tRNALys-Derived Fragment Alleviates Cisplatin-Induced Apoptosis in Prostate Cancer Cells



Abstract
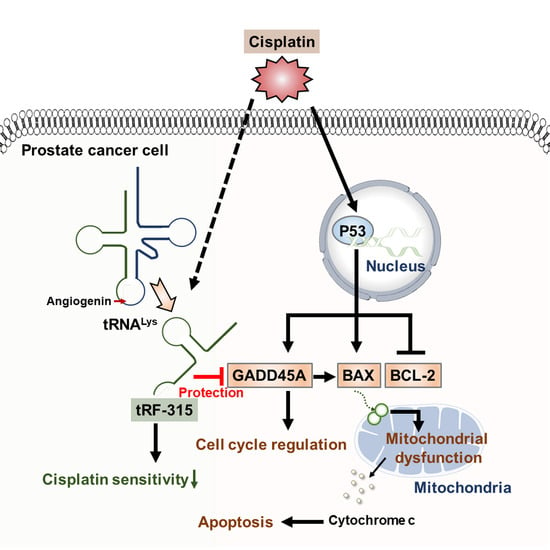
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Detection of tRFs
2.3. Quantitative RT-PCR for tRFs
2.4. Transfection
2.5. BrdU Incorporation Analysis
2.6. Annexin V and Propidium Iodide Staining
2.7. Quantitative RT-PCR
2.8. Western Blotting
2.9. Analysis of Mitochondrial Membrane Potential
2.10. Cell Cycle Analysis
2.11. Statistical Analysis
3. Results
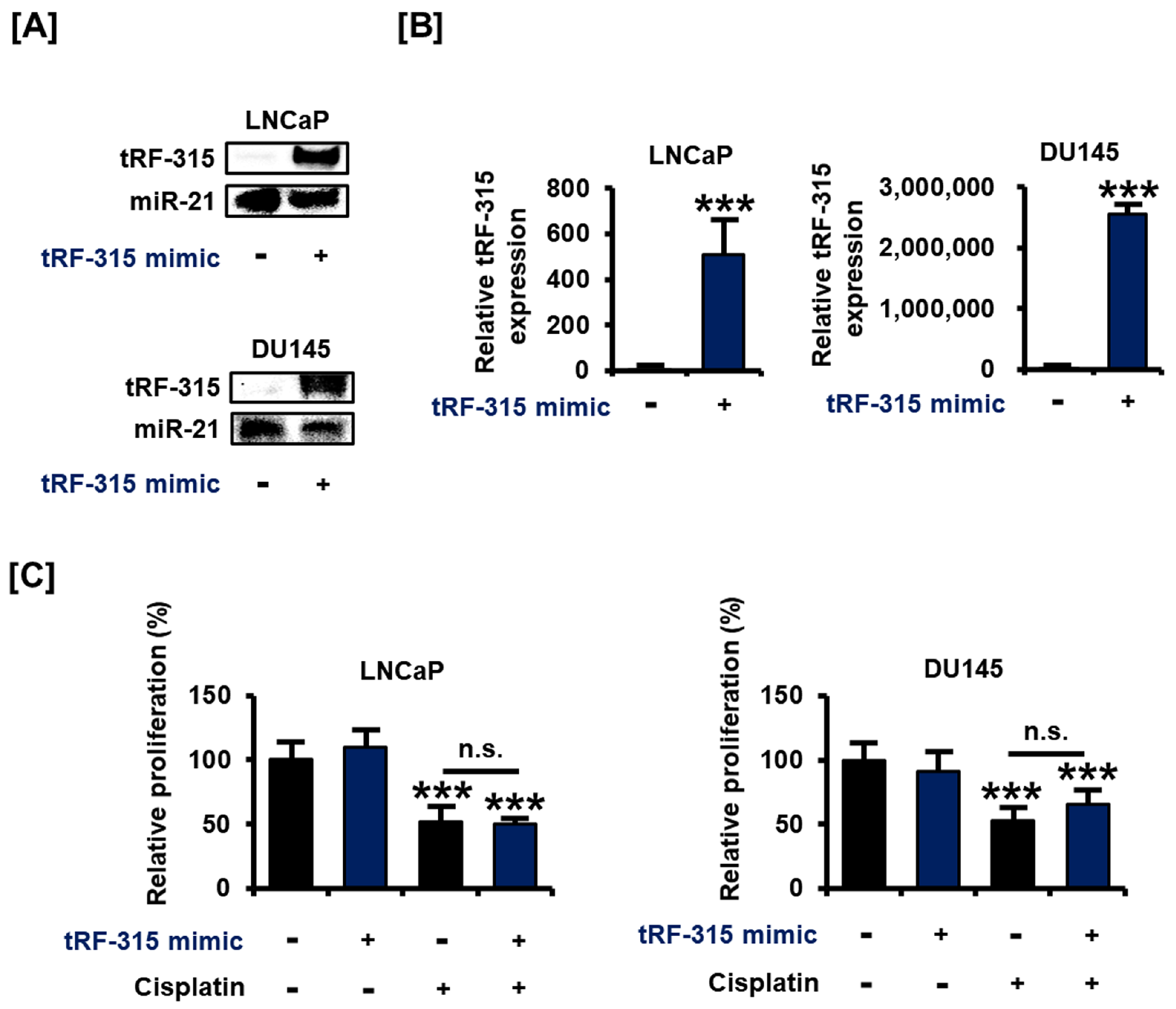
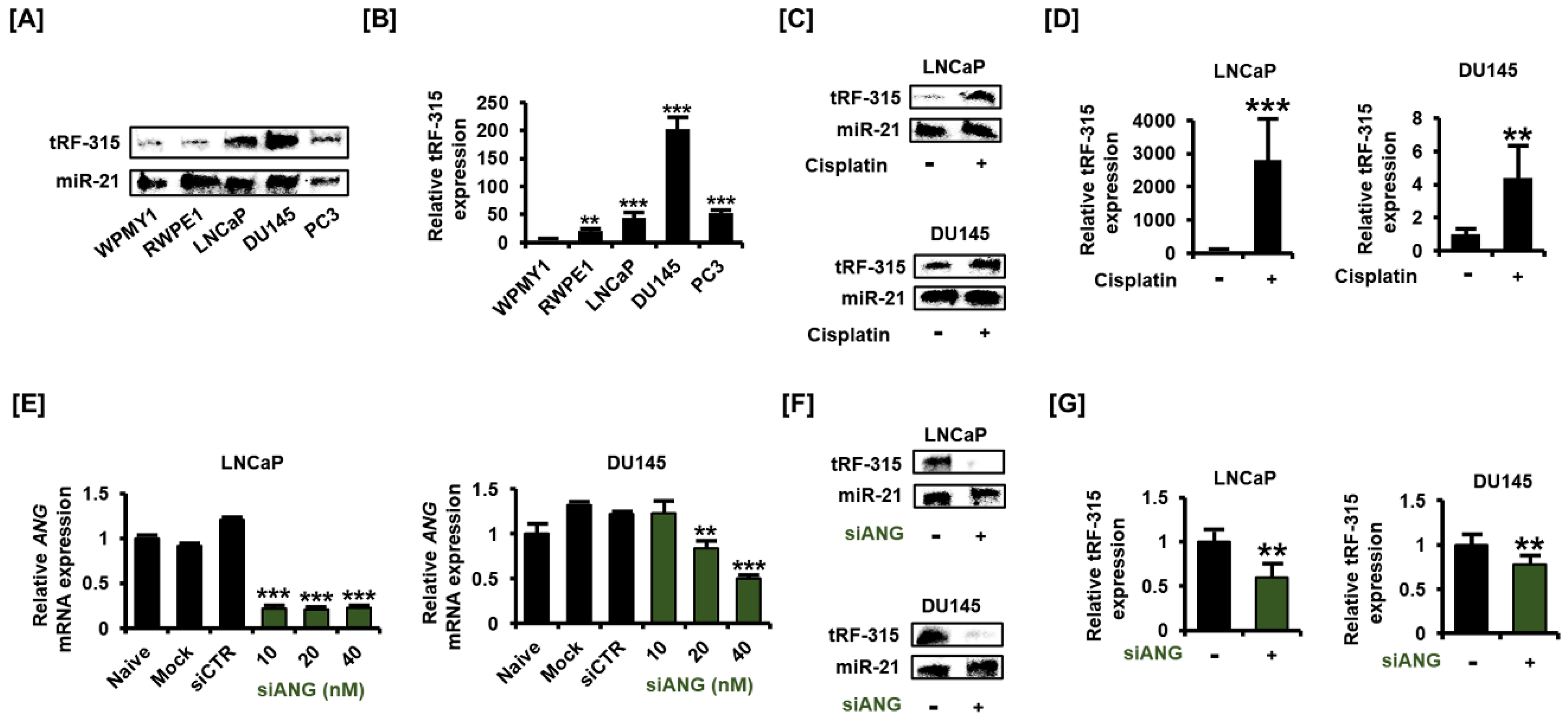
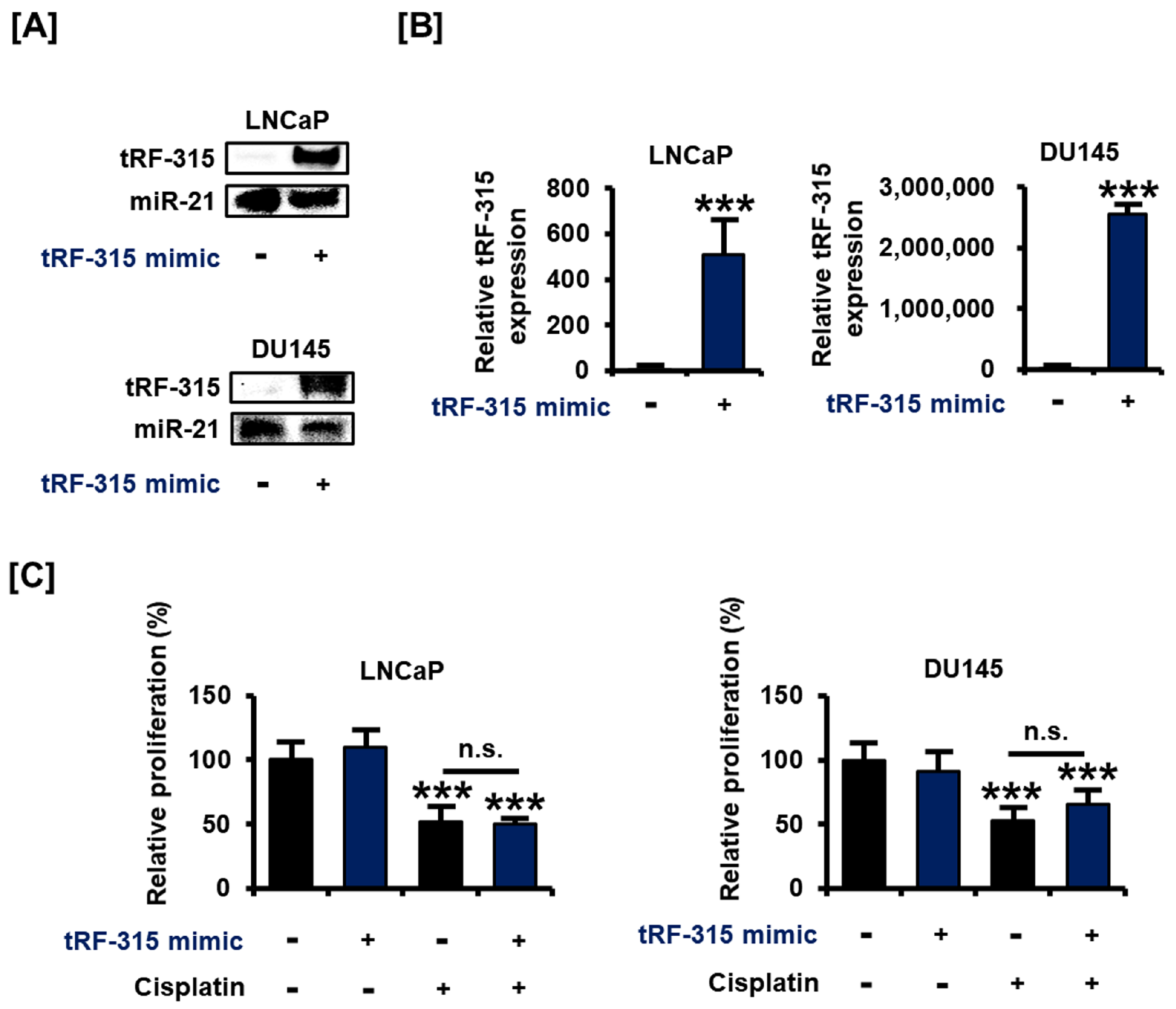
3.1. tRF-315 Is More Abundant in Prostate Cancer Cells than in Normal Prostate Cells
3.2. Transfection with tRF-315 Mimics Does Not Affect Prostate Cancer Cell Proliferation but Leads to the Induction of Prostate Cancer Cell Death
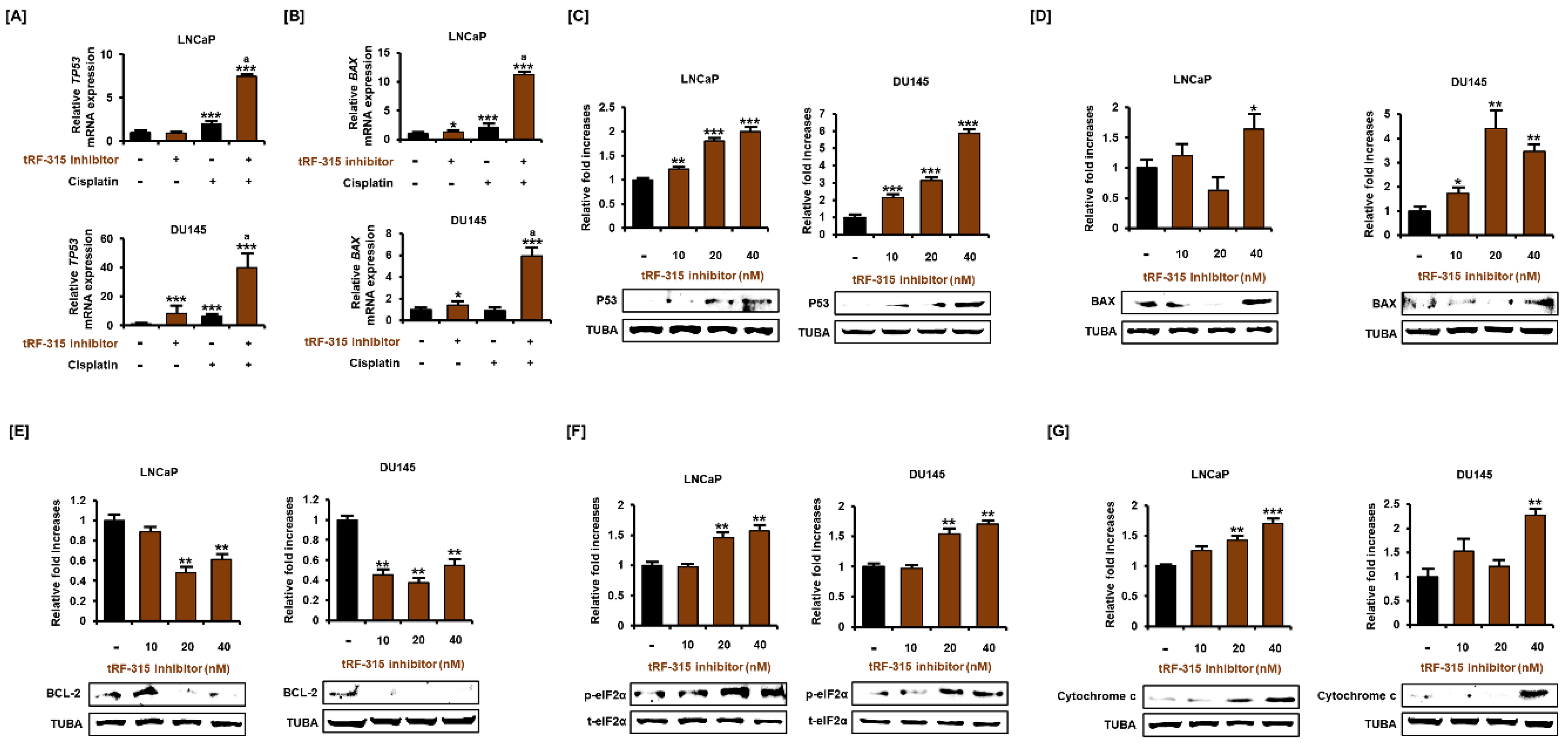
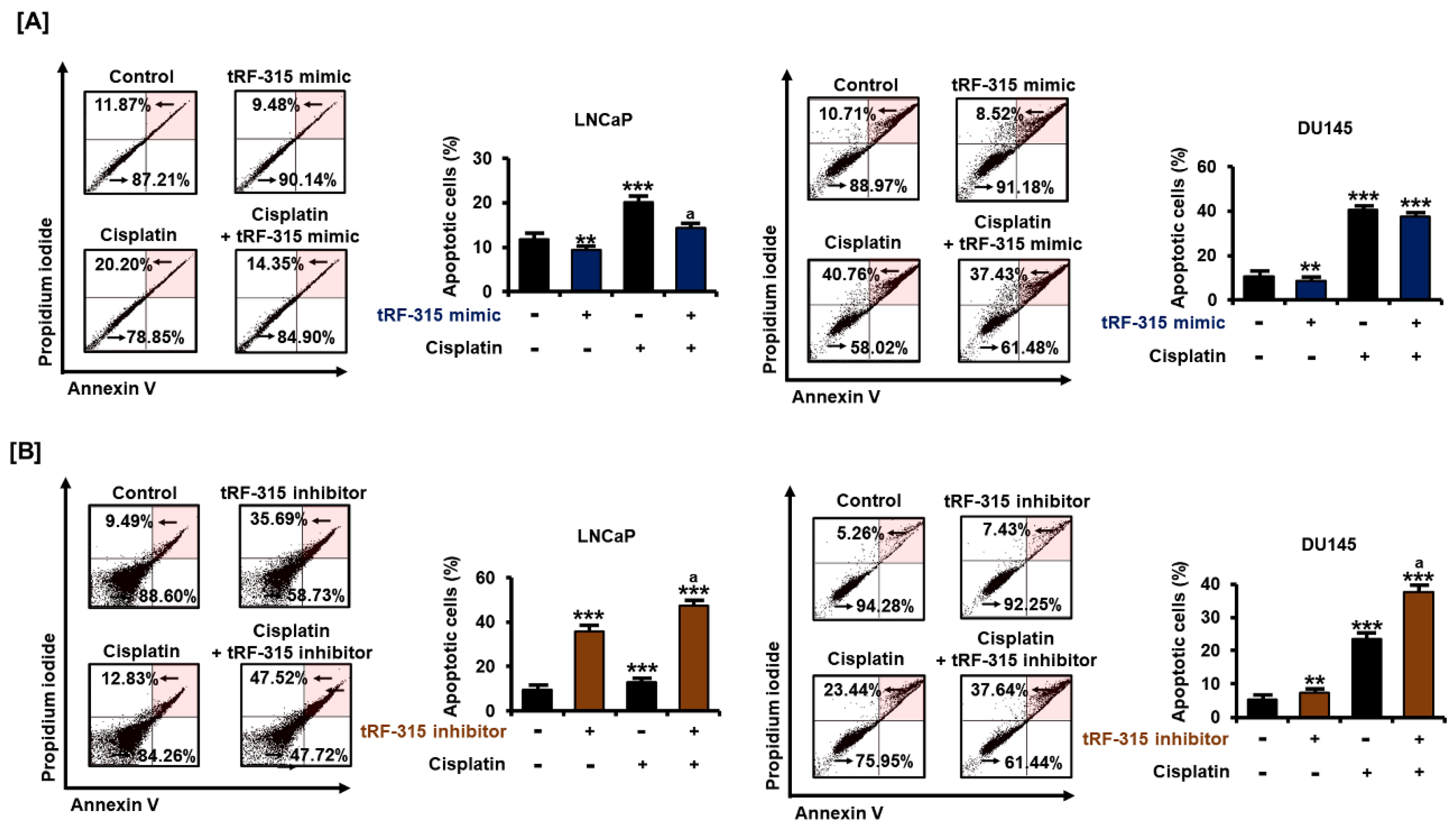
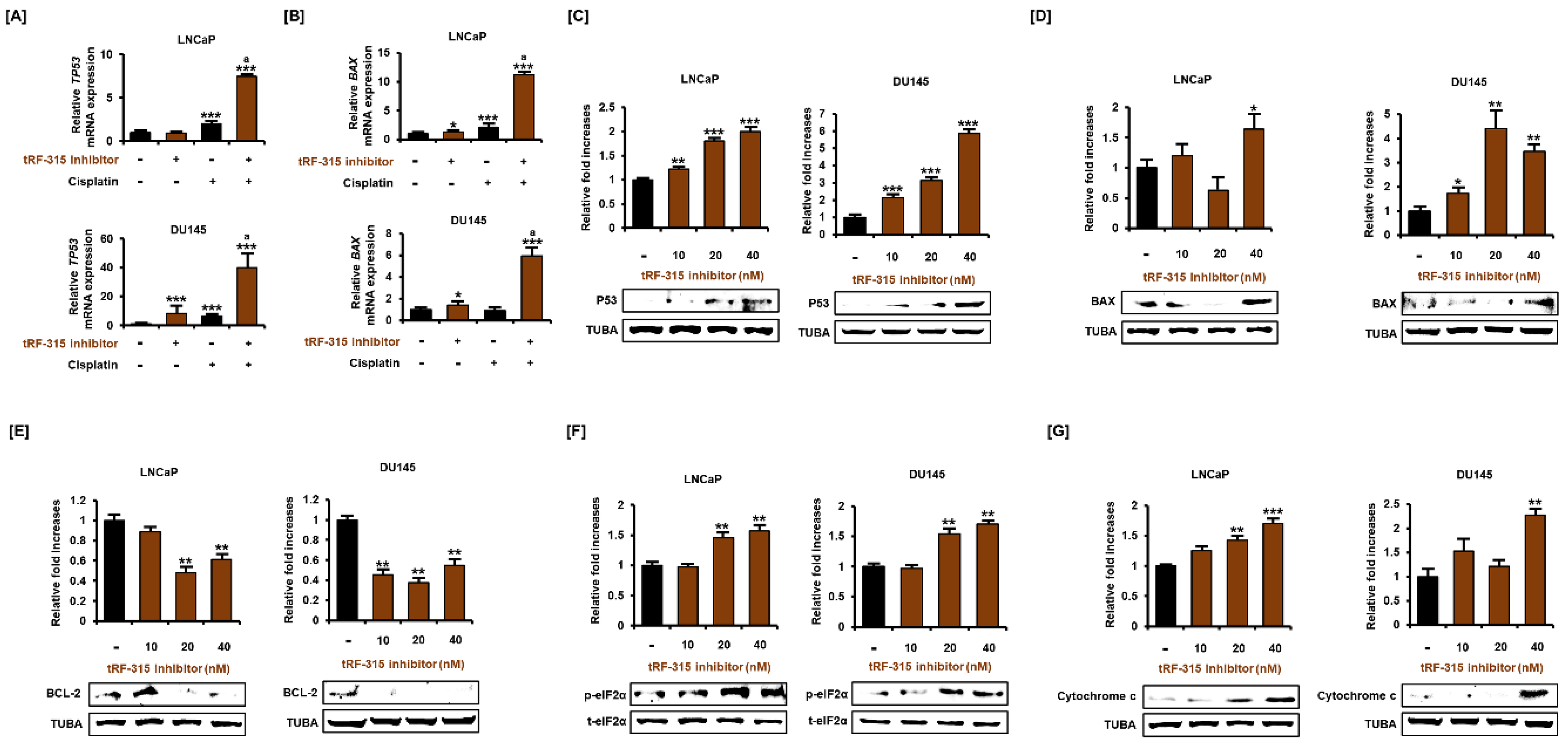
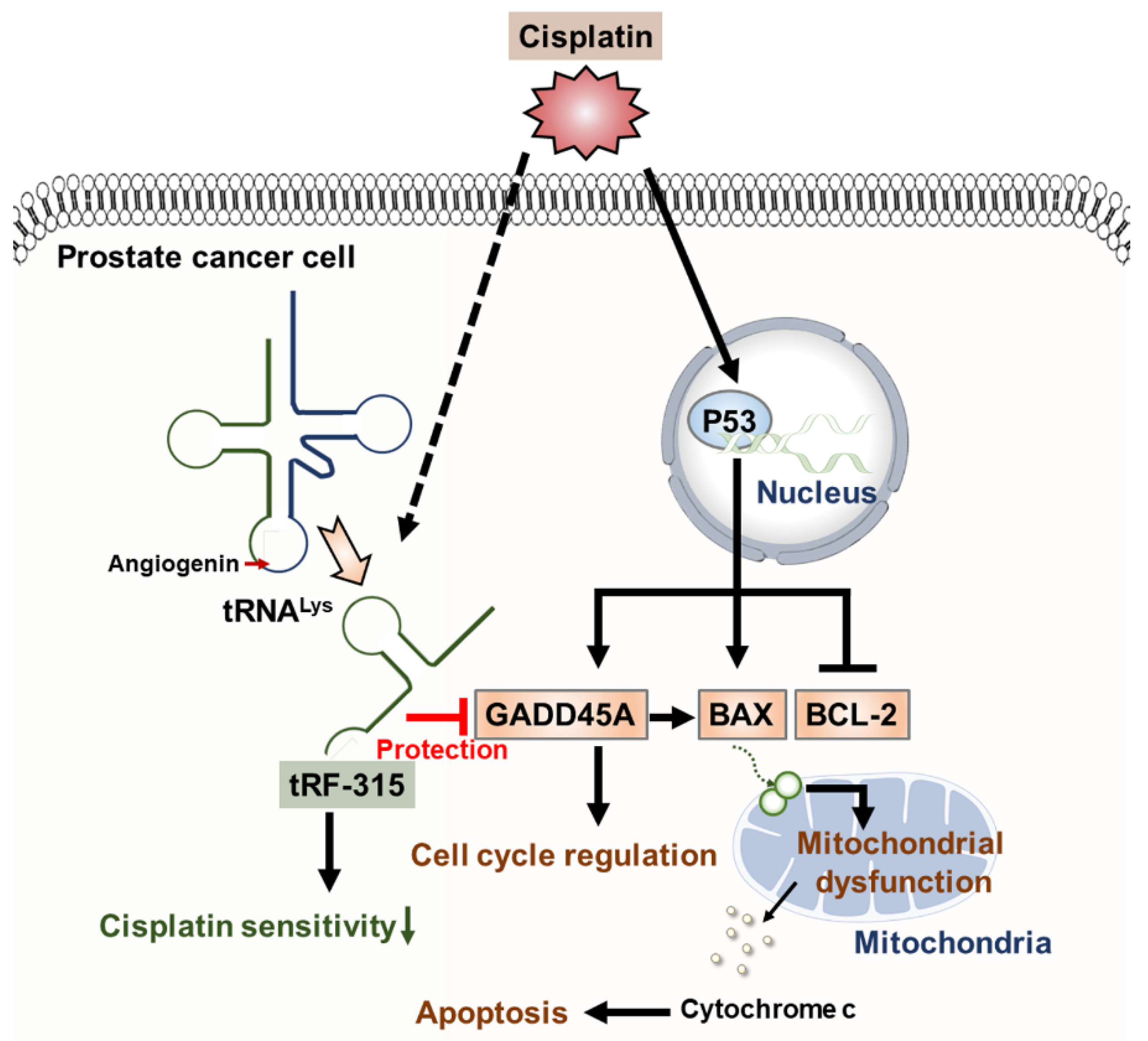
3.3. Expression of tRF-315 Is Involved in the Sensitivity to Cisplatin-Induced Apoptotic Pathway in Prostate Cancer Cells
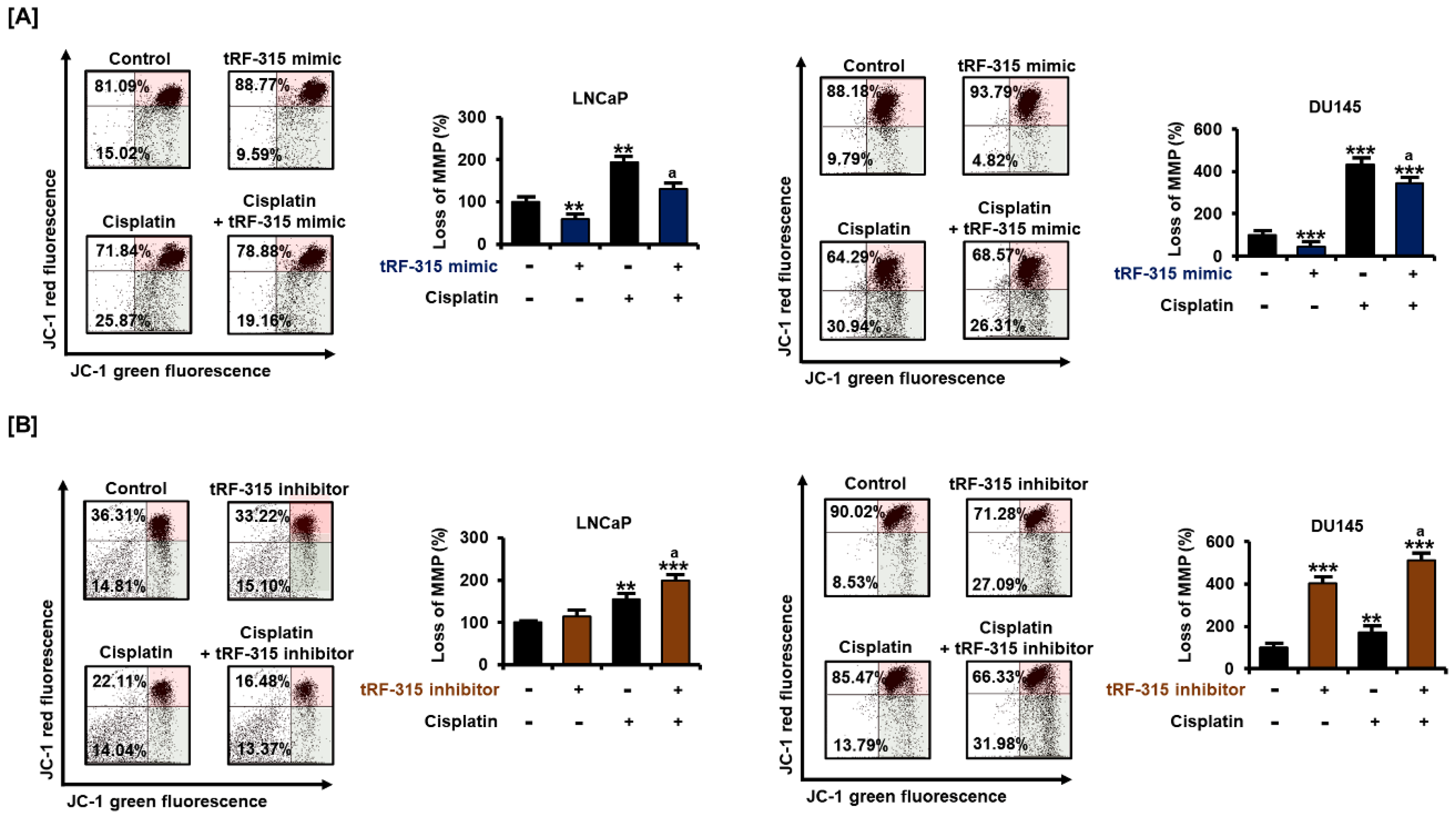
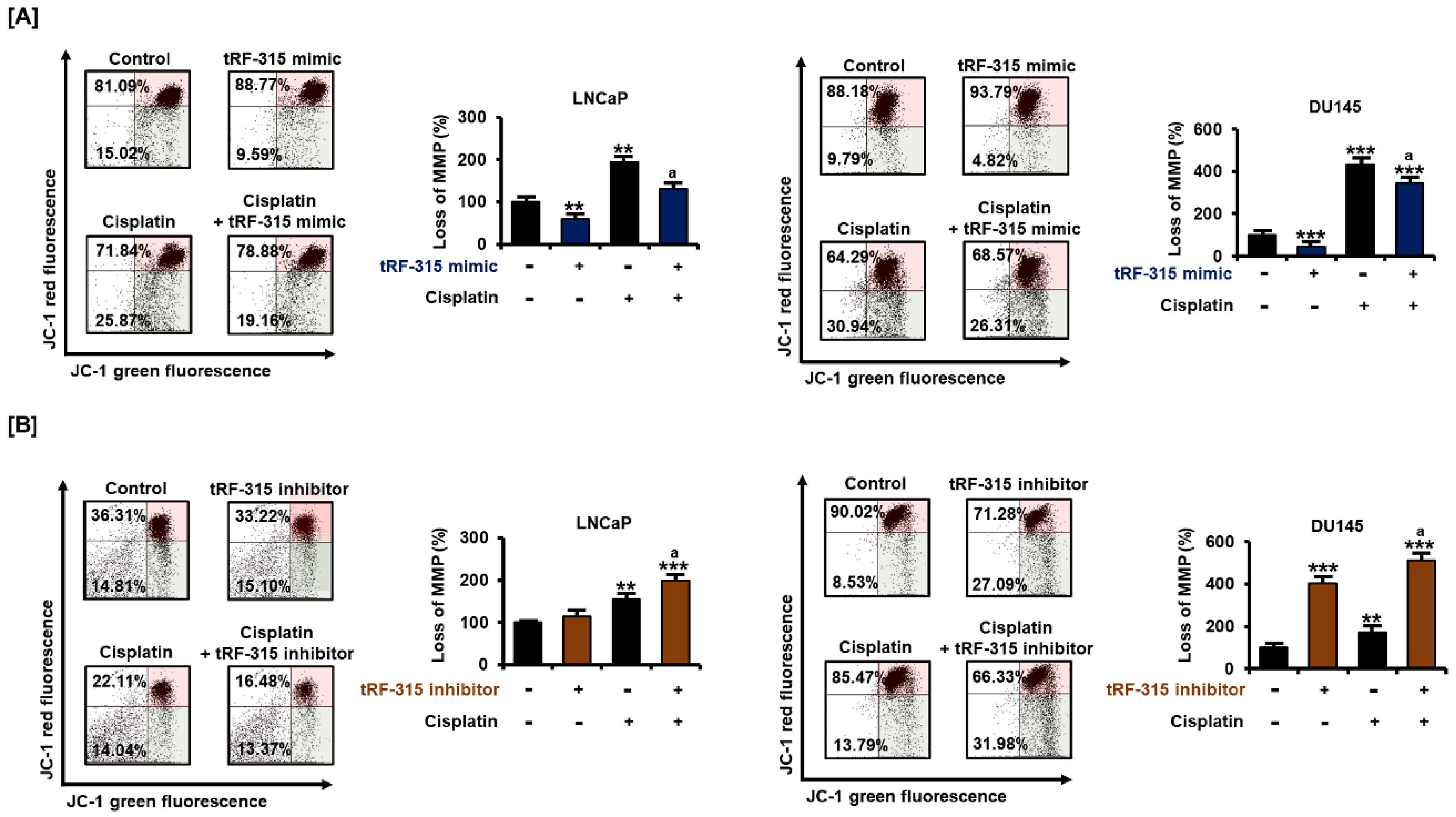
3.4. tRF-315 Alleviates Mitochondrial Dysfunction Induced by Cisplatin in Prostate Cancer Cells
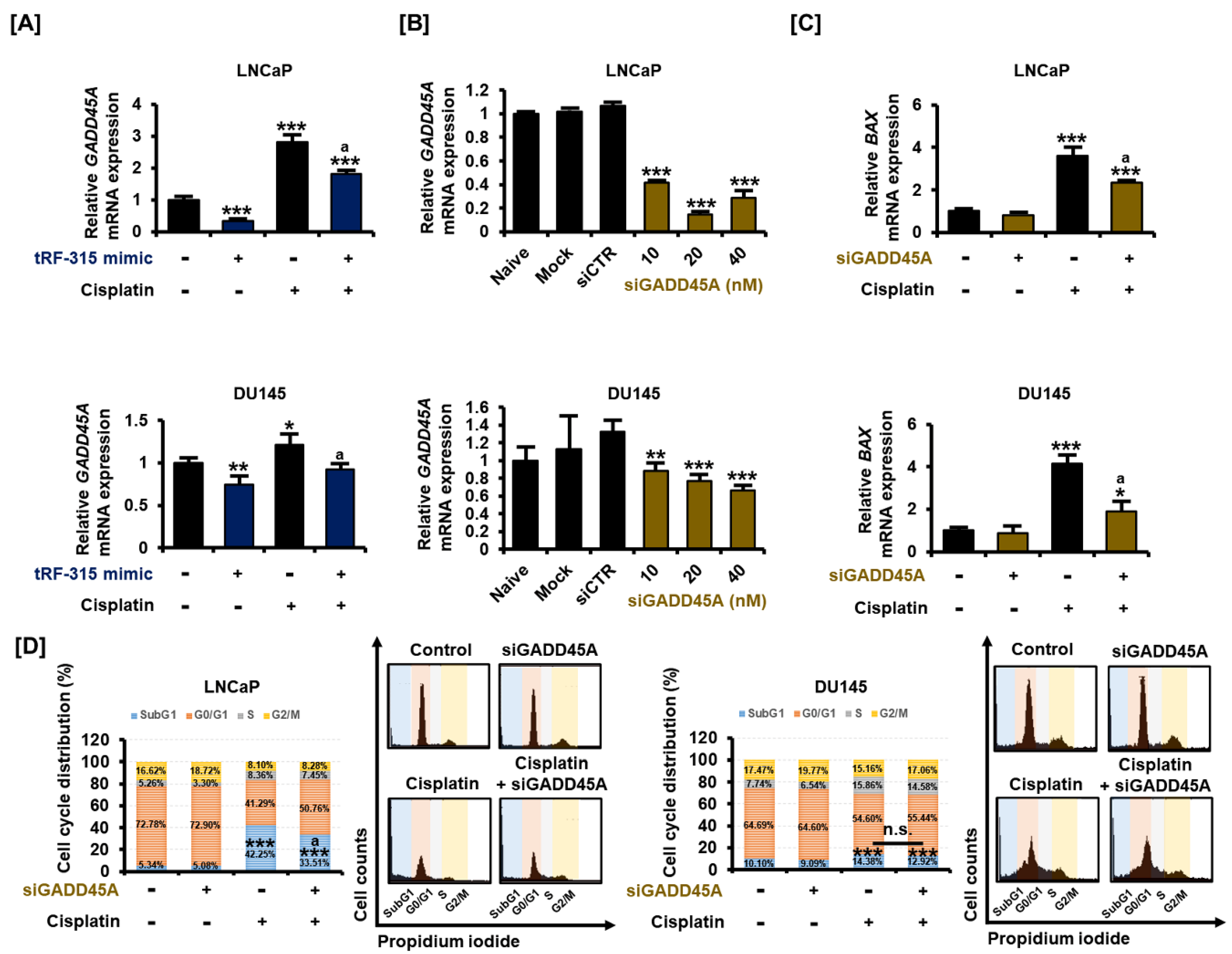
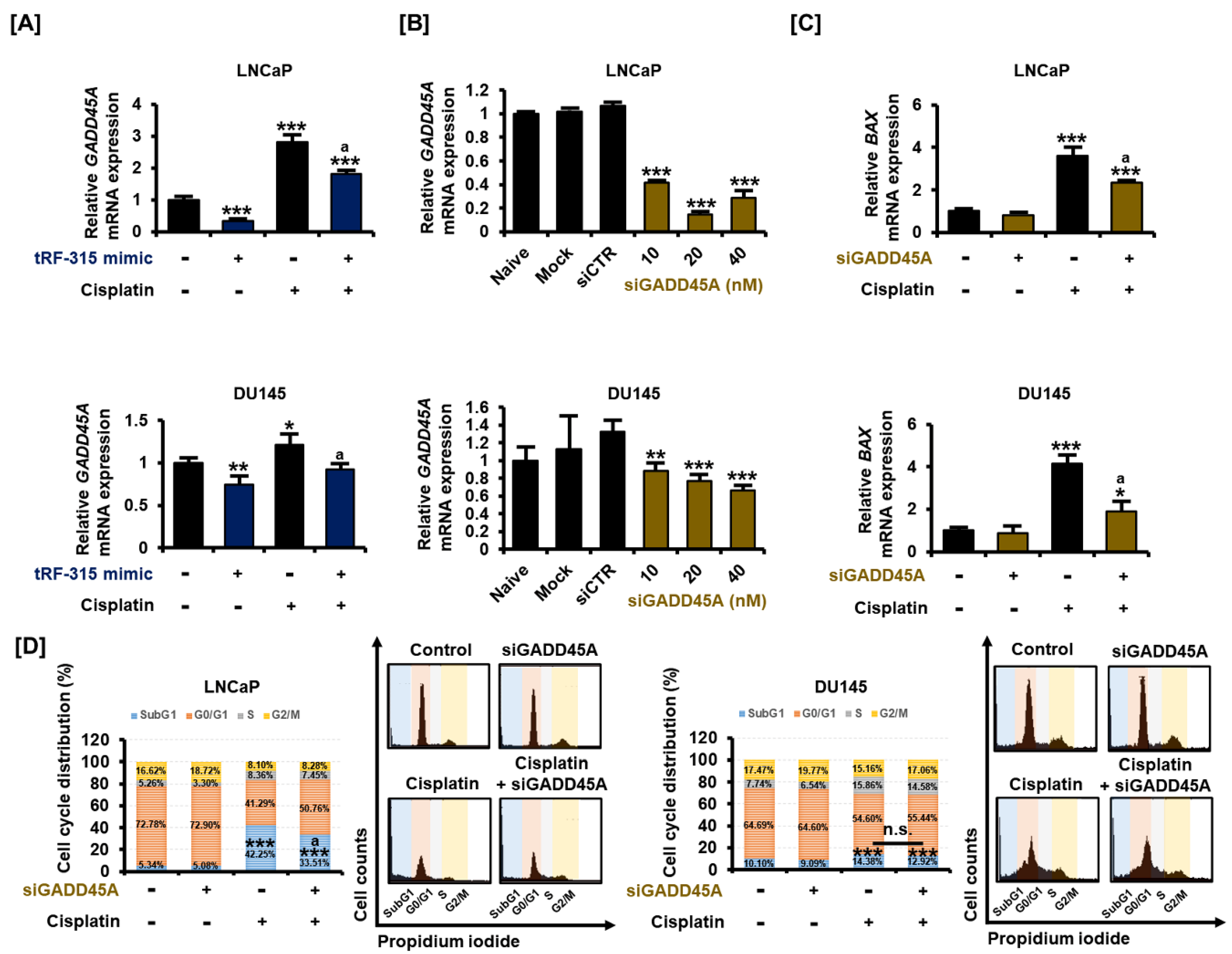
3.5. As a Target of tRF-315, GADD45A Is Involved in Cell Cycle Regulation by Cisplatin in Prostate Cancer Cells
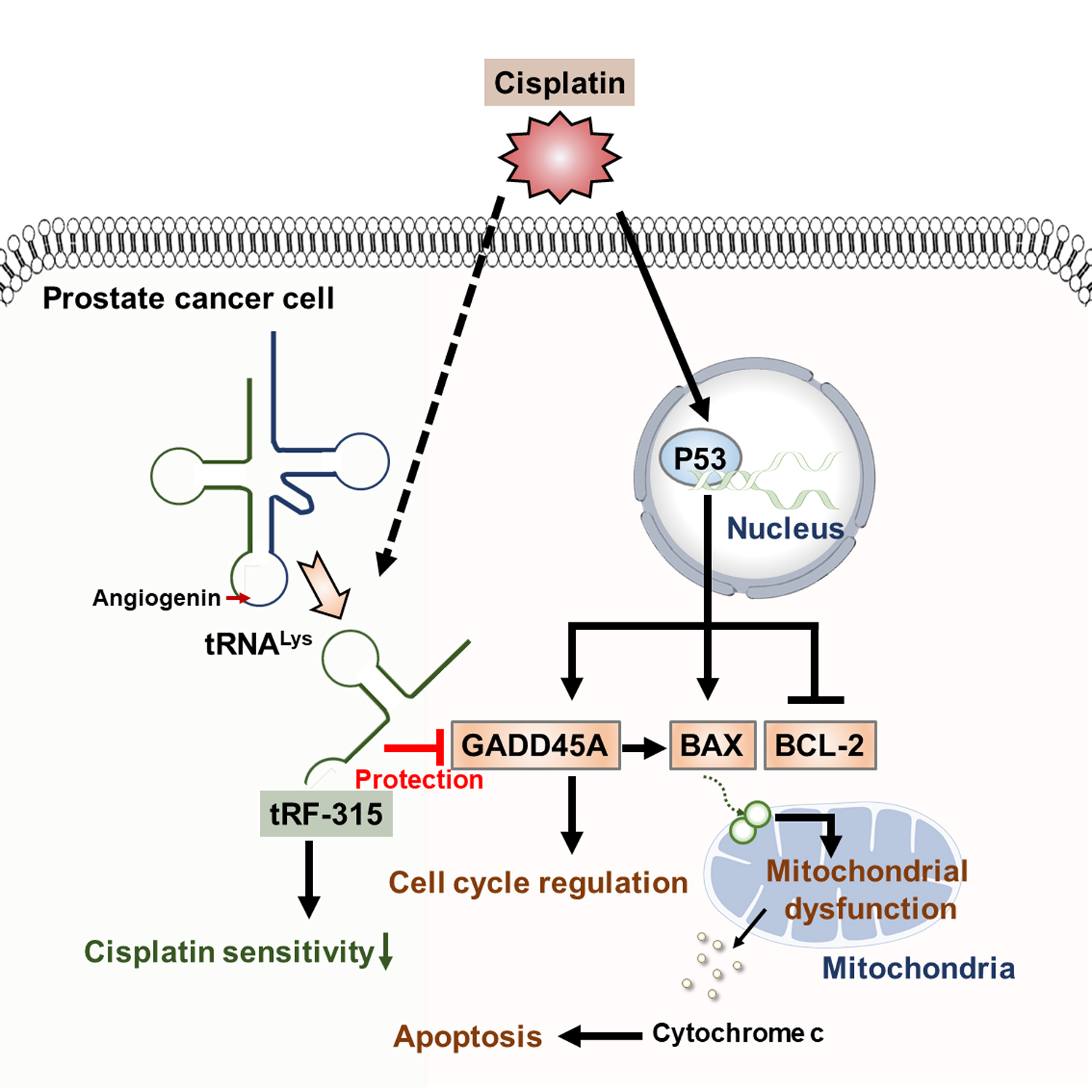
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, K.C.; Tsui, K.H.; Chung, L.C.; Yeh, C.N.; Feng, T.H.; Chen, W.T.; Chang, P.L.; Chiang, H.Y.; Juang, H.H. Cisplatin modulates B-cell translocation gene 2 to attenuate cell proliferation of prostate carcinoma cells in both