1. Introduction
Nanotechnology is referred to as the area of science focused on the study of the synthesis, characterization, and application of materials and functional systems of particles with sizes ranging between 1 and 100 nm. Nowadays, the interest in these materials is not only due to their small size, but also at these dimensions the material properties change in comparison to the same material at the macroscopic scale and make possible highly specific cellular and molecular interactions. The application of nanotechnology to biomedical research (nanobiotechnology) will improve, in the next few years, the development of new and more effective tools for the detection and treatment of diseases like diabetes, Parkinson’s disease, or cancer [
1,
2]. This science will progress the monitorization, reparation, and control of biological functions by using systems on a nanoscale. Despite the development of nanoparticles for different biomedical applications, the exposition to certain types of particles could promote cell damage. Nowadays, the accepted paradigms of the toxicity of nanomaterials are oxidative stress and inflammation. In fact, our research group described the damage produced by Au (gold) nanoparticles in DNA structure, ROS production, and lipid in vitro and in vivo conditions and its relation with the nanoparticle size [
3], but other mechanisms are still not well-defined.
Iron oxide nanoparticles are widely used in biomedical research, especially in cancer research, due to their properties of magnetism, biocompatibility and due to their drug delivery and multi-imaging functions [
4]. Even the treatment of anemia using iron nanoparticles have been developed in the last years to reduce the side effects of traditional treatments due to the reduced size of nanoparticles that could increase its absorption [
5]. Moreover, iron nanoparticles, especially the iron oxide, have been developed as an alternative in gene therapy and as a drug delivery system [
6]. Recent studies have been conducted to evaluate some more specific and relevant aspects related to toxicity mechanisms of iron oxide nanoparticles like mitochondrial damage, ROS production or autophagy [
7]. Khan, M.I. and co-workers [
8] showed that certain iron oxide nanoparticles promoted cell death in lung cancer cells through ROS production and autophagy, but these nanoparticles did not promote cell death in normal lung cells. On the other hand, other authors have proposed the role of iron nanoparticles in autophagosome induction and oxidative damage in mammary gland cells [
9]. Nonetheless, more studies are necessary on the possible interaction between iron nanoparticles and mitochondria. Autophagy is an intracellular degradation process that delivers cytoplasmic constituents to lysosomes for degradation in response to a high variety of stimuli [
10]. This process is related to create new cellular structures [
11], so it represents a catabolic process of cytosolic renovation, but it is also able to induce autophagy-dependent cell death [
12]. There are defined three types of autophagy depending on how the autophagic substrate move to the lysosome: macro-autophagy, micro-autophagy, and chaperone-mediated autophagy. One of the effects of iron nanoparticles could be associated with the induction of autophagy due to most endocytic routes of nanomaterial cell uptake by phagocytic and non-phagocytic mechanisms converge upon the lysosome.
In the present study, the effect of ultra-small iron nanoparticles on autophagy induction and mitochondrial function are evaluated using an in vitro model. Two types of ultra-small iron nanoparticles have been tested, a commercial preparation used in clinical practice (Venofer
®) and one synthesized in the laboratory (FeNPs). This work addresses, for the first time, a comprehensive study about the traffic of iron and other elements in the subcellular fractions, the integrity of the mtDNA, the mitochondrial functionality and the induction of the autophagy process that will increase the knowledge about the metabolism of nanoparticles and its possible application in therapy focused on mitochondria. To perform the study HT-29 cell line was chosen based on previous studies in which the implications of nanomaterials in therapy have been evaluated [
13,
14,
15].
2. Materials and Methods
2.1. Synthesis and Characterization of Nanoparticles: Synthesis of Ultra-Small Nanoparticles (FeNPs)
Iron nanoparticles (4 nm core Fe
2O
3 coated with tartaric/adipic acid) were synthesized following a slightly modified protocol from Pereira et al. [
16]. This method is based on the precipitation of Fe
3+ in the presence of highly basic medium (5 mol L
−1 NaOH solution) with the addition of tartrate and adipic acid solution for the iron core coating as described somewhere else [
15]. The molar ratio tartaric: adipic: Fe used corresponds to 1:1:2, which has given best performance in previous experiments. The three components are mixed and constantly stirred in a buffer media (ammonium acetate 50 mmol L
−1 at pH 4). The initial pH of the mixture is increased stepwise until reaching pH 8. When mixture turns dark brown/blackish, centrifugation and ultrafiltration (30,000 Da; 3000 Da Ultra-15 MWCO centrifugal filter units, Millipore, Darmstadt, Germany) steps are needed to separate the microparticulate and nanoparticulate iron fractions from the supernatant and remove excess of soluble ligands and the rest of reagents. Centrifuge Biofuge Stratos Heraeus (Thermo Scientific
TM, Waltham, MA, USA) was used for these purposes. Size and shape characterization of the particles has been conducted by TEM, DLS, and UV-VIS. The characterization of these nanoparticles was published in previous articles of our research group [
15].
2.2. Cell Culture Conditions
HT-29 cell lines were obtained from the Cell Culture Resource Centre at the University of Granada, Spain. The cells were precultured in 25 cm2 culture flasks in RPMI-1640 medium supplemented with 10% (v/v) FBS and 2 mmol L−1 L-glutamine. The culture flasks were maintained in a cell incubator at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. Medium was replaced every 2–3 days after rinsing with PBS. Upon reaching confluence, cells were treated with trypsin-EDTA solution (Sigma, St Louis, MO, USA) and split 1/10 to allow for continuous growth. Cells were exposed to 0.5 mmol L−1 Fe NPs concentration over 48 h. Cells cultured were treated with 0.5 mmol L−1 FeNPs or Venofer® for 48 h. Every week, the absence of mycoplasma was evaluated employing a commercial kit (Sigma, St Louis, MO, USA).
2.3. TEM Analysis
Once the exposure time had finished, cells were prepared to be visualized by TEM. Cells samples were fixed with fresh primary fixative (1.5% glutaraldehyde, 1.0% formaldehyde in 0.05 mol L−1 sodium cacodylate buffer, pH 7.4) and post-fixed with secondary fixative (1% osmium tetroxide, 1% potassium ferrocyanide in Milli Q water) followed by dehydration with ascending series of alcohol before embedding samples in epoxy resin. Ultra-thin sections were cut and doubly stained with uranyl acetate and lead citrate. A transmission electron microscope LIBRA 120 PLUS microscope at 120 kV (Carl Zeiss SMT, Oberkochen, Germany) was used to determine the distribution of ultra-small iron nanoparticles.
2.4. Subcellular Fractionation
HT-29 cells were previously washed with physiologic saline solution. Mitochondria isolation was carried out employing a commercial kit according to the manufacturer’s instruction (Abcam, Cambridge, UK). Briefly, cells were trypsinized, collected by centrifugation and frozen. Then, cells were homogenized and centrifuged at 1000× g, 10 min, 4 °C, two times. Both supernatants were collected and the pellet with the nucleus portion was preserved. Afterward, the supernatants were centrifuged at 12,000× g during 10 min, 4 °C, obtaining the pellet with the isolated of mitochondria. Supernatant was also preserved in which ERP, Golgi, and cytosol was included.
