Improved Manufacturability and In Vivo Comparative Pharmacokinetics of Dapagliflozin Cocrystals in Beagle Dogs and Human Volunteers


Abstract
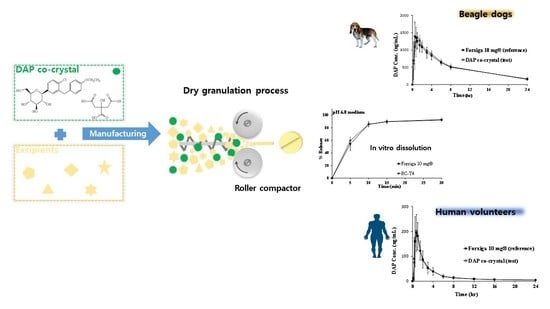
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. DAP Cocrystal Characterization
2.2.1. X-ray Diffraction (XRD)
2.2.2. Fourier Transform Infrared Spectroscopy (FTIR)
2.2.3. Differential Scanning Calorimetry (DSC)
2.2.4. Thermogravimetric Analysis (TGA)
2.2.5. Dynamic Vapor Sorption (DVS)
2.3. Solubility Test
2.4. Appearance and Stability Test of API
2.5. Scanning Electron Microscopy (SEM)
2.6. Manufacturing DAP Cocrystal Tablets by Direct Compression
2.6.1. DAP Cocrystal Tablets Preparation
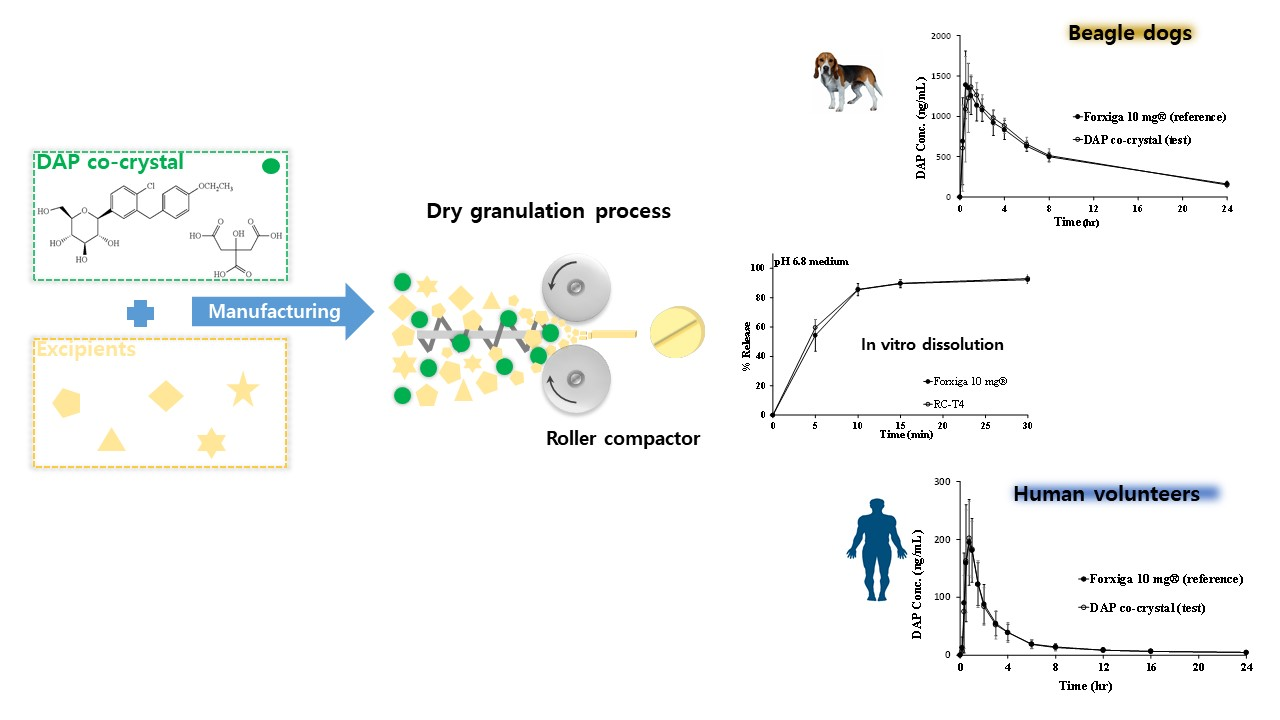
2.6.2. DAP Cocrystal Milling Effect
2.7. Manufacturing DAP Cocrystal Tablets by Roller Compaction
2.7.1. Preparation Process of DAP Cocrystal Tablets
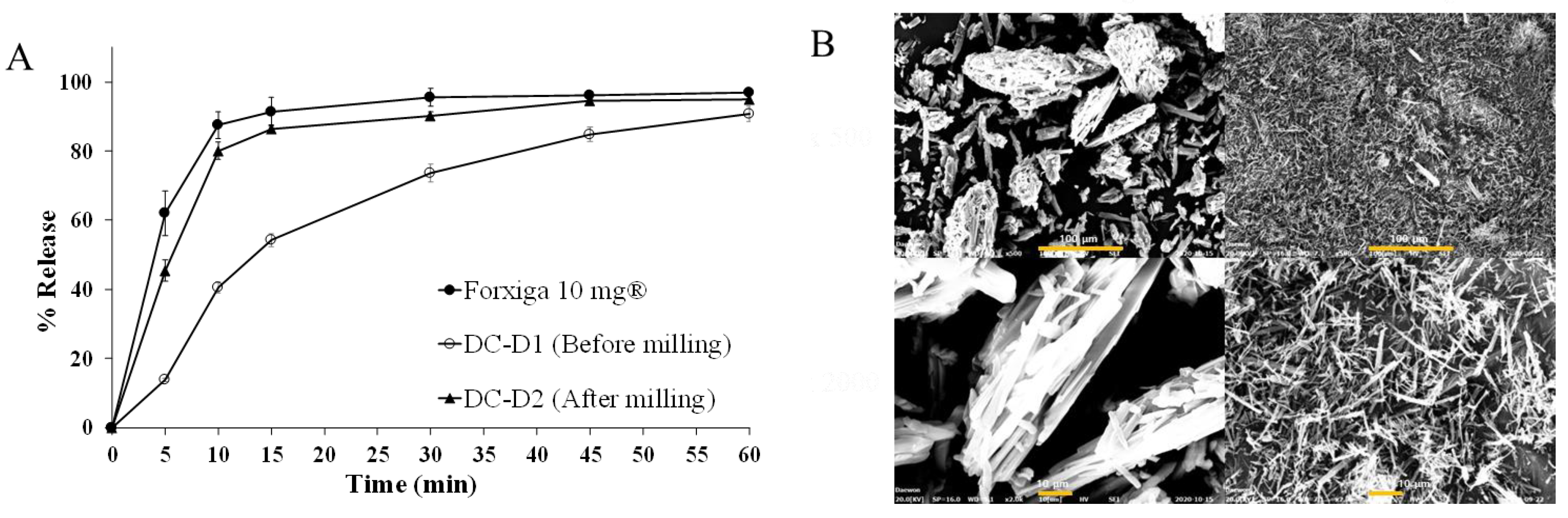
2.7.2. RC Manufacturing Optimization
2.7.3. Effect of Tablet Hardness on DAP Cocrystal Release
2.7.4. Amount of Disintegrant
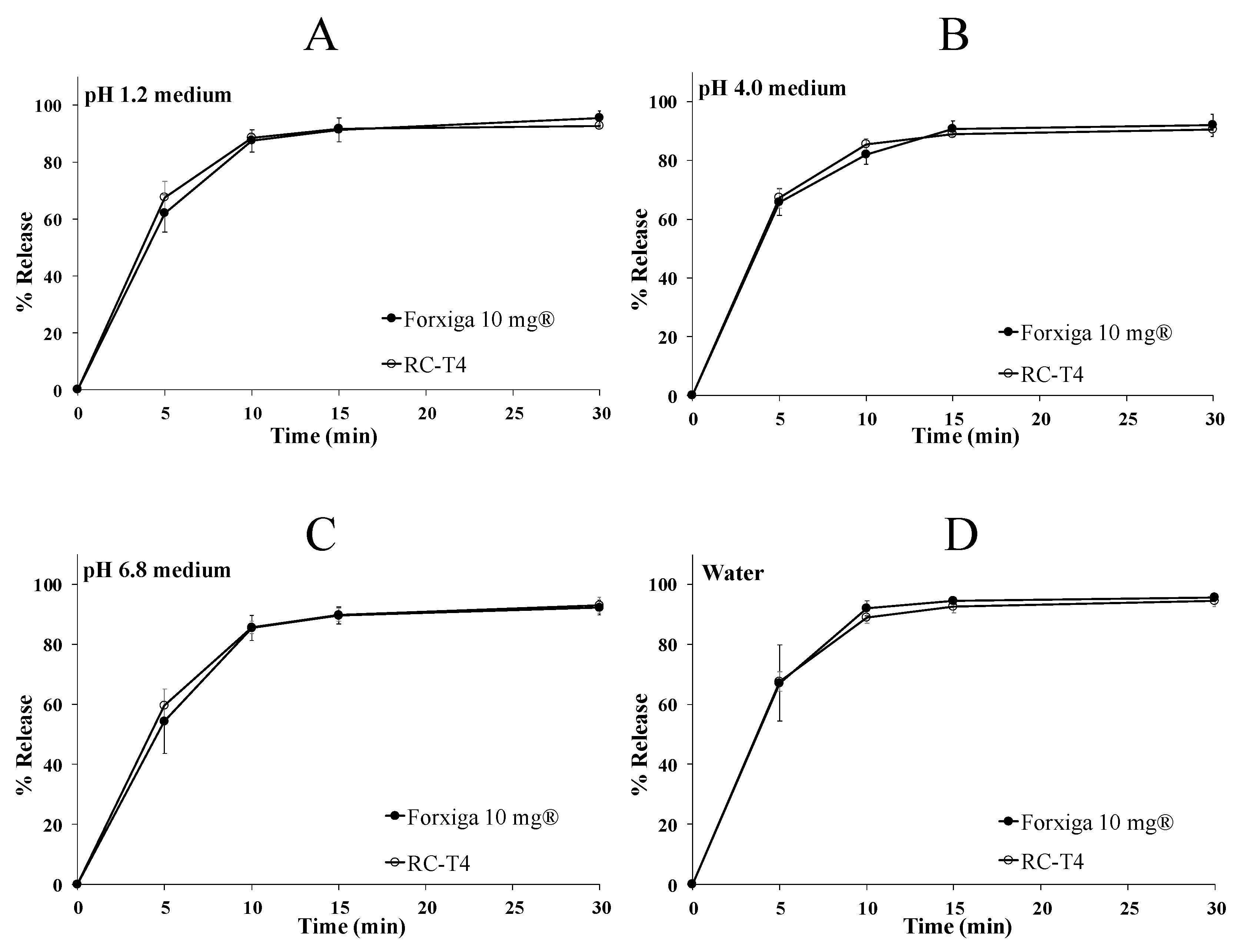
2.7.5. In Vitro Dissolution Test of DAP Cocrystal Tablet and Forxiga® 10 mg
2.8. Stability Assessment
2.8.1. Stability Assessment of DAP Cocrystal Tablets
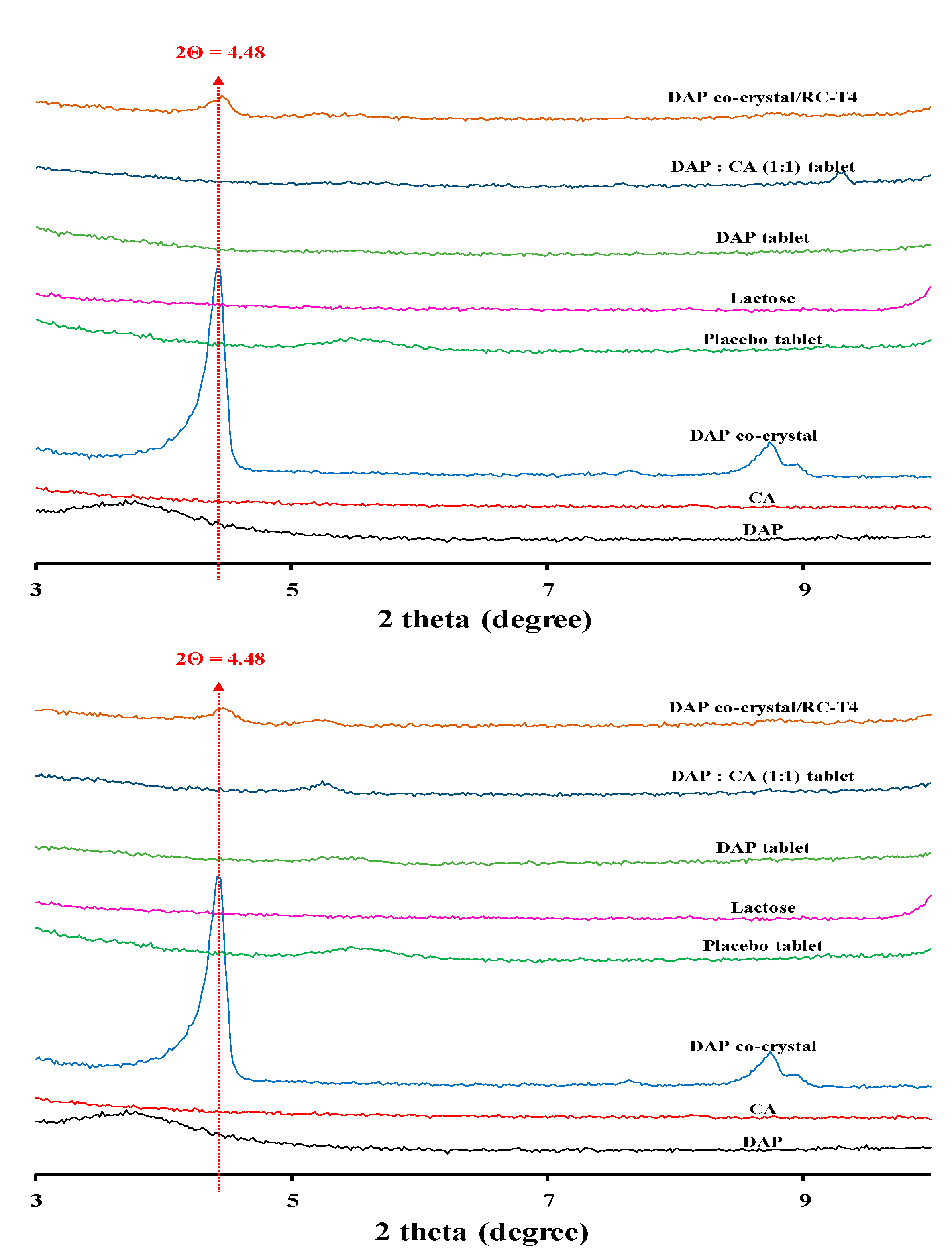
2.8.2. Stability Assessment of Cocrystal Structure Maintenance
2.9. HPLC Determination of DAP Content and Impurities
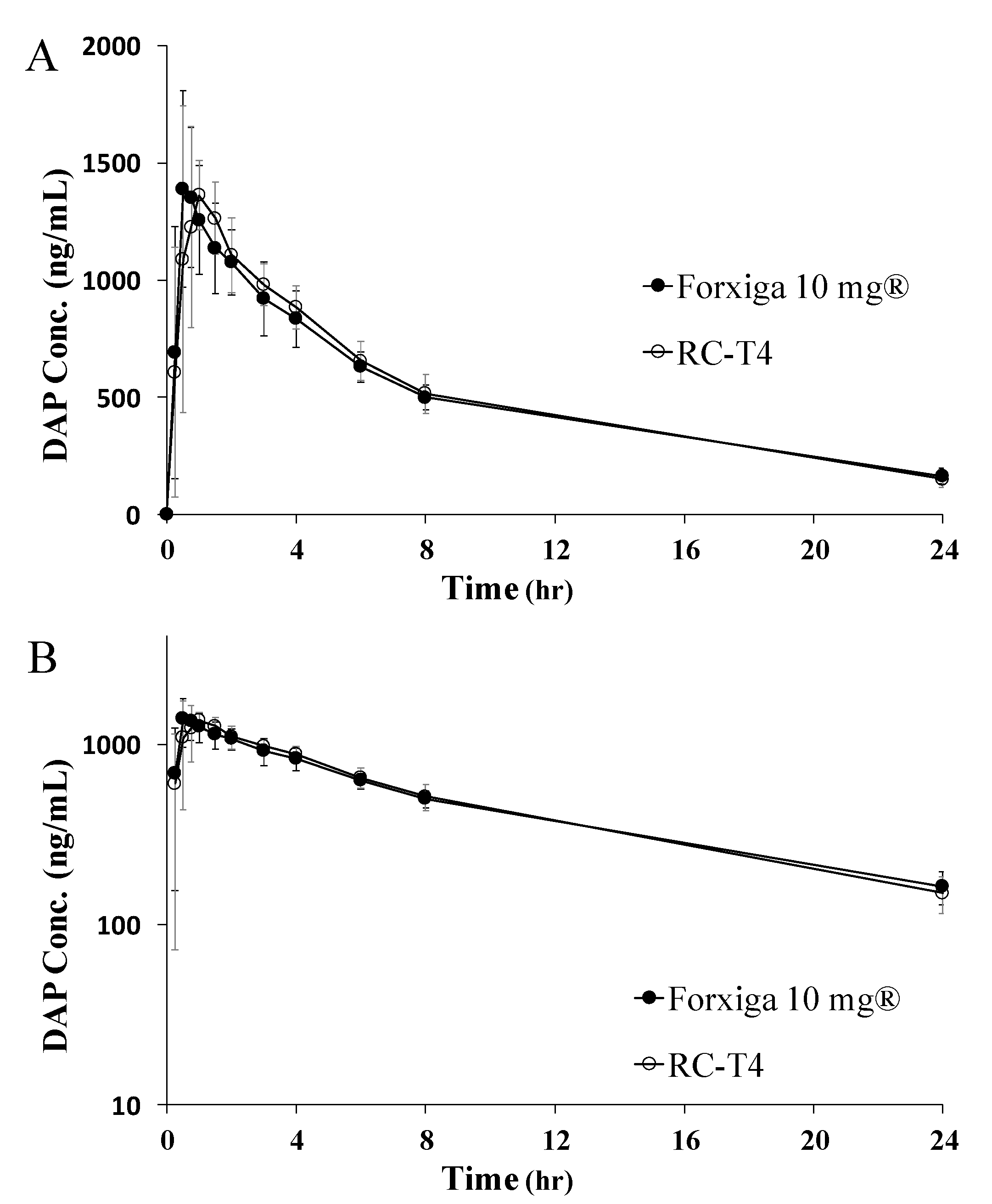
2.10. PK Study in Beagle Dogs
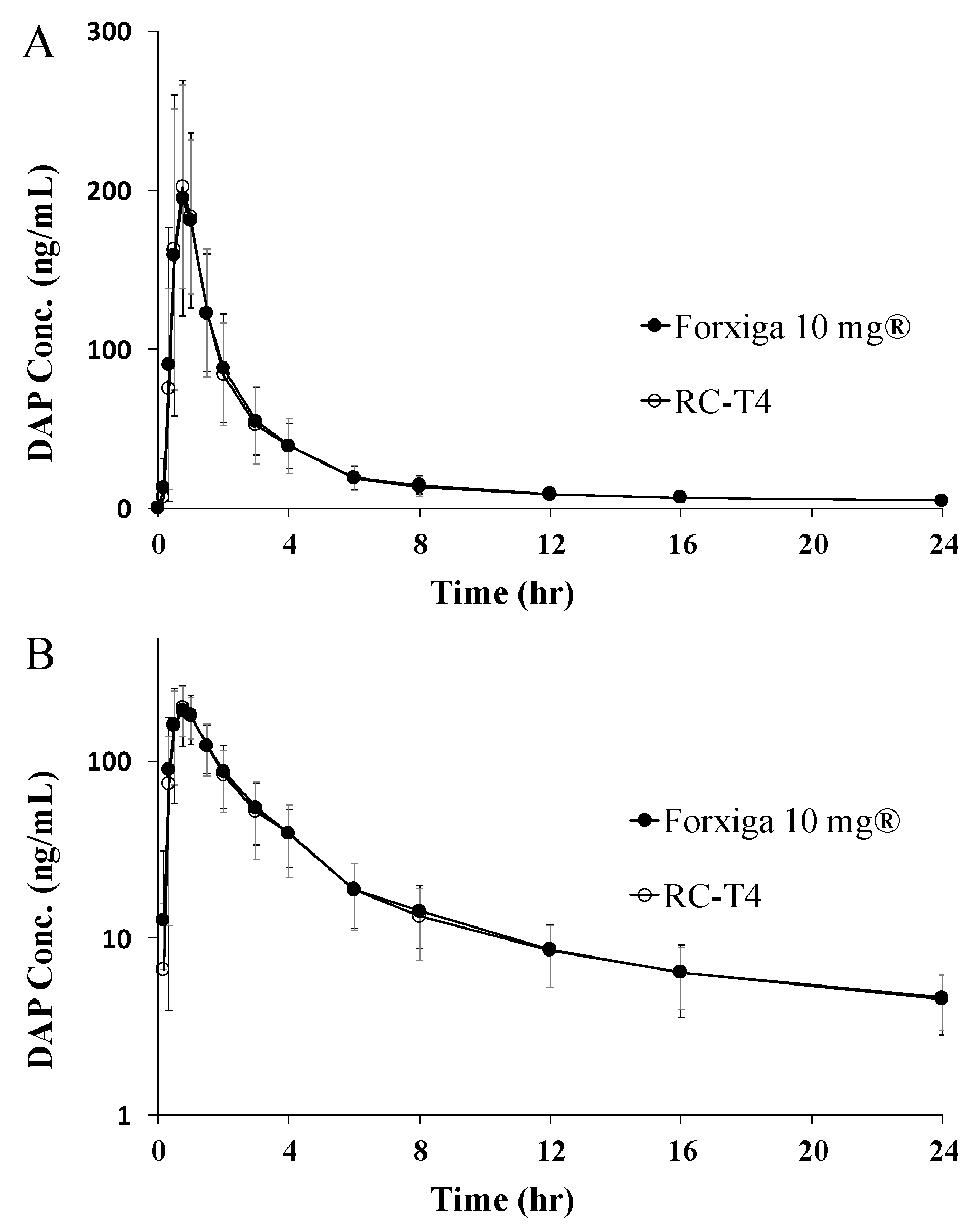
2.11. PK Study in Human Volunteers
2.12. PK Analysis
3. Results
3.1. DAP Cocrystal Characterization
3.2. Solubility Test