p53-dependent and p53-independent pathways. Sci. Rep. 2014, 4, 5511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gumulec, J.; Balvan, J.; Sztalmachova, M.; Raudenska, M.; Dvorakova, V.; Knopfova, L.; Polanska, H.; Hudcova, K.; Ruttkay-Nedecky, B.; Babula, P.; et al. Cisplatin-resistant prostate cancer model: Differences in antioxidant system, apoptosis and cell cycle. Int. J. Oncol. 2014, 44, 923–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, M.; Bajo-Santos, C.; Hessvik, N.P.; Lorenz, S.; Fromm, B.; Berge, V.; Sandvig, K.; Line, A.; Llorente, A. Identification of non-invasive miRNAs biomarkers for prostate cancer by deep sequencing analysis of urinary exosomes. Mol. Cancer 2017, 16, 156. [Google Scholar] [CrossRef]
- Kanwal, R.; Plaga, A.R.; Liu, X.; Shukla, G.C.; Gupta, S. MicroRNAs in prostate cancer: Functional role as biomarkers. Cancer Lett. 2017, 407, 9–20. [Google Scholar] [CrossRef]
- Martens-Uzunova, E.S.; Olvedy, M.; Jenster, G. Beyond microRNA—Novel RNAs derived from small non-coding RNA and their implication in cancer. Cancer Lett. 2013, 340, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Romano, G.; Veneziano, D.; Acunzo, M.; Croce, C.M. Small non-coding RNA and cancer. Carcinogenesis 2017, 38, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Qin, D.; Li, H.; Xie, H. Ultrasoundtargeted microbubble destructionmediated miR205 enhances cisplatin cytotoxicity in prostate cancer cells. Mol. Med. Rep. 2018, 18, 3242–3250. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Ma, L.; Zhou, J.; Jiang, M.; Rao, E.; Zhao, Y.; Guo, F. miR-17-92 plays an oncogenic role and conveys chemo-resistance to cisplatin in human prostate cancer cells. Int. J. Oncol. 2016, 48, 1737–1748. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Wang, Q.; Liu, Y.; Xia, Z.Y. miR-425-5p suppresses tumorigenesis and DDP resistance in human-prostate cancer by targeting GSK3beta and inactivating the Wnt/beta-catenin signaling pathway. J. Biosci. 2019, 44, 102. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Shibata, Y.; Malhotra, A.; Dutta, A. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev. 2009, 23, 2639–2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, W.; Wang, X.; Cai, X.; Xiong, W.; Liu, Y.; Li, C.; Liu, Q.; Qin, J.; Li, Y. Identification of tRNAderived fragments in colon cancer by comprehensive small RNA sequencing. Oncol. Rep. 2019, 42, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zhu, C.; Qin, X. Expression profile of tRNA-derived fragments in pancreatic cancer. Oncol. Lett. 2019, 18, 3104–3114. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, Y.; Tan, X.; Mao, X.; Wei, D.; Yao, Y.; Jiang, P.; Mo, D.; Wang, T.; Yan, F. Identification of tRNA-Derived Fragments Expression Profile in Breast Cancer Tissues. Curr. Genom. 2019, 20, 199–213. [Google Scholar] [CrossRef]
- Olvedy, M.; Scaravilli, M.; Hoogstrate, Y.; Visakorpi, T.; Jenster, G.; Martens-Uzunova, E.S. A comprehensive repertoire of tRNA-derived fragments in prostate cancer. Oncotarget 2016, 7, 24766–24777. [Google Scholar] [CrossRef]
- Magee, R.G.; Telonis, A.G.; Loher, P.; Londin, E.; Rigoutsos, I. Profiles of miRNA Isoforms and tRNA Fragments in Prostate Cancer. Sci. Rep. 2018, 8, 5314. [Google Scholar] [CrossRef] [Green Version]
- Honda, S.; Loher, P.; Shigematsu, M.; Palazzo, J.P.; Suzuki, R.; Imoto, I.; Rigoutsos, I.; Kirino, Y. Sex hormone-dependent tRNA halves enhance cell proliferation in breast and prostate cancers. Proc. Natl. Acad. Sci. USA 2015, 112, E3816–E3825. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Gong, P.; Li, J.; Yang, J.; Zhang, G.; Li, H.; Yang, Z.; Zhang, X. Simple and nonradioactive detection of microRNAs using digoxigenin (DIG)-labeled probes with high sensitivity. RNA 2014, 20, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Lai, Y.; Li, Y.; Shu, N.; Wang, Z.; Wang, Y.; Li, Y.; Chen, Z. Antineoplastic activity of isoliquiritigenin, a chalcone compound, in androgen-independent human prostate cancer cells linked to G2/M cell cycle arrest and cell apoptosis. Eur. J. Pharmacol. 2018, 821, 57–67. [Google Scholar] [CrossRef]
- Yang, C.; Lim, W.; Bazer, F.W.; Song, G. Oleic acid stimulation of motility of human extravillous trophoblast cells is mediated by stearoyl-CoA desaturase-1 activity. Mol. Hum. Reprod. 2017, 23, 755–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chappell, W.H.; Lehmann, B.D.; Terrian, D.M.; Abrams, S.L.; Steelman, L.S.; McCubrey, J.A. p53 expression controls prostate cancer sensitivity to chemotherapy and the MDM2 inhibitor Nutlin-3. Cell Cycle 2012, 11, 4579–4588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.K.; Kim, Y.S. Phosphorylation of eIF2alpha attenuates statin-induced apoptosis by inhibiting the stabilization and translocation of p53 to the mitochondria. Int. J. Oncol. 2013, 42, 810–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, S.M.; Jo, Y.S.; Jeon, Y.M.; Pae, H.O.; Kang, D.G.; Lee, H.S.; Bae, J.S.; Jeon, B.H. Phosphorylation of eIF2alpha suppresses cisplatin-induced p53 activation and apoptosis by attenuating oxidative stress via ATF4-mediated HO-1 expression in human renal proximal tubular cells. Int. J. Mol. Med. 2017, 40, 1957–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florea, A.M.; Busselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef]
- Moriyama-Gonda, N.; Shiina, H.; Terashima, M.; Satoh, K.; Igawa, M. Rationale and clinical implication of combined chemotherapy with cisplatin and oestrogen in prostate cancer: Primary evidence based on methylation analysis of oestrogen receptor-alpha. BJU Int. 2008, 101, 485–491. [Google Scholar] [CrossRef]