2.5. Determination of Total Metal Concentrations
Determination of sodium (Na), magnesium (Mg), phosphorus (P), potassium (K), calcium (Ca), iron (Fe), copper (Cu), manganese (Mn), zinc (Zn) and selenium (Se) in subcellular fractions, diluted with a basic solution containing ammonium hydroxide, butanol, EDTA, and triton X-100, was performed by means an ICP-MS instrument (Agilent 7500, Agilent Technologies, Tokyo, Japan) fitted with a Meinhard type nebulizer (Glass Expansion, Romainmotier, Switzerland) and equipped with a He collision cell. A Milli-Q system (Millipore, Bedford, MA, USA) was used to obtain deionized water (18 MΩ cm). All reagents (ammonium hydroxide solution, butanol, EDTA, Triton X-100) used were of the highest available purity. A standard solution of 100 µg L−1 of Li, Mg, Sc, Co, Y, In, Ce, Ba, Pb, Bi, and U in 1% (v/v) HNO3 was prepared from a 1.000 mg L−1 multi-element stock standard solution (Merck, Darmstadt, Germany) and used for daily optimizing of the ICP parameters. Single-element standard solutions for ICP-MS containing 1.000 µg mL−1 of each analyte were also purchased from Merck. Calibration curves were prepared using Ga as an internal standard and by the dilution of stock solutions of 1.000 mg L−1 in 1% HNO3. The accuracy of this method was evaluated by comparison with a certified reference material SeronormTM Trace Elements Serum (Billingstad, Norway) and by recovery studies of spiked samples with multi-element standards. The calculated recoveries for each element were between 95% and 105% in all cases. For each element, we used the mean of five separate determinations.
2.6. Proportion of Deleted mtDNA and mtDNA Copy Number Assessment
The proportion of deleted mtDNA was determined using a quantitative reverse-transcription polymerase chain reaction (QRT-PCR) to amplify two mitochondrial genes, MT-ND4 and MT-ND1. Relative levels of mtDNA copy number were also determined by QRT-PCR by amplifying the nuclear single-copy nuclear gene acidic ribosomal phosphoprotein PO (36B4) and using the amplification data of mitochondrial MT-ND1 gene previously obtained. DNA amplification was performed in a MicroAmp Optical 384-well Reaction Plate (Applied Biosystems, Foster City, CA, USA) using the Applied Biosystem’s 7900HT Fast Real-Time PCR system. Primers for the assays were as follows: MT-ND1 forward primer 5′-CCCTAAAACCCGCCACATCT′-3, MT-ND1 reverse primer GAGCGATGGTGAGAGCTAAGGT-3′, MT-ND4 forward primer 5′-CCATTCTCCTCCTATCCCTCAAC-3′, MT-ND4 reverse primer 5′-CACAATCTGATGTTTTGGTTAAACTATATTT-3′, 36B4 forward primer 5′-CTGCAGATTGGCTACCCGAC-3′ and 36B4 reverse primer 5′-CACAGACAAAGCCAGGACCC-3′. Final primer concentrations were 200 nmol L−1 except for the last two that were 300 and 500 nmol L−1, respectively. Reactions were performed in 10 µL volumes. Reaction mixture in each well included genomic DNA (5 ng), 2× QuantiNova SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) (5 μL), ROX dye (Applied Biosystems, Foster City, CA, USA) (1 μL), Mili-Q sterilized H2O and the proper primer oligonucleotides. Reaction conditions were set at 95 °C for 2 min followed by 40 cycles of data collection consisting of a denaturation step at 95 °C for 8 s and an annealing/extension at the 58 °C step for 15 s. An additional phase to obtain a melting curve was performed at the end of each reaction to verify specific amplification. Standard curves were included for analysis of data. Mean quantification cycle (Cq) for each sample were calculated using Sequence Detector Systems version 2.4 software. The proportion of deleted mtDNA was calculated for each well dividing MT-ND4 by MT-ND1 amplification data taking into account reaction efficiency values. The resulting values were divided by the average value obtained from control samples (untreated cells) analysis. This allowed to obtain a relative MT-ND4:MT-ND1 ratio. In turn, the MT-ND1:36B4 ratio was calculated and multiplied by two to obtain mtDNA copy number for each sample. DNA used in the aforementioned assays was isolated using a NucleoSpin Tissue kit (Macherey-Nagel GmbH & Co. KG, Düren, Germany) according to manufacturers.
2.7. Determination of Energetic Metabolism
2.7.1. Mitochondrial Respiration
The oxygen consumption rate (OCR) in HT-29 cells was measured in real-time using a XF-24 Extracellular Flux Analyzer (Seahorse Bioscience, Billerica, MA, USA) as previously reported [
17]. For this, 3 × 10
4 cells were seeded for 16 h in the XF-24 plate and treated for 48 h on 0.5 mmol L
−1 of FeNPs or VENOFER
®. The medium was replaced with 450 μL/well of XF-24 running media (Seahorse Bioscience, Billerica, MA, USA), supplemented with 25 mmol L
−1 glucose, 2 mmol L
−1 glutamine, 1 mmol L
−1 sodium pyruvate, without serum and pre-incubated at 37 °C for 20 min in the XF Prep Station incubator (Seahorse Bioscience, Billerica, MA, USA) in the absence of CO
2. The plate was then transferred to the XF-24 Extracellular Flux Analyzer, and after an OCR baseline measurement, four sequential injections of compounds that affect bioenergetics were performed, as follows: 55 μL of oligomycin (1 μg mL
−1), 61 μL of 2,4-Dinitrophenol (2,4 DNP) (1 mmol L
−1), and 68 μL of antimycin A/rotenone (10 μmol L
−1/1 μmol L
−1) at injection in port C. Each treatment was carried out in three replicates and the final results were expressed as pmol of O
2 consumed per 105 cells per minute (pmol O
2/105 cells/min).
2.7.2. Glycolysis
To evaluate glycolysis, extracellular acidification rate (ECAR) was measured in real-time using a XF-24 Extracellular Flux Analyzer (Seahorse Bioscience, Billerica, MA, USA) as previously reported [
17]. ECAR was measured after addition of 55 μL of rotenone (1 μmol L
−1), 61 μL of glucose (30 mmol L
−1) and 68 μL 2-Deoxy-d-glucose (2-DG) (100 mmol L
−1). Prior to measure ECAR, HT-29 cells were treated as in previous assay per triplicate and average ECAR value, expressed as milli-pH per minute (mpH/min) per 3.0 × 10
4 cells, was calculated for each treatment.