3.3. Appearance and Stability Test of Active Pharmaceutical Ingredient (API)
3.4. Manufacturing DAP Cocrystal Tablets by Direct Compression
3.5. Manufacturing DAP Cocrystal Tablets by Roller Compaction
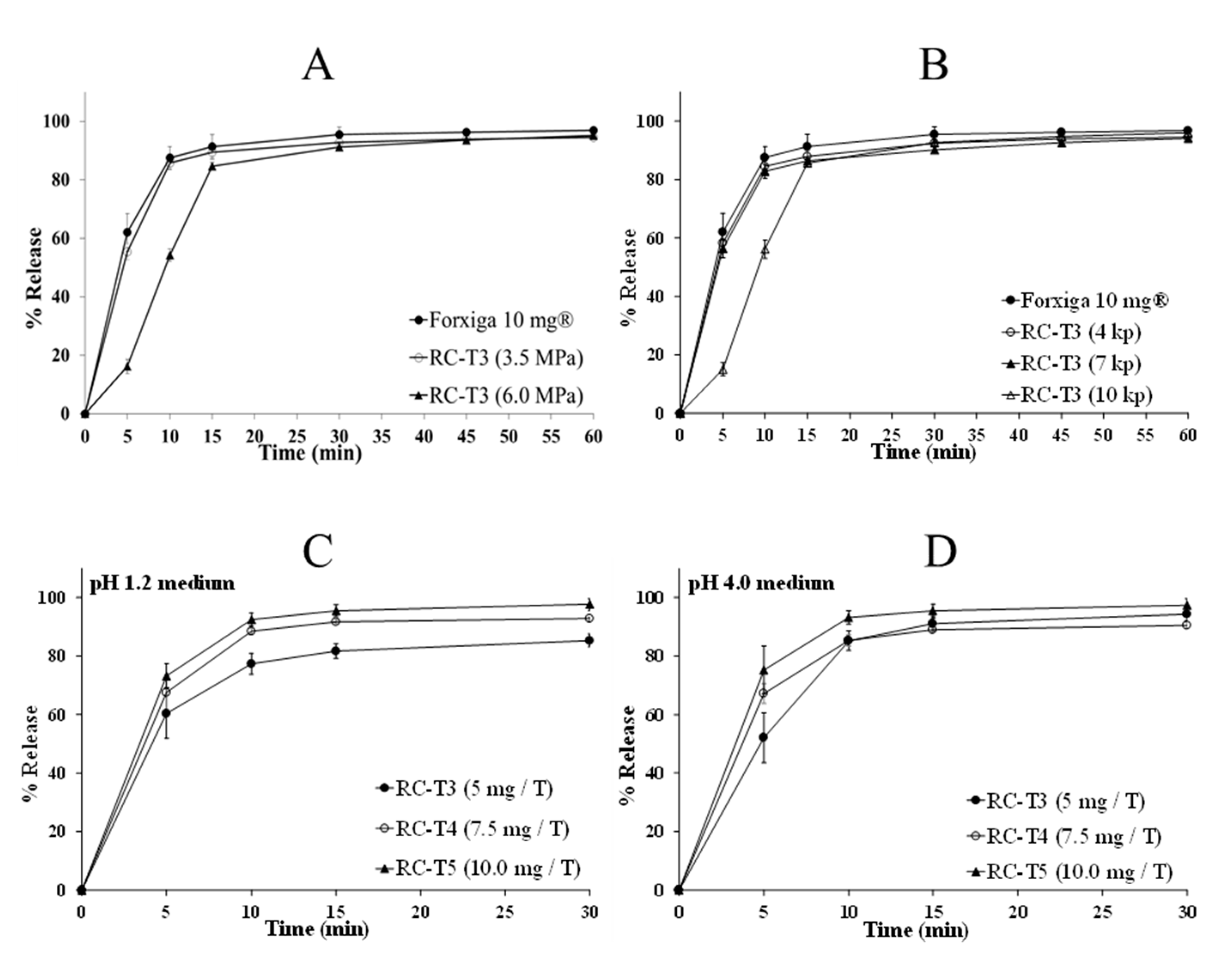
3.5.1. Effect of RC Manufacturing Parameters
3.5.2. Effect of Tablet Hardness
3.5.3. In Vitro Dissolution Test of DAP Cocrystal and Forxiga® 10 mg Tablets
3.6. Stability Assessment
3.6.1. Stability Assessment of DAP Cocrystal Tablets
3.6.2. Stability Assessment of Cocrystal Structure Maintenance
3.7. In Vivo Comparative PK Studies in Beagle Dogs and Healthy Human Volunteers
3.7.1. PK Study in Beagle Dogs
3.7.2. PK Study in Healthy Human Volunteers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| API | Active pharmaceutical ingredient |
| CA | Citric acid |
| DAP | Dapagliflozin |
| DAP-PH | Dapagliflozin propanediol monohydrate |
| DC | Direct compression |
| RC | Roller compaction |
| XRD | X-ray diffraction |
| FTIR | Fourier transform infrared spectroscopy |
| DSC | Differential scanning calorimetry |
| TGA | Thermogravimetric analysis |
| DVS | Dynamic vapor sorption |
| SGLTs | Sodium glucose transport proteins |
| SEM | Scanning electron microscopy |
| RH | Relative humidity |
| PK | Pharmacokinetics |
| AUC | Area under the plasma concentration-time curve |
| Cmax | Maximum plasma concentration |
| Tmax | Time to reach peak concentration |
| T/R ratio | Test/reference ratio |
References
- Shin, S.J.; Chung, S.; Kim, S.J.; Lee, E.M.; Yoo, Y.H.; Kim, J.W.; Ahn, Y.B.; Kim, E.S.; Moon, S.D.; Kim, M.J.; et al. Effect of Sodium-Glucose Co-Transporter 2 Inhibitor, Dapagliflozin, on Renal Renin-Angiotensin System in an Animal Model of Type 2 Diabetes. PLoS ONE 2016, 11, e0165703. [Google Scholar] [CrossRef]