- Barabas, K.; Milner, R.; Lurie, D.; Adin, C. Cisplatin: A review of toxicities and therapeutic applications. Vet. Comp. Oncol. 2008, 6, 1–18. [Google Scholar] [CrossRef]
- Wade, C.A.; Kyprianou, N. Profiling Prostate Cancer Therapeutic Resistance. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [Green Version]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, J.; Afridi, A.; Vatsia, S.; Joshi, G.; Joshi, G.; Kaplan, S.A.; Smith, N.L.; Khan, S.A. The molecular biology of prostate cancer: Current understanding and clinical implications. Prostate Cancer Prostatic Dis. 2018, 21, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.P.; Tong, S.J.; Wu, Y.P.; Qu, L.X.; Ding, Q. miR-181 regulation of BAX controls cisplatin sensitivity of prostate cancer cells. Int. J. Clin. Exp. Pathol. 2017, 10, 10127–10133. [Google Scholar] [PubMed]
- Zhang, L.; Li, X.; Chao, Y.; He, R.; Liu, J.; Yuan, Y.; Zhao, W.; Han, C.; Song, X. KLF4, a miR-32-5p targeted gene, promotes cisplatin-induced apoptosis by upregulating BIK expression in prostate cancer. Cell Commun. Signal. 2018, 16, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Ge, J.; Li, T.; Shen, Y.; Guo, J. tRNA-derived fragments and tRNA halves: The new players in cancers. Cancer Lett. 2019, 452, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ender, C.; Meister, G.; Moore, P.S.; Chang, Y.; John, B. Extensive terminal and asymmetric processing of small RNAs from rRNAs, snoRNAs, snRNAs, and tRNAs. Nucleic Acids Res. 2012, 40, 6787–6799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Anaya, J.; Mudunuri, S.B.; Dutta, A. Meta-analysis of tRNA derived RNA fragments reveals that they are evolutionarily conserved and associate with AGO proteins to recognize specific RNA targets. BMC Biol. 2014, 12, 78. [Google Scholar] [CrossRef]