2.8. Autophagy
2.8.1. Autophagy Induction
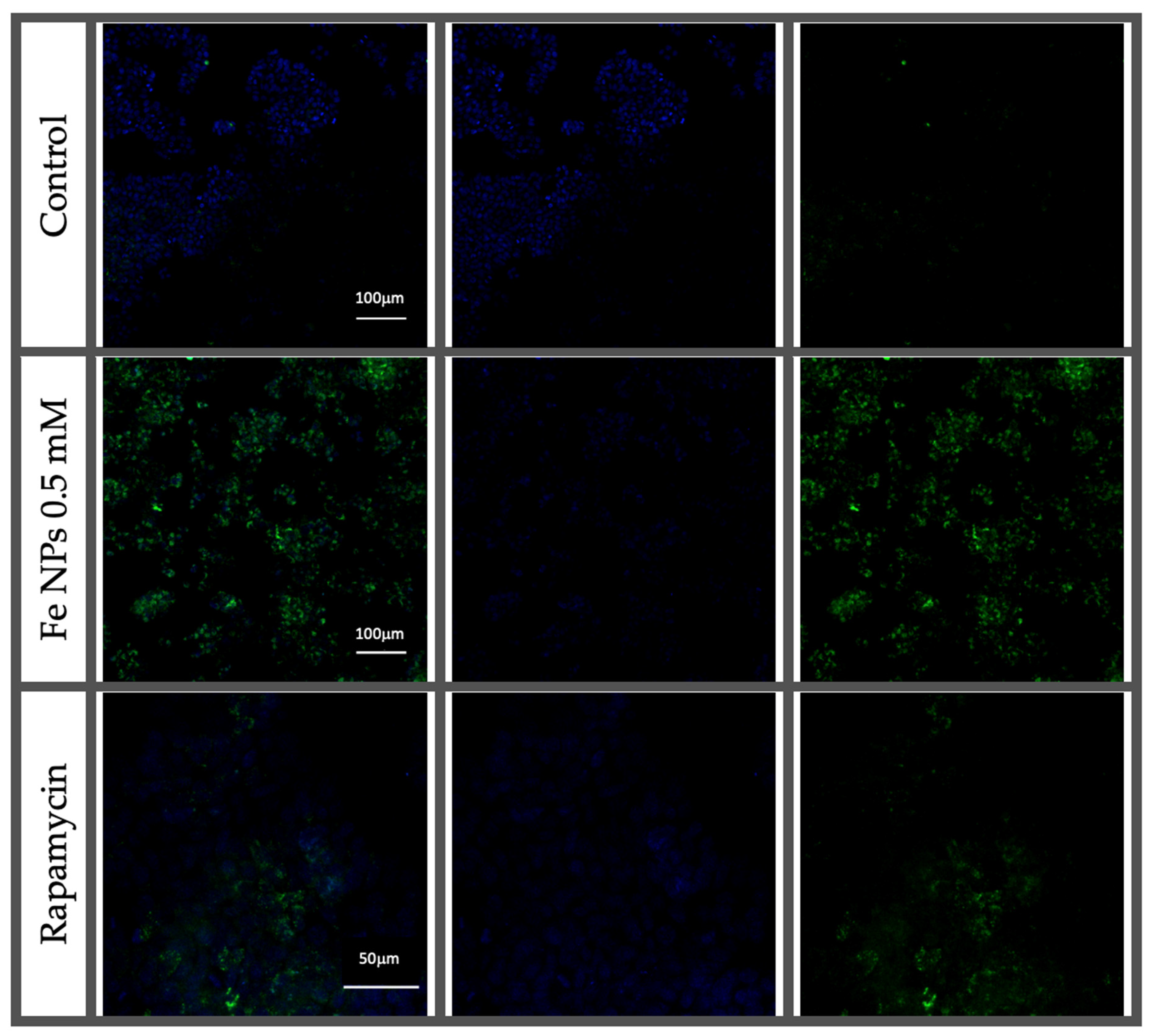
The CYTO-ID ENZKIT175 Autophagy Detection Kit 2.0 (Enzo Life Sciences, Lausen, Switzerland) was used for the detection of autophagy in HT-29 cells. Briefly, HT-29 cells were seeded at a density of 0.75 × 105 cells/mL and left to adhere for 24 h. Then, cells were treated with 0.5 mM FeNPs for 48 h, untreated (control group) or rapamycin 0.5 µmol L−1 (autophagy inductor) for 12 h. Then, the supernatant was removed and the cells were washed twice with 200 µL of PBS supplemented with 5% (v/v) FBS and stained with an autophagy staining solution containing CYTO-ID® Green Detection Reagent and Hoechst 33342 Nuclear Stain following the protocol provided by the manufacturer. Fluorescence was determined using a microplate reader (Biotek, VT, USA), excitation 480 nm-emission 530 nm and also a confocal fluorescence microscope. For confocal fluorescence microscopy, staining solution was removed after 30 min of incubation at 37 °C in a humidified incubator, and cells were washed with 200 µL of PBS supplemented with 5% (v/v) FBS three times. Then, cells were fixed with PFA 4% (v/v), at room temperature in the absence of light for 20 min, and washed with 200 µL of PBS supplemented with 5% (v/v) FBS three times. Then microscope slide preparation with glycerol and cells were visualized and photographed in a confocal microscopy (Nikon, Tokyo, Japan) with a coupled camera (ref) Nis-Elements microscope imaging software (version 4.13).