- Jabbour, S.; Goldstein, B. Sodium glucose co-transporter 2 inhibitors: Blocking renal tubular reabsorption of glucose to improve glycaemic control in patients with diabetes. Int. J. Clin. Pract. 2008, 62, 1279–1284. [Google Scholar] [CrossRef]
- Singh, D.; Tiwary, A.K.; Bedi, N. Canagliflozin loaded SMEDDS: Formulation optimization for improved solubility, permeability and pharmacokinetic performance. J. Pharm. Investig. 2019, 49, 67–85. [Google Scholar] [CrossRef]
- Kasichayanula, S.; Chang, M.; Hasegawa, M.; Liu, X.; Yamahira, N.; LaCreta, F.; Imai, Y.; Boulton, D. Pharmacokinetics and pharmacodynamics of dapagliflozin, a novel selective inhibitor of sodium-glucose co-transporter type 2, in Japanese subjects without and with type 2 diabetes mellitus. Diabetesobes. Metab. 2011, 13, 357–365. [Google Scholar] [CrossRef]
- Filippatos, T.D.; Liberopoulos, E.N.; Elisaf, M.S. Dapagliflozin in patients with type 2 diabetes mellitus. Ther. Adv. Endocrinol. Metab. 2015, 6, 29–41. [Google Scholar] [CrossRef] [Green Version]
- El-Bagory, I.; Alruwaili, N.K.; Elkomy, M.H.; Ahmad, J.; Afzal, M.; Ahmad, N.; Elmowafy, M.; Alharbi, K.S.; Alam, M.S. Development of novel dapagliflozin loaded solid self-nanoemulsifying oral delivery system: Physiochemical characterization and in vivo antidiabetic activity. J. Drug Deliv. Sci. Technol. 2019, 54, 101279. [Google Scholar]
- Deng, J.-H.; Lu, T.-B.; Sun, C.C.; Chen, J.-M. Dapagliflozin-citric acid cocrystal showing better solid state properties than dapagliflozin. Eur. J. Pharm. Sci. 2017, 104, 255–261. [Google Scholar] [CrossRef]