- Eastman, A. Improving anticancer drug development begins with cell culture: Misinformation perpetrated by the misuse of cytotoxicity assays. Oncotarget 2017, 8, 8854–8866. [Google Scholar] [CrossRef] [Green Version]
- Nawrot, B.; Sochacka, E.; Duchler, M. tRNA structural and functional changes induced by oxidative stress. Cell Mol. Life Sci. 2011, 68, 4023–4032. [Google Scholar] [CrossRef] [Green Version]
- Thompson, D.M.; Lu, C.; Green, P.J.; Parker, R. tRNA cleavage is a conserved response to oxidative stress in eukaryotes. RNA 2008, 14, 2095–2103. [Google Scholar] [CrossRef] [Green Version]
- Falconi, M.; Giangrossi, M.; Zabaleta, M.E.; Wang, J.; Gambini, V.; Tilio, M.; Bencardino, D.; Occhipinti, S.; Belletti, B.; Laudadio, E.; et al. A novel 3′-tRNA(Glu)-derived fragment acts as a tumor suppressor in breast cancer by targeting nucleolin. FASEB J. 2019, 33, 13228–13240. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Hada, K.; Shiraishi, H.; Yatsuka, H.; Fujinami, H.; Morisaki, I.; Nishida, Y.; Matsubara, E.; Ishitani, T.; Hanada, R.; et al. Tyrosine pre-transfer RNA fragments are linked to p53-dependent neuronal cell death via PKM2. Biochem. Biophys. Res. Commun. 2020, 525, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Tan, Z.; Gan, M.; Li, Q.; Chen, L.; Niu, L.; Jiang, D.; Zhao, Y.; Wang, J.; Li, X.; et al. tRNA-Derived Small Non-Coding RNAs as Novel Epigenetic Molecules Regulating Adipogenesis. Biomolecules 2019, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodarzi, H.; Liu, X.; Nguyen, H.C.; Zhang, S.; Fish, L.; Tavazoie, S.F. Endogenous tRNA-Derived Fragments Suppress Breast Cancer Progression via YBX1 Displacement. Cell 2015, 161, 