2.8.2. Autophagic Flux
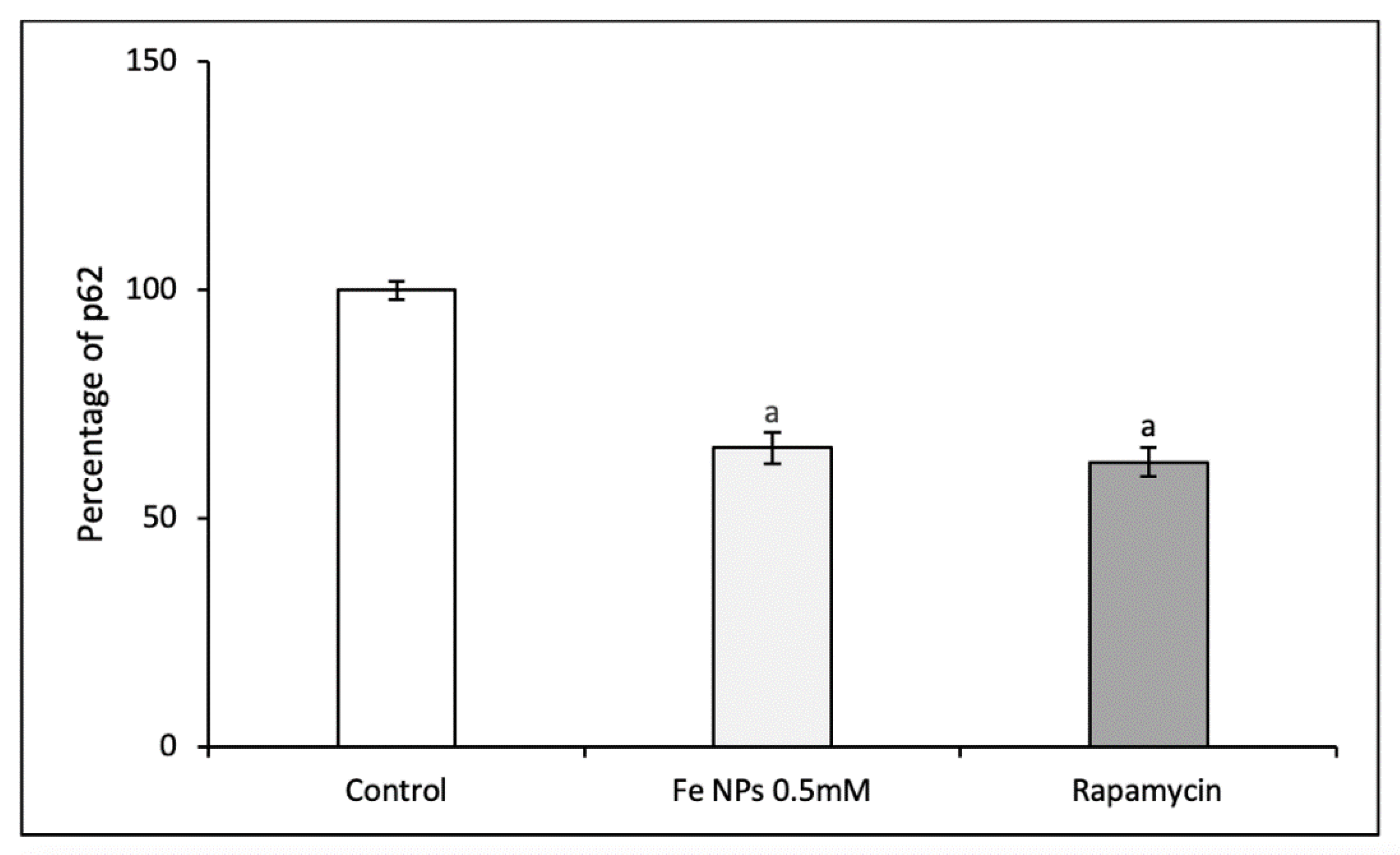
The levels of p62 were assessed in cells by the p62 Elisa kit (Enzo Life Sciences, Lausen, Switzerland) according to the manufacturer’s protocol. Briefly, after 48 h exposure to iron nanoparticles and rapamycin 0.5 µmol L−1 (as positive control), cells were collected and protein were extracted and seeded on a pre-coated plate with p62 antibody. After incubating samples in presence of a second anti-p62 antibody (rabbit polyclonal), the amount of p62 was revealed by adding a secondary donkey anti-rabbit IgG antibody conjugated to horseradish peroxidase and a mix composed by TMB and hydrogen peroxide. The plate was read in a microplate reader (Biotek, Winooski, VT, USA) and the amount of p62 normalized for the total protein amount.
2.9. Apoptosis/Necrosis
To quantify if nanoparticles induced apoptosis or necrosis in HT-29 cells, a double staining with annexin V and propidium iodide was performed. Cells were seeded in 35 mm culture dishes (VWR, Radnor, PA, USA) at 2 × 10
5 cells/mL and incubated in the conditions described in
Section 2.2 Culture medium was removed after 24 h and replaced with 1 mL of fresh medium containing either the IC50 of extracts or 0.4 µM of doxorubicin (positive control). Cells were incubated for 48 h, collected by trypsinization and centrifuged at 1000×
g for 5 min at room temperature (RT). The obtained pellet was rinsed twice with 1 mL of cold PBS 1× intercalated with 5 min of centrifugations at 1000×
g. Following centrifugations, 100 μL of annexin binding buffer 1×, 5 μL of annexin V-FITC and 2 μL of PI (Annexin V-FITC Apoptosis Detection Kit; Invitrogen, Waltham, MA, USA) was added to all samples and incubated for 15 min at RT in the absence of light. Afterwards, to these cellular suspensions were added 400 μL of annexin binding buffer 1× and 500 μL of cold PBS 1×. The analysis and quantification of apoptotic events and necrosis were performed by flow cytometry on Attune Acoustic Focusing Flow Cytometer (Life Technologies, Carlsbad, CA, USA) using an Attune Cytometric software (Life Technologies), with the acquisition of at least 10,000 events per sample. Data presented as the mean of two independent experiments.
2.10. Statistical Analysis