- Domingos, S.; André, V.; Quaresma, S.; Martins, I.C.; Minas da Piedade, M.F.; Duarte, M.T. New forms of old drugs: Improving without changing. J. Pharm. Pharmacol. 2015, 67, 830–846. [Google Scholar] [CrossRef]
- Matsumoto, K.; Hasegawa, T.; Ohara, K.; Takei, C.; Akimoto, M. Roles of CYP2C9 and its variants (CYP2C9* 2 and CYP2C9* 3) in the metabolism of 6-methoxy-2-napthylacetic acid, an active metabolite of the prodrug nabumetone. J. Pharm. Investig. 2020, 50, 71–79. [Google Scholar] [CrossRef]
- Kim, P.; Kim, G.-Y.; Cho, M.-Y.; Lee, M.-J.; Choi, G.J. Manufacture and characterization of two distinct quasi-polymorphs of empagliflozin. J. Cryst. Growth 2020, 534, 125489. [Google Scholar] [CrossRef]
- Saha, S.; Desiraju, G.R. Acid···Amide supramolecular synthon in cocrystals: From spectroscopic detection to property engineering. J. Am. Chem. Soc. 2018, 140, 6361–6373. [Google Scholar] [CrossRef] [PubMed]
- Tothadi, S.; Desiraju, G.R. Synthon modularity in 4-hydroxybenzamide–dicarboxylic acid cocrystals. Cryst. Growth Des. 2012, 12, 6188–6198. [Google Scholar] [CrossRef]
- Aitipamula, S.; Wong, A.B.; Chow, P.S.; Tan, R.B. Cocrystallization with flufenamic acid: Comparison of physicochemical properties of two pharmaceutical cocrystals. CrystEngComm 2014, 16, 5793–5801. [Google Scholar] [CrossRef]
- Yadav, A.; Shete, A.; Dabke, A.; Kulkarni, P.; Sakhare, S. Co-crystals: A novel approach to modify physicochemical properties of active pharmaceutical ingredients. Indian J. Pharm. Sci. 2009, 71, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diniz, L.F.; Souza, M.S.; Carvalho, P.S., Jr.; da Silva, C.C.; D’Vries, R.F.; Ellena, J. Novel Isoniazid cocrystals with aromatic carboxylic acids: Crystal engineering, spectroscopy and thermochemical investigations. J. Mol. Struct. 