790–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, K.; Gopisetty, G.; Gordian, E.; Navarro, L.; Hader, C.; Reis, I.M.; Schulz, W.A.; Singal, R. Methylation-mediated repression of GADD45alpha in prostate cancer and its role as a potential therapeutic target. Cancer Res. 2009, 69, 1527–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tront, J.S.; Huang, Y.; Fornace, A.J., Jr.; Hoffman, B.; Liebermann, D.A. Gadd45a functions as a promoter or suppressor of breast cancer dependent on the oncogenic stress. Cancer Res. 2010, 70, 9671–9681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, L.Y.; Xin, H.Y.; Liu, Y.L.; Zhang, J.L.; Xin, H.W.; Su, X.L. Anticancer bioactive peptide (ACBP) inhibits gastric cancer cells by upregulating growth arrest and DNA damage-inducible gene 45A (GADD45A). Tumour. Biol. 2014, 35, 10051–10056. [Google Scholar] [CrossRef]
- Jin, S.; Mazzacurati, L.; Zhu, X.; Tong, T.; Song, Y.; Shujuan, S.; Petrik, K.L.; Rajasekaran, B.; Wu, M.; Zhan, Q. Gadd45a contributes to p53 stabilization in response to DNA damage. Oncogene 2003, 22, 8536–8540. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Q. Gadd45a, a p53- and BRCA1-regulated stress protein, in cellular response to DNA damage. Mutat. Res. 2005, 569, 133–143. [Google Scholar] [CrossRef]
- Liu, L.Q.; Tian, F.J.; Xiong, Y.; Zhao, Y.; Song, J.B. Gadd45a gene silencing by RNAi promotes cell proliferation and inhibits apoptosis and senescence in skin squamous cell carcinoma through the p53 signaling pathway. J. Cell Physiol. 2018, 233, 7424–7434. [Google Scholar] [CrossRef]
- Reis, I.M.; Ramachandran, K.; Speer, C.; Gordian, E.; Singal, R. Serum GADD45a methylation is a useful biomarker to distinguish benign vs. malignant prostate disease. Br. J. Cancer 2015, 113, 460–468. [Google Scholar] [CrossRef] [Green Version]