Descriptive statistical parameters (means and standard deviations of 8 samples for each experiment) were obtained for each of the variables studied. Statistical comparisons among the groups were performed by a comparison of means between independent variables. All analyses were performed using SPSS 20.0 (SPSS, Chicago, IL, USA). Differences were considered statistically significant at a probability level <5%.
4. Discussion
The application of nanoparticles in biomedical therapies will make possible the development of new diagnosis and treatment tools. One of the most promising aspect related to the use of nanoparticles in biomedicine therapy is its employ as carriers in gene therapy or drug delivery, due to its facility of synthesis, good biocompatibility and low toxicity [
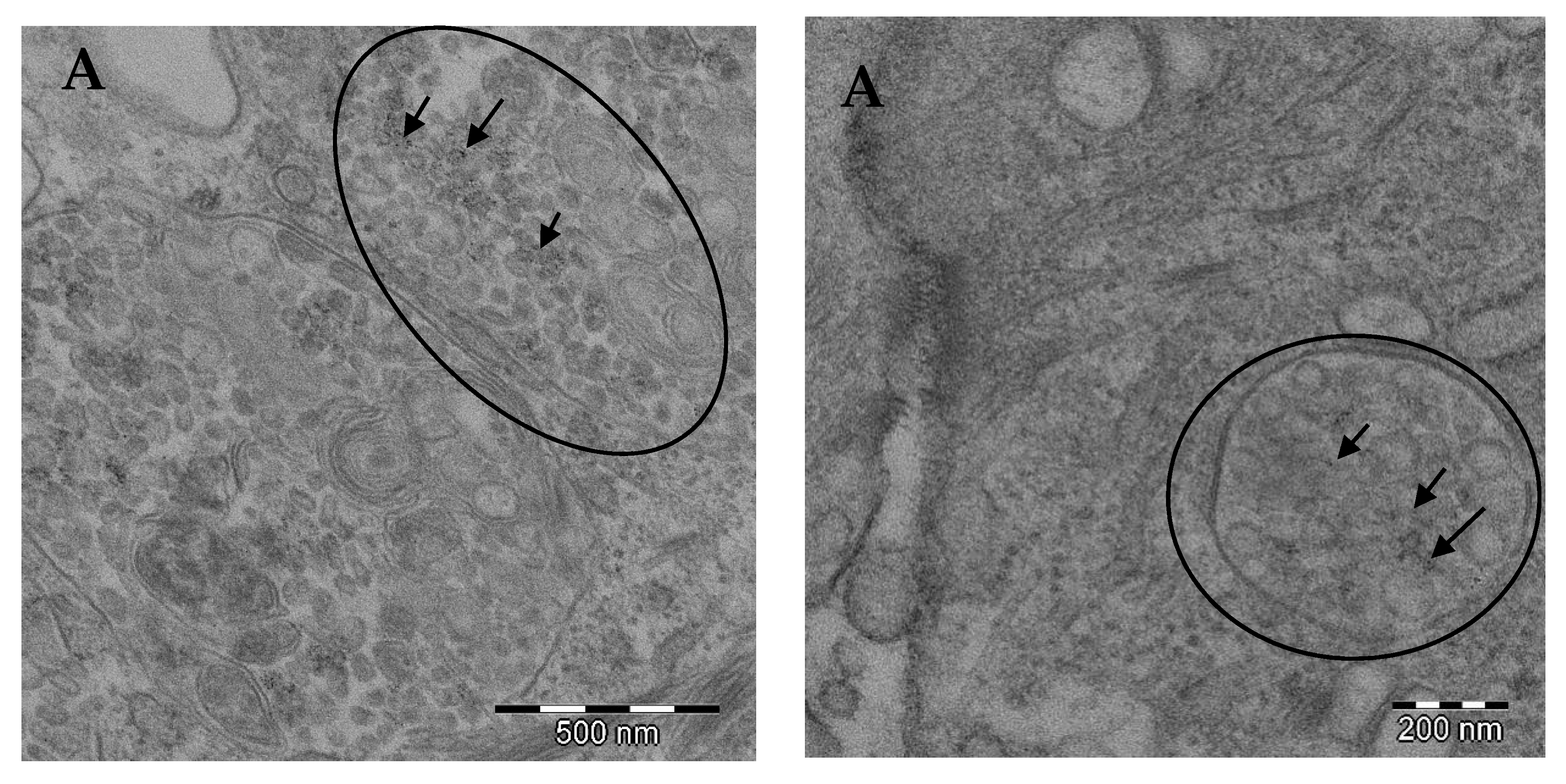
18]. In the present study, the uptake and distribution of the iron nanoparticles in the different compartments of colorectal cells were qualitatively visualized by TEM and quantified by ICP-MS. Although there is an increasing number of studies focused on understanding the interaction between the particles and the biological systems, publications about the mechanism of uptake and biodistribution are not yet clear [
19]. The results of this study show that exposure of cells to ultra-small iron nanoparticles produced an increase in the levels of all the elements studied in the subcellular fraction corresponding to the mitochondria except copper (which seems to be directed towards the nucleus where an increase in its levels was detected). This traffic of elements towards this fraction could indicate their participation in the cellular processes involving mitochondria and lysosomes induced by iron nanoparticles that are detailed below.
The results revealed that the greatest abundance of iron at baseline levels resides in the subcellular fraction corresponding to the mitochondria. It is possible that FeNP went to the mitochondria fraction through the endosome-lysosome-autolysosome pathway, instead of going to mitochondria directly. After the exposure of the cells to the different treatments with NPs, the results showed that iron levels increased in all subcellular fractions, but especially in the fraction corresponding to the mitochondria, where levels increased 62 fold when the cells were exposed to FeNP, and 94 fold when they were exposed to Venofer
®. These results are consistent with those obtained by TEM and with previous experiments that revealed a faster solubilization of Venofer
