107–21133. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Zhou, Y.; Liu, X.; Chen, Y.; Duan, S.; Zhu, R.; Liu, Y.; Yin, L. Recent Advances on Reactive Oxygen Species-Responsive Delivery and Diagnosis System. Biomacromolecules 2019, 20, 2441–2463. [Google Scholar] [CrossRef] [PubMed]
- Burhans, W.C.; Heintz, N.H. The cell cycle is a redox cycle: Linking phase-specific targets to cell fate. Free Radic. Biol. Med. 2009, 47, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Ballatori, N.; Krance, S.M.; Notenboom, S.; Shi, S.; Tieu, K.; Hammond, C.L. Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem. 2009, 390, 191–214. [Google Scholar] [CrossRef] [Green Version]
- Balendiran, G.K.; Dabur, R.; Fraser, D. The role of glutathione in cancer. Cell Biochem. Funct. 2004, 22, 343–352. [Google Scholar] [CrossRef]
- Quinn, J.F.; Whittaker, M.R.; Davis, T.P. Glutathione responsive polymers and their application in drug delivery systems. Polym. Chem. 2017, 8, 97–126. [Google Scholar] [CrossRef]
- Wang, X.; Liu, L.; Luo, Y.; Shi, H.; Li, J.; Zhao, H. Comb-Shaped Glycopolymer/Peptide Bioconjugates by Combination of RAFT Polymerization and Thiol-Ene “Click” Chemistry. Macromol. Biosci. 2012, 12, 1575–1582. [Google Scholar] [CrossRef] [PubMed]
- Linsley, C.S.; Wu, B.M. Recent advances in light-responsive on-demand drug-delivery systems. Ther. Deliv. 2017, 8, 89–107. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Ye, H.; Chen, Y.; Zhu, R.; Yin, L. Photoresponsive Drug/Gene Delivery Systems. Biomacromolecules 2018, 19, 1840–1857. [Google Scholar] [CrossRef]
- Wei, H.; Cheng, S.-X.; Zhang, X.-Z.; Zhuo, R.-X. Thermo-sensitive polymeric micelles based on poly(N-isopropylacrylamide) as drug carriers. Prog. Polym. Sci. 2009, 34, 893–910. [Google Scholar] [CrossRef]
- Corrie, J.E.; Barth, A.; Munasinghe, V.R.; Trentham, D.R.; Hutter, M.C. Photolytic cleavage of 1-(2-nitrophenyl)ethyl ethers involves two parallel pathways and product release is rate-limited by decomposition of a common hemiacetal intermediate. J. Am. Chem. Soc. 2003, 125, 8546–8554. [Google Scholar] [CrossRef]
- Gaplovsky, M.; Il’ichev, Y.V.; Kamdzhilov, Y.; Kombarova, S.V.; Mac, M.; Schwörer, M.A.; Wirz, J. Photochemical reaction mechanisms of 2-nitrobenzyl compounds: 2-Nitrobenzyl alcohols form 2-nitroso hydrates by dual proton transfer. Photochem. Photobiol. Sci. 2005, 4, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xu, Y.; Wang, B.; Qiao, W.; Liu, D.; Li, Z. Cationic compounds used in lipoplexes and polyplexes for gene delivery. J. Control. Release 2004, 100, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Blencowe, A.; Tan, J.F.; Goh, T.K.; Qiao, G.G. Core cross-linked star polymers via controlled radical polymerisation. Polymer 2009, 50, 5–32. [Google Scholar] [CrossRef] [Green Version]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymeric Arms 1,2 | Architecture | Stimulus | Cargo | Encapsulation Efficiency (%) | Loading Capacity (%) | Year | Citation |
|---|---|---|---|---|---|---|---|
| PNIPAM, PUA | AB2 | Temperature | Prednisone acetate | 27.7 | N/A | 2006 | [27] |
| PEG, PCL, P2VP | ABC | pH | Nile Red | N/A | N/A | 2006, 2009, 2012 | [28,29,30] |
| PMMA, PNIPAM | AB3 | Temperature | Prednisone acetate | 55 | N/A | 2007 | [31] |
| PEG, PTMC | AB2 | Indomethacin | 27.3–56.6 | 9.1–21.4 | 2008 | [32] | |
| PEG, PCL | A2B2 | Ibuprofen | 26.8–89.7 | 7.3–20.3 | 2009 | [33] | |
| PEG, PLLA | AB2 | Doxorubicin Hydrochloride | 72 | N/A | 2009 | [34] | |
| PEG, PLLA, PDLA | ABC, AB2 | Paclitaxel | N/A | 5.0–11.6 | 2009 | [35] | |
| PCL, PEG | A14B7 | Ibuprofen | 7.7–46.0 | 2.3–13.8 | 2010 | [36] | |
| PEG, PCL | A2B | Nimodipine | 23–70 | 2.3–7.0 | 2010 | [24] | |
| PEG, PCL | AB10 | Prednisone acetate | 21.1–44.5 | 2.1–4.3 | 2010 | [37] | |
| PNIPAM, PLL | AB2 | Prednisone acetate | 18.6–21.4 | 2.1–2.5 | 2010 | [38] | |