2018, 1153, 58–68. [Google Scholar] [CrossRef]
- Mitchell, S.A.; Reynolds, T.D.; Dasbach, T.P. A compaction process to enhance dissolution of poorly water-soluble drugs using hydroxypropyl methylcellulose. Int. J. Pharm. 2003, 250, 3–11. [Google Scholar] [CrossRef]
- Kleinebudde, P. Roll compaction/dry granulation: Pharmaceutical applications. Eur. J. Pharm. Biopharm. 2004, 58, 317–326. [Google Scholar] [CrossRef]
- Guigon, P.; Simon, O. Roll press design—Influence of force feed systems on compaction. Powder Technol. 2003, 130, 41–48. [Google Scholar] [CrossRef]
- Park, M.S.; Choi, D.H. Application of mechanism-based modeling to predict drug quality during the pharmaceutical unit operations of granulation and compression: A review. J. Pharm. Investig. 2020, 50, 445–467. [Google Scholar] [CrossRef]
- Pishnamazi, M.; Casilagan, S.; Clancy, C.; Shirazian, S.; Iqbal, J.; Egan, D.; Edlin, C.; Croker, D.M.; Walker, G.M.; Collins, M.N. Microcrystalline cellulose, lactose and lignin blends: Process mapping of dry granulation via roll compaction. Powder Technol. 2019, 341, 38–50. [Google Scholar] [CrossRef]
- Rambali, B.; Baert, L.; Jans, E.; Massart, D. Influence of the roll compactor parameter settings and the compression pressure on the buccal bio-adhesive tablet properties. Int. J. Pharm. 2001, 220, 129–140. [Google Scholar] [CrossRef]