® than the synthetic FeNPs. Such solubilization might increase the bioavailability of ionic Fe to get into the mitochondria once the NPs have been internalized as such, as can be observed in
Figure 1. It is known that iron is transported to the mitochondria, where it is utilized for synthesis of cofactors essentials for the function of enzymes involved in oxidation-reduction reactions, DNA synthesis and repair, and a variety of other cellular processes. Nowadays, the trafficking of iron to the mitochondria and normal mitochondrial iron metabolism, including heme synthesis and iron-sulfur cluster biogenesis, are being investigated [
20].
There are different factors that could modify the clearance of this type of particles into the cells, like size, shape, or surface properties [
21]. Recently, Feng and co-workers [
7] studied the intracellular traffic of iron nanoparticles and the uptake in in vitro models. In their study, the iron nanoparticles were deposited in the cell structure within membrane-bound structures like lysosomes or endosome after the exposition, but not in mitochondria. This fact could be due to the exposition time of these experiments (2 h). In this way, the exposition time used in the present study (48 h) may explain a more compartmentalized distribution of this type of particles. On the other hand, the reduced size of the particles employed in the present study could promote the particles to cross the membranes and their transport to the mitochondria. Nevertheless, other authors that studied the intracellular traffic of other types of metallic nanoparticles, in particular, silver nanoparticles, suggested that they could be located close to the mitochondria [
22,
23].
At the present time, iron nanoparticles are being developed as an effective carrier in gene therapy [
18]. In that way, some approaches based on metallic nanoparticles, like iron oxide nanoparticles [
24] or gold silver alloy nanoparticles [
25] have been employed to deliver genetic material to mitochondrial tissue. Therefore, ultra-small iron nanoparticles could be employed as a new tool due to their small size to be directed to the mitochondria.
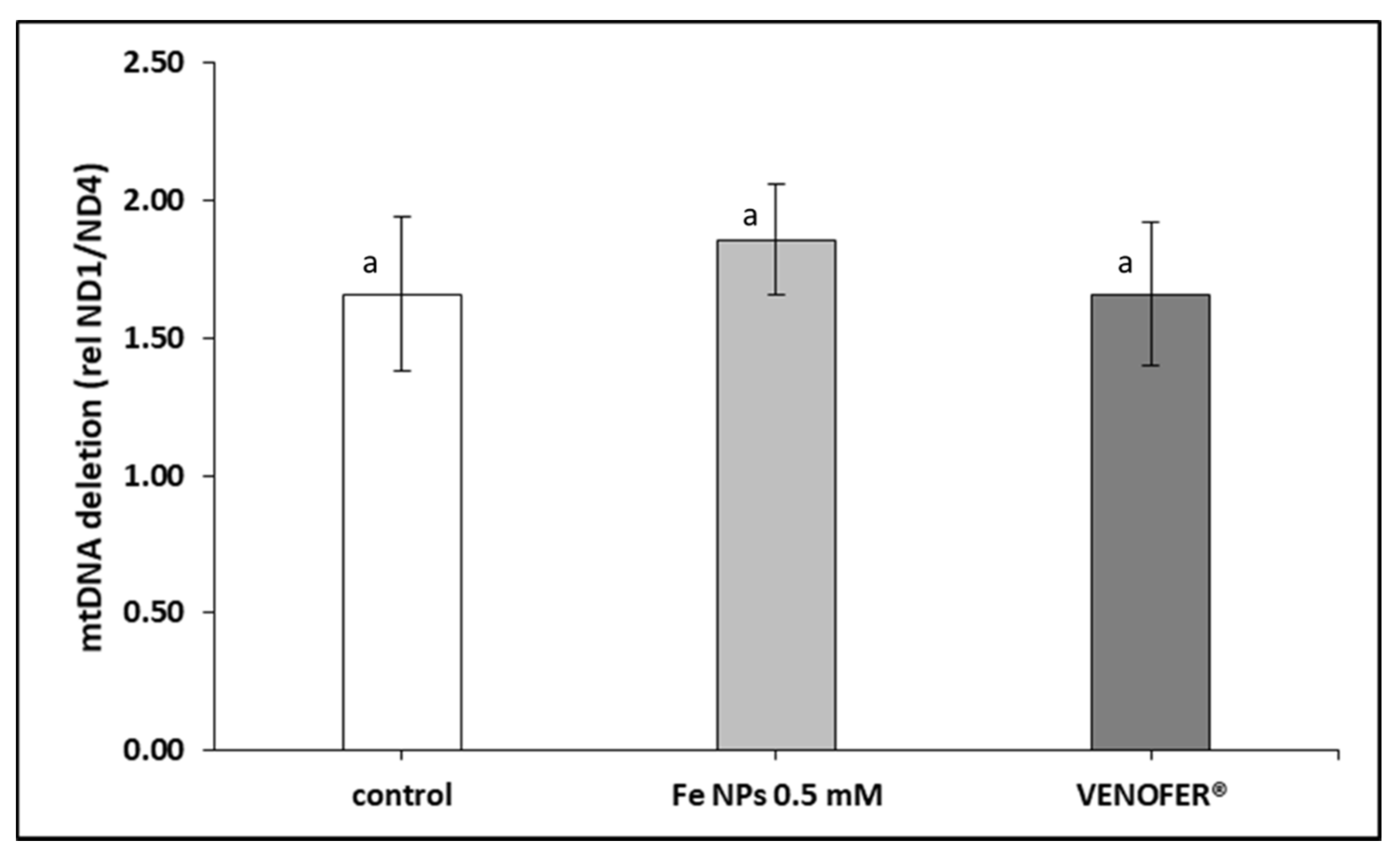
Therefore, after confirming the nanoparticles localization, we evaluated the integrity of mitochondrial DNA and the respiratory activity of mitochondria. The effects of the FeNPs on mitochondria was elucidated measuring mtDNA copy number and deletions, and the respiratory activity. The ratio between the mtDNA genes ND4 and ND1 is considered as a marker of the so-called common deletion 20. This is because ND1 is in a very stable region of the mtDNA. Meanwhile, ND4 is in the middle of the region that suffer the common deletion [