- Arencibia, J.M.; Del Rio, M.; Bonnin, A.; Lopes, R.; Lemoine, N.R.; Lopez-Barahona, M. Doxazosin induces apoptosis in LNCaP prostate cancer cell line through DNA binding and DNA-dependent protein kinase down-regulation. Int. J. Oncol. 2005, 27, 1617–1623. [Google Scholar] [PubMed]
- Satomi, Y. Fucoxanthin induces GADD45A expression and G1 arrest with SAPK/JNK activation in LNCap human prostate cancer cells. Anticancer Res. 2012, 32, 807–813. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibodies | Dilution | Supplier | Catalog Number |
|---|---|---|---|
| P53 | 1:1000 | Cell Signaling Technology | 2527 |
| BAX | 1:1000 | Cell Signaling Technology | 2772 |
| BCL-2 | 1:1000 | Cell Signaling Technology | 2876 |
| Phosphor-eIF2α | 1:1000 | Cell Signaling Technology | 3398 |
| eIF2α | 1:1000 | Cell Signaling Technology | 5324 |
| Cytochrome c | 1:1000 | Cell Signaling Technology | 11940 |
| TUBA | 1:1000 | Santa Cruz | sc-5286 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.; Lee, M.; Song, G.; Lim, W. tRNALys-Derived Fragment Alleviates Cisplatin-Induced Apoptosis in Prostate Cancer Cells. Pharmaceutics 2021, 13, 55. https://doi.org/10.3390/pharmaceutics13010055
Yang C, Lee M, Song G, Lim W. tRNALys-Derived Fragment Alleviates Cisplatin-Induced Apoptosis in Prostate Cancer Cells. Pharmaceutics. 2021; 13(1):55. https://doi.org/10.3390/pharmaceutics13010055
Chicago/Turabian StyleYang, Changwon, Minkyeong Lee, Gwonhwa Song, and Whasun Lim. 2021. "tRNALys-Derived Fragment Alleviates Cisplatin-Induced Apoptosis in Prostate Cancer Cells" Pharmaceutics 13, no. 1: 55. https://doi.org/10.3390/pharmaceutics13010055
APA StyleYang, C., Lee, M., Song, G., & Lim, W. (2021). tRNALys-Derived Fragment Alleviates Cisplatin-Induced Apoptosis in Prostate Cancer Cells. Pharmaceutics, 13(1), 55. https://doi.org/10.3390/pharmaceutics13010055