- Kasichayanula, S.; Liu, X.; LaCreta, F.; Griffen, S.C.; Boulton, D.W. Clinical pharmacokinetics and pharmacodynamics of dapagliflozin, a selective inhibitor of sodium-glucose co-transporter type 2. Clin. Pharmacokinet. 2014, 53, 17–27. [Google Scholar] [CrossRef]
- Jang, J.-H.; Jeong, S.-H.; Cho, H.-Y.; Lee, Y.-B. Comparison of UPLC-MS/MS and HPLC-UV methods for the determination of zaltoprofen in human plasma. J. Pharm. Investig. 2019, 49, 613–624. [Google Scholar] [CrossRef]
- Jeong, S.-H.; Jang, J.-H.; Cho, H.-Y.; Oh, I.-J.; Lee, Y.-B. A sensitive UPLC–ESI–MS/MS method for the quantification of cinnamic acid in vivo and In Vitro: Application to pharmacokinetic and protein binding study in human plasma. J. Pharm. Investig. 2020, 50, 159–172. [Google Scholar] [CrossRef]
- Akimoto, M.; Nagahata, N.; Furuya, A.; Fukushima, K.; Higuchi, S.; Suwa, T. Gastric pH profiles of beagle dogs and their use as an alternative to human testing. Eur. J. Pharm. Biopharm. 2000, 49, 99–102. [Google Scholar] [CrossRef]
- Kosugi, Y.; Yamamoto, S.; Sano, N.; Furuta, A.; Igari, T.; Fujioka, Y.; Amano, N. Evaluation of acid tolerance of drugs using rats and dogs controlled for gastric acid secretion. J. Pharm. Sci. 2015, 104, 2887–2893. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredients | Function | DC-D1 | DC-D2 | DC-D3 | DC-D4 | DC-D5 |
|---|---|---|---|---|---|---|
| DAP cocrystal (before milling) | API | 14.7 | - | - | - | - |
| DAP cocrystal (after milling) | API | - | 14.7 | 14.7 | 14.7 | 14.7 |
| Microcrystalline cellulose | Filler | 141.3 | 141.3 | 141.3 | 76.3 | 141.3 |
| Lactose anhydrous | Filler | - | - | 65.0 | 65.0 | - |
| Mannitol | Filler | 65.0 | 65.0 | - | 65.0 | 65.0 |