26]. mtDNA deletions may be considered as markers of mtDNA damage of oxidative origin or other [
27]. In the present study, cells exposed to FeNPs and Venofer
® did not cause changes in this ratio, which can be interpreted as the absence of damage at the mtDNA. However, results showed a decrease in copy number of mtDNA. mtDNA copy number is considered as an indirect marker of mitochondrial function [
28]. Also, some authors related the low copy number with the decrease of mitochondrial replicative activity [
29] or with an increase in mitochondrial destruction for example through autophagy 11.
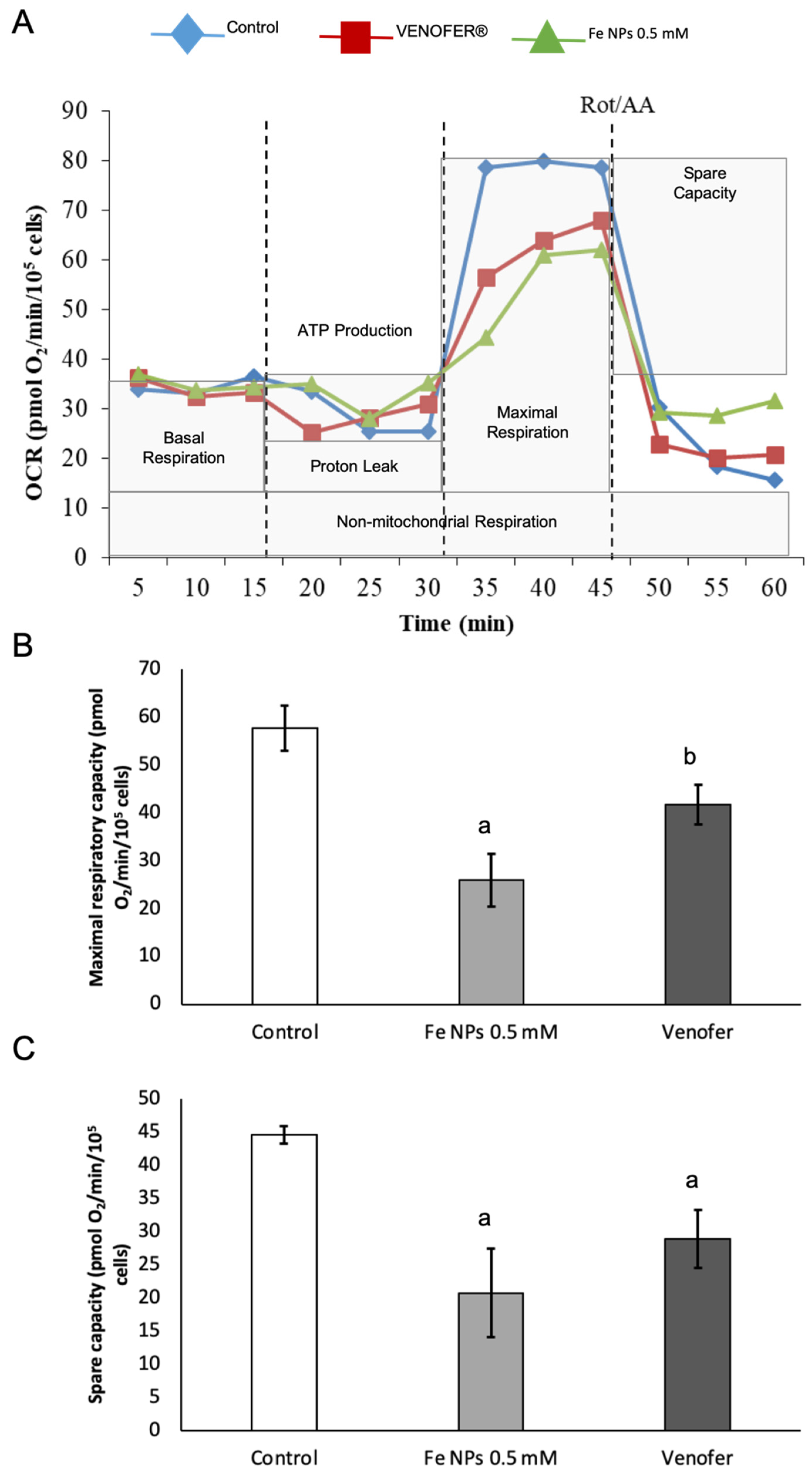
Results found in an mtDNA copy study were confirmed after analyzing mitochondrial respiration by Seahorse technology. In fact,
Figure 5 shows how the ultra-small FeNPs affect maximal respiration and mitochondrial spare respiratory capacity, which is regarded as an important aspect of mitochondrial function and is defined as the difference between basal ATP production and its maximal activity. When cells are subjected to stress, energy demand increases, requiring more ATP to maintain cellular functions. A cell with a larger spare respiratory capacity can produce more ATP and overcome more stress, including oxidative stress [
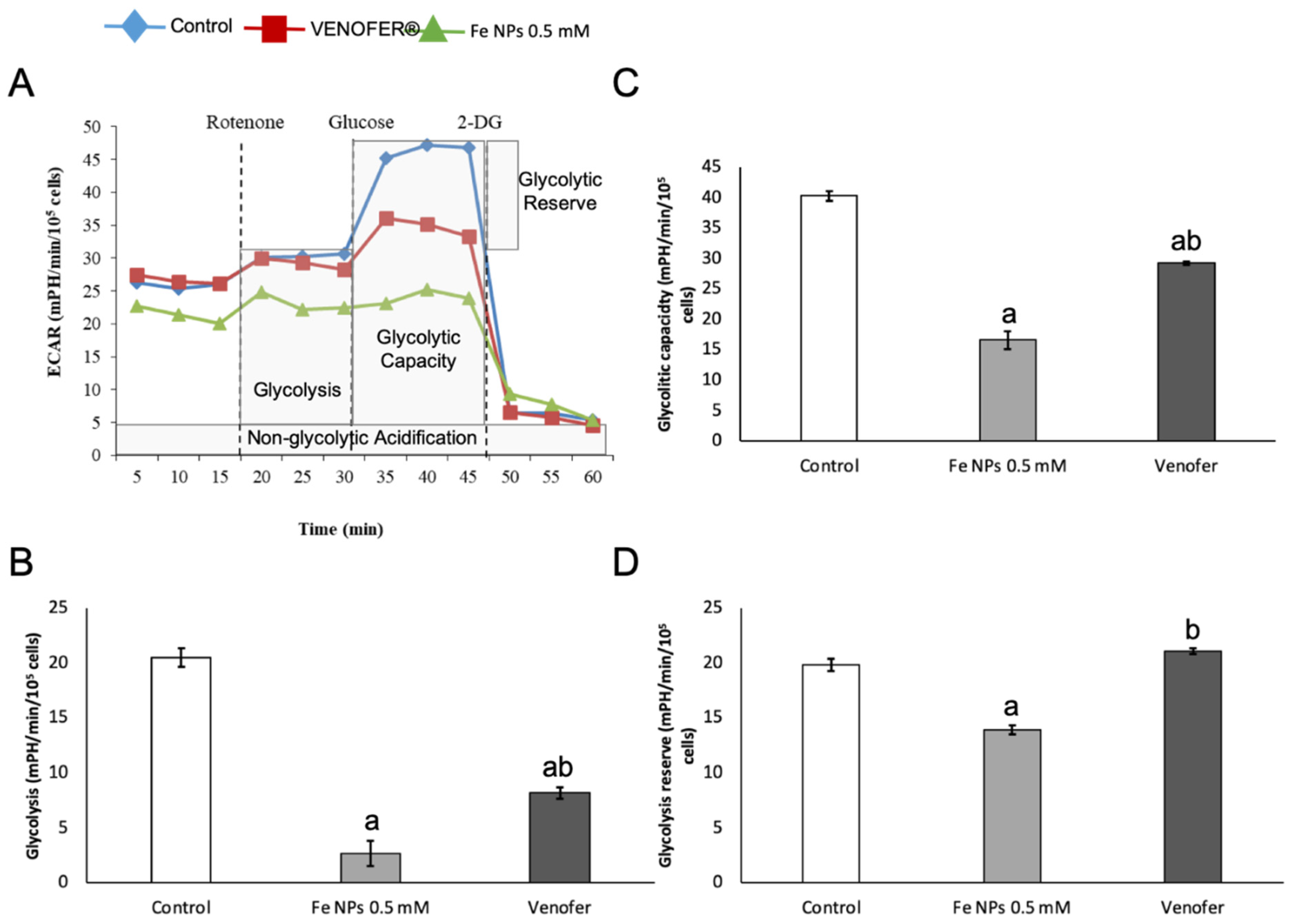
30]. Overall, changes found in mitochondrial function after nanoparticles administration present a very interesting opportunity for ultra-small iron nanoparticles as antiproliferative agents in cancer, since they would allow the reduction of metabolic activity in cancer cells when administered locally in the tumor, without causing alterations in mitochondrial DNA from adjacent healthy cells.
Then, from the study, it can be demonstrated that iron particles decreased mitochondria functionality without increasing mtDNA damage. This could be used to promote cell death using the mitochondria as a target, when necessary, without affecting healthy neighbor cells. Preserving the integrity of the mtDNA of adjacent cells is a very important advantage of this possible therapeutic tool when it is administered locally, since the strategy of producing cell death through oxidative damage is not very selective and could compromise neighboring cells. Some of these effects of nanomaterials have been proposed previously by other authors [