| PEG, PS, PCL | ABC | Disperse Red 1 | N/A | 0.4–2.1 | 2010 | [39] | |
| PEG, PCL | AB2, A(BA)2 | Rhodamine B isothiocyanate-Dextran | 40–57 | N/A | 2011 | [40] | |
| PEG, Niacin, BODIPY | A2B, ABC | Niacin | N/A | N/A | 2011 | [41] | |
| PEG, PCL, TPPBr | ABC | Coenzyme Q10 | 83–85 | 8.3–8.5 | 2012 | [42] | |
| PEG, PHis | AB2 | pH | 5(6)-carboxyfluorescein | N/A | 0.92–1.42 μL mg−1 | 2012, 2014 | [43,44] |
| PCL, PEG, PLL | ABC | Paclitaxel, plasmid DNA | N/A | 5.0 | 2013 | [45] | |
| PAzo, PDEAA | AB3 | UV light, Temperature | Nile Red | N/A | N/A | 2013 | [46] |
| PEG, PCL | AB2 | Curcumin | 48.5–65.0 | 4.6–6.5 | 2014 | [47] | |
| PCL, PBLA, PEG | ABC | Doxorubicin | 56.2 | 11.2 | 2014 | [48] | |
| PEG, PCL, TIF | ABC | Curcumin | 39.2–55.9 | 3.9–5.6 | 2014 | [49] | |
| PCL, PDEAEMA-b-PPEGMA | A2(BC)2 | pH | Doxorubicin | N/A | 10 | 2014 | [50] |
| PCL, PDEAEMA-b-PPEGMA | A3(BC)3 | pH | Doxorubicin | 29.4–71.4 | 9.5–19.6 | 2014 | [51] |
| PEG, PGA | AB2 | pH | Doxorubicin | N/A | 16.2–18.2 | 2014 | [52] |
| PAzo, PEG | AB3 | UV light | Nile Red, Rhodamine B, | N/A | N/A | 2014 | [53] |
| PEG, PCL | AB2 | Doxorubicin | 41.6–72.7 | 6.1–16.1 | 2015 | [54] | |
| PCL, PEG | AB3 | Curcumin | 17.3–27.6 | 11.4–13.3 | 2015 | [55] | |
| PEG, PAA, PCL | ABC | pH | Naproxen | 58.2–72.8 | 9.7–12.1 | 2015 | [56] |
| PEG, PEG-b-PCL | A(AB)3 A2(AB)2 A3(AB) | Redox | Camptothecin | N/A | 3.6–10.8 | 2015 | [57] |
| PEG, PMMA | AB2 | Redox | Methotrexate | 64 | 16 | 2015 | [58] |
| PCL, PCL-b-PEG | A(AB)3 | Doxorubicin | 52.8–54.6 | 8.8–9.1 | 2016 | [59] | |
| PEG, PCL | AB2 | Doxorubicin | 24.7 | 5.6 | 2016 | [60] | |
| PEG, PHLG | A16B23 CCS | siRNA | N/A | N/A | 2016 | [61] | |
| PEG, P(MMA-co-MAA) | AB2 | pH | Methotrexate | 48.7–82.3 | 10.3–16.5 | 2016 | [62] |
| PCL, P(MAA-co-MMA) | A2B6 | pH | Doxorubicin, Camptothecin | DOX: 42–60 CPT: 6 | DOX: 14–20 CPT: 2 | 2016 | [63] |
| PEG, qPDMAEMA | AB4 | pH, Ionic strength | Rhodamine B, FITC-dextran | N/A | N/A | 2016 | [64] |
| PPEGMA, PMMA, PNIPAM | (BA)(AC)2 | Temperature | Celecoxib | N/A | 8.8 | 2016 | [65] |
| PEG, PNBM, PNIPAM | ABC | UV light, Temperature | Nile Red | N/A | N/A | 2017 | [66] |
| PEG, PGA | AB3 | Lysozyme | N/A | N/A | 2017, 2018 | [67,68] | |
| PVDF, PS, PEG | (AB)2C2 | Nile Red | N/A | N/A | 2018 | [69] | |
| PCL, POEGMA | A2B, A2B2, AB3, A3B | Doxorubicin | 34.3–62.9 | 4.4–8.1 | 2018 | [70] | |
| PEG, PCL | AB2 | Curcumin, Methotrexate | CUR: 93.8–94.2 MTX: 72.9–75.7 | CUR: 14.1 MTX: 10.9–11.4 | 2018 | [71] | |
| PLLA, PNAM | AB2 | Doxorubicin | 55.6–78.7 | 9.4–13.6 | 2018 | [72] | |
| PEG, PLL | AB2 | Redox | pDNA | N/A | N/A | 2018 | [73] |
| PEG, PCL | AB2 | Redox | Chlorin e6, Doxorubicin | Ce6: 77.7–82.1 DOX: 16.2–29.2 | Ce6: 15.5–16.4 DOX: 3.2–5.8 | 2018 | [74] |
| PLGA, PEG | AB2 | Ibuprofen | 51.5–76.5 | 1.7–7.0 | 2019 | [75] | |
| PEG, PGA | AB3 | Lysozyme | N/A | N/A | 2019 | [76] | |
| OA, PEG | AB2 | pH | Vancomycin | 39.6 | 3.6 | 2020 | [77] |
| PEG, PCL | AB2 | Baicalein | 94.3–94.7 | 12.4 | 2020 | [78] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lotocki, V.; Kakkar, A. Miktoarm Star Polymers: Branched Architectures in Drug Delivery. Pharmaceutics 2020, 12, 827. https://doi.org/10.3390/pharmaceutics12090827
Lotocki V, Kakkar A. Miktoarm Star Polymers: Branched Architectures in Drug Delivery. Pharmaceutics. 2020; 12(9):827. https://doi.org/10.3390/pharmaceutics12090827
Chicago/Turabian StyleLotocki, Victor, and Ashok Kakkar. 2020. "Miktoarm Star Polymers: Branched Architectures in Drug Delivery" Pharmaceutics 12, no. 9: 827. https://doi.org/10.3390/pharmaceutics12090827
APA StyleLotocki, V., & Kakkar, A. (2020). Miktoarm Star Polymers: Branched Architectures in Drug Delivery. Pharmaceutics, 12(9), 827. https://doi.org/10.3390/pharmaceutics12090827