| Crospovidone | Disintegrant | 5.0 | 5.0 | 5.0 | 5.0 | 5.0 |
| Colloidal silicon dioxide | Lubricant | 4.0 | 4.0 | 4.0 | 10.0 | 10.0 |
| Magnesium stearate | Lubricant | 5.0 | 5.0 | 5.0 | 5.0 | 5.0 |
| Opadry 03B62323 | Coating agent | 10.0 | 10.0 | 10.0 | 10.0 | 10.0 |
| Ingredients | Function | RC-T1 | RC-T2 | RC-T3 | RC-T4 | RC-T5 |
|---|---|---|---|---|---|---|
| DAP cocrystal | API | 14.7 | 14.7 | 14.7 | 14.7 | 14.7 |
| Microcrystalline cellulose | Filler | 142.5 | 76.3 | 150.0 | 147.5 | 145 |
| Lactose anhydrous | Filler | - | 65.0 | 61.3 | 61.3 | 61.3 |
| Mannitol | Filler | 63.8 | 65.0 | - | - | - |
| Crospovidone | Disintegrant | 5.0 | 5.0 | 5.0 | 7.5 | 10.0 |
| Colloidal silicon dioxide | Lubricant | 4.0 | 4.0 | 4.0 | 4.0 | 4.0 |
| Magnesium stearate | Lubricant | 5.0 | 5.0 | 5.0 | 5.0 | 5.0 |
| Opadry 03B62323 | Coating agent | 10.0 | 10.0 | 10.0 | 10.0 | 10.0 |
| Active Ingredients | Media | Equilibrium Solubility (CS, mg/mL) | D0 * |
|---|---|---|---|
| DAP | pH 1.2 | 0.90 | 0.04 |
| pH 4.0 | 0.94 | 0.04 | |
| pH 6.8 | 0.81 | 0.05 | |
| Water | 0.86 | 0.05 | |
| DAP-PH | pH 1.2 | 1.68 | 0.02 |
| pH 4.0 | 1.74 | 0.02 | |
| pH 6.8 | 1.60 | 0.03 | |
| Water | 1.70 | 0.02 | |
| DAP cocrystal | pH 1.2 | 2.20 | 0.02 |
| pH 4.0 | 2.62 | 0.02 | |
| pH 6.8 | 1.73 | 0.02 | |
| Water | 2.33 | 0.02 |
| Parameter | Forxiga® 10 mg | RC-T4 | T/R ratio f | CV-ANOVA g |
|---|---|---|---|---|
| AUC0–24 (ng·h/mL) a | 11919 ± 1469 | 12116 ± 1550 | 1.017 (0.9899~1.045) | 4.044 |
| AUC0–∞ (ng·h/mL) b | 13617 ± 2013 | 13364 ± 1965 | ||
| Cmax (ng/mL) c | 1500 ± 308 | 1504 ± 233 | 1.019 (0.9310~1.115) | 11.729 |
| tmax (h) d | 0.50 ± 0.75 | 0.75 ± 0.50 | ||
| t1/2 (h) e | 9.47 ± 1.40 | 8.50 ± 0.72 |
| Parameter | Forxiga® 10 mg | RC-T4 | T/R Ratio f | CV-ANOVA g |
|---|---|---|---|---|
| AUC0–24 (ng·h/mL) a | 592.01 ± 165.56 | 618.74 ± 184.71 | 1.0413 (1.0121~1.0713) | 6.425 |
| AUC0–∞ (ng·h/mL) b | 626.21 ± 160.53 | 651.65 ± 183.18 | ||
| Cmax (ng/mL) c | 223.44 ± 54.75 | 229.41 ± 69.13 | 1.0135 (0.9113~1.1270) | 27.032 |
| tmax (h) d | 0.75 ± 0.75 | 0.75 ± 0.75 | ||
| t1/2 (h) e | 12.64 ± 4.47 | 11.27 ± 3.14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, S.; Lee, J.; Yoo, Y.; Cho, M.; Sohn, S.; Lee, B.-J. Improved Manufacturability and In Vivo Comparative Pharmacokinetics of Dapagliflozin Cocrystals in Beagle Dogs and Human Volunteers. Pharmaceutics 2021, 13, 70. https://doi.org/10.3390/pharmaceutics13010070
Cho S, Lee J, Yoo Y, Cho M, Sohn S, Lee B-J. Improved Manufacturability and In Vivo Comparative Pharmacokinetics of Dapagliflozin Cocrystals in Beagle Dogs and Human Volunteers. Pharmaceutics. 2021; 13(1):70. https://doi.org/10.3390/pharmaceutics13010070
Chicago/Turabian StyleCho, Sangho, Jeongwook Lee, Yongwon Yoo, Minyong Cho, Seil Sohn, and Beom-Jin Lee. 2021. "Improved Manufacturability and In Vivo Comparative Pharmacokinetics of Dapagliflozin Cocrystals in Beagle Dogs and Human Volunteers" Pharmaceutics 13, no. 1: 70. https://doi.org/10.3390/pharmaceutics13010070
APA StyleCho, S., Lee, J., Yoo, Y., Cho, M., Sohn, S., & Lee, B.-J. (2021). Improved Manufacturability and In Vivo Comparative Pharmacokinetics of Dapagliflozin Cocrystals in Beagle Dogs and Human Volunteers. Pharmaceutics, 13(1), 70. https://doi.org/10.3390/pharmaceutics13010070