9], but the effect on the respiratory function have not been elucidated yet. Consequently, the present work could contribute to understanding the relation between iron nanoparticles and mitochondrial function.
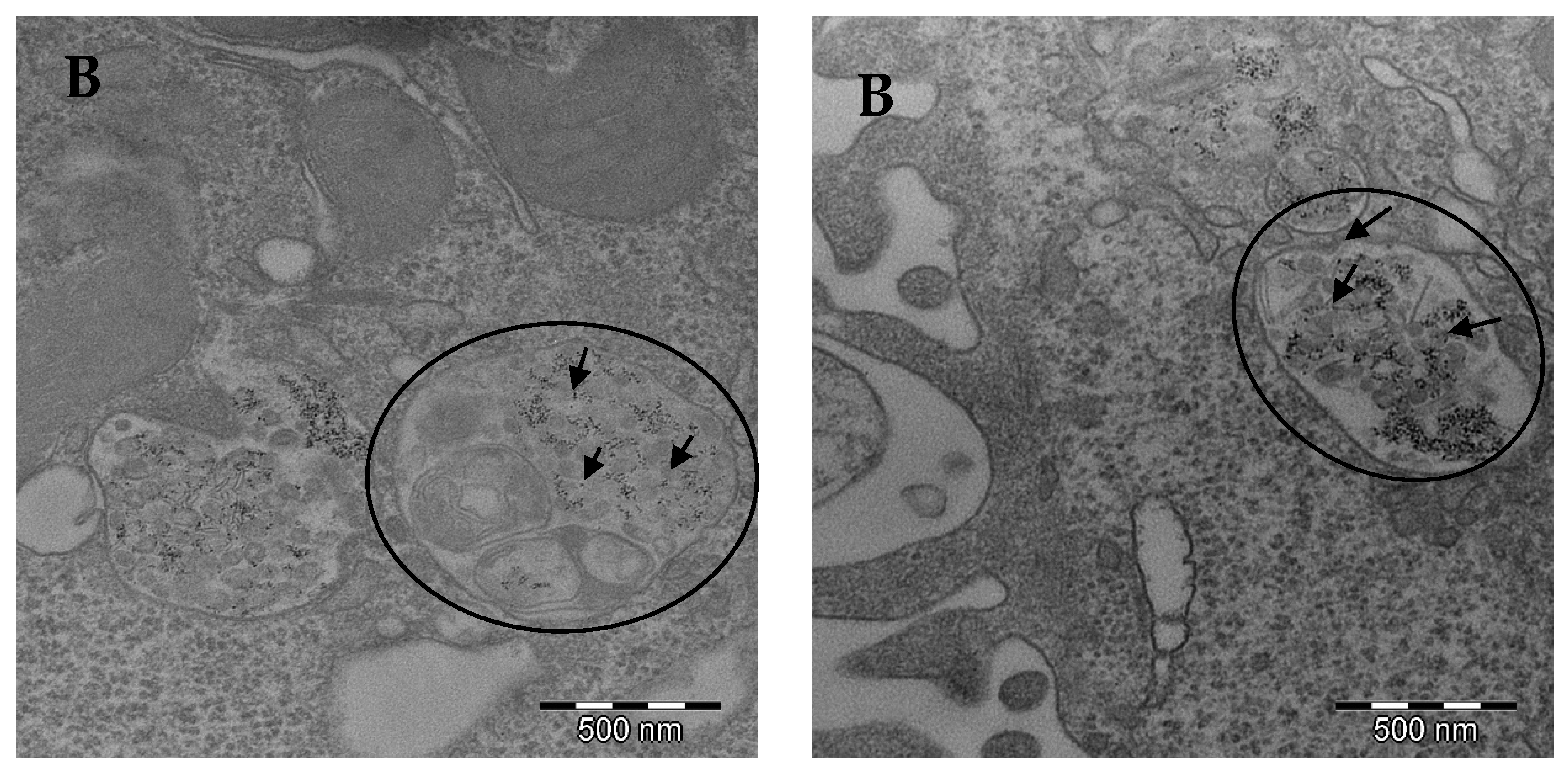
Autophagy is an intracellular degradation process associated with an appropriate replacement of certain types of cellular organelles, and it is important for the cell [
12]. Some authors have proposed autophagy as one of the possible mechanisms of the nanoparticle mechanism of action in cells [
31]. In that way, the exposition of different cell lines to metallic nanoparticles has been related to the activation of the autophagic process [
4]. Certain authors [
32] showed that rare-earth elements nanoparticles, samarium (Sm)/europium (Eu) and gadolinium (Gd)/terbium(Tb), induced severe autophagy in cervical cancer cells (HeLa). Sun and co-workers [
33] demonstrated the autophagic process and the mitochondrial damage of silica particles in hepatocytes. The relation of autophagy in mitochondria has not been deeply studied. Yoo and co-workers [
34] suggested that one link between autophagy and mitochondria is the process called mitophagy, the selective degradation of mitochondria by autophagy. By means of this process, the excess or defective mitochondria following damage or stress (i.e., oxidative stress) is removed. Damaged mitochondria cause a depletion in ATP and a release of cytochrome c, which leads to activation of caspases and onset of apoptosis. This process courses with the sequestration and hydrolytic degradation by lysosomes [
35,
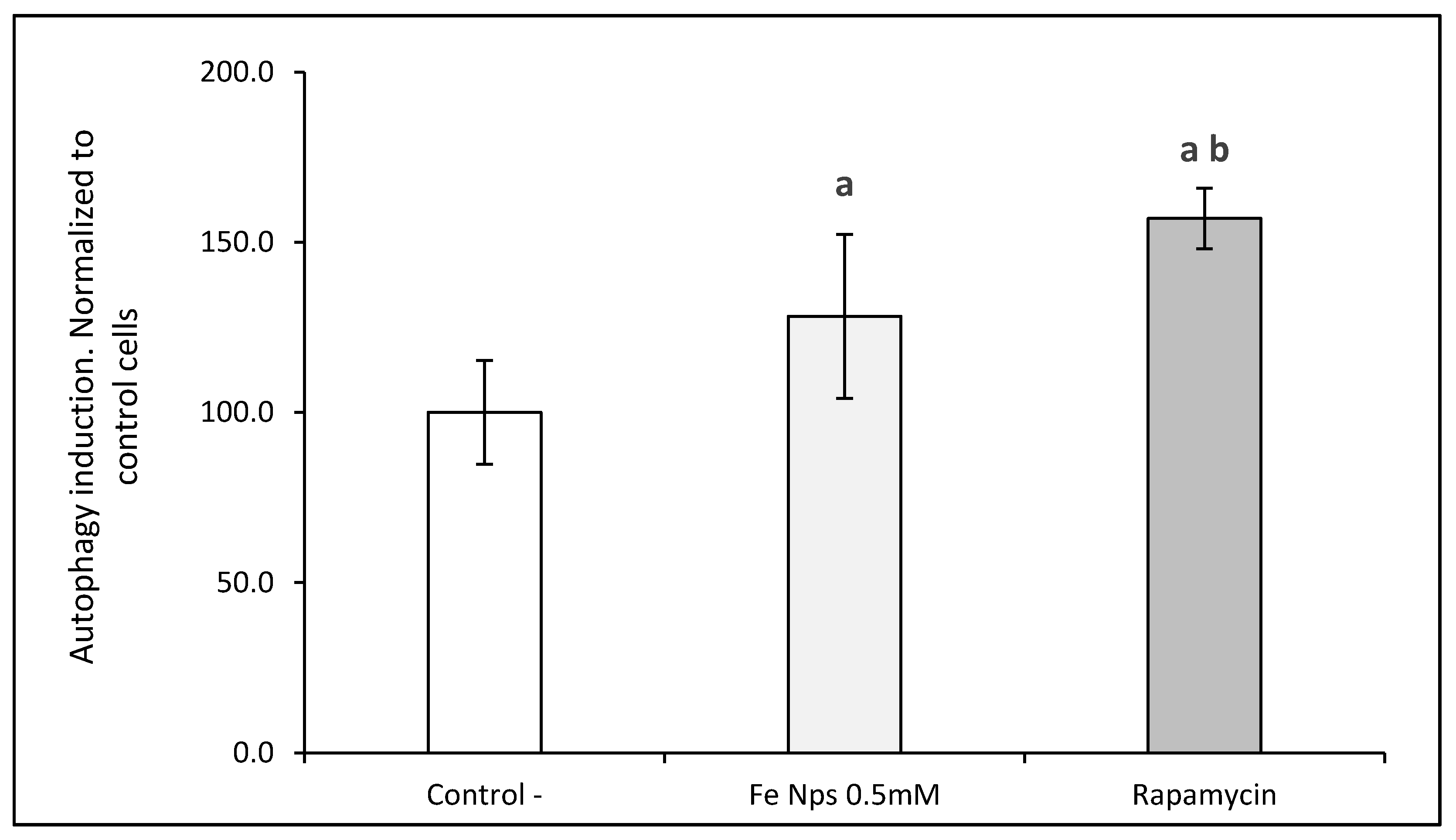
36]. In our study, confocal microscopy results suggested a greater autophagic activity in FeNP-treated cells. This result agrees with those obtained from the TEM images.
Moreover, to determine if the autophagic flux was initiated by the FeNPs, p62 was determined. In that way, p62 decreased when cells were treated with nanoparticles producing a similar effect of rapamycin. In that way, p62 low levels and high levels of autophagy induction could suggest that the beginning of the autophagic process was mediated by the nanomaterials [
37]. Consequently, these nanoparticles induced the autophagy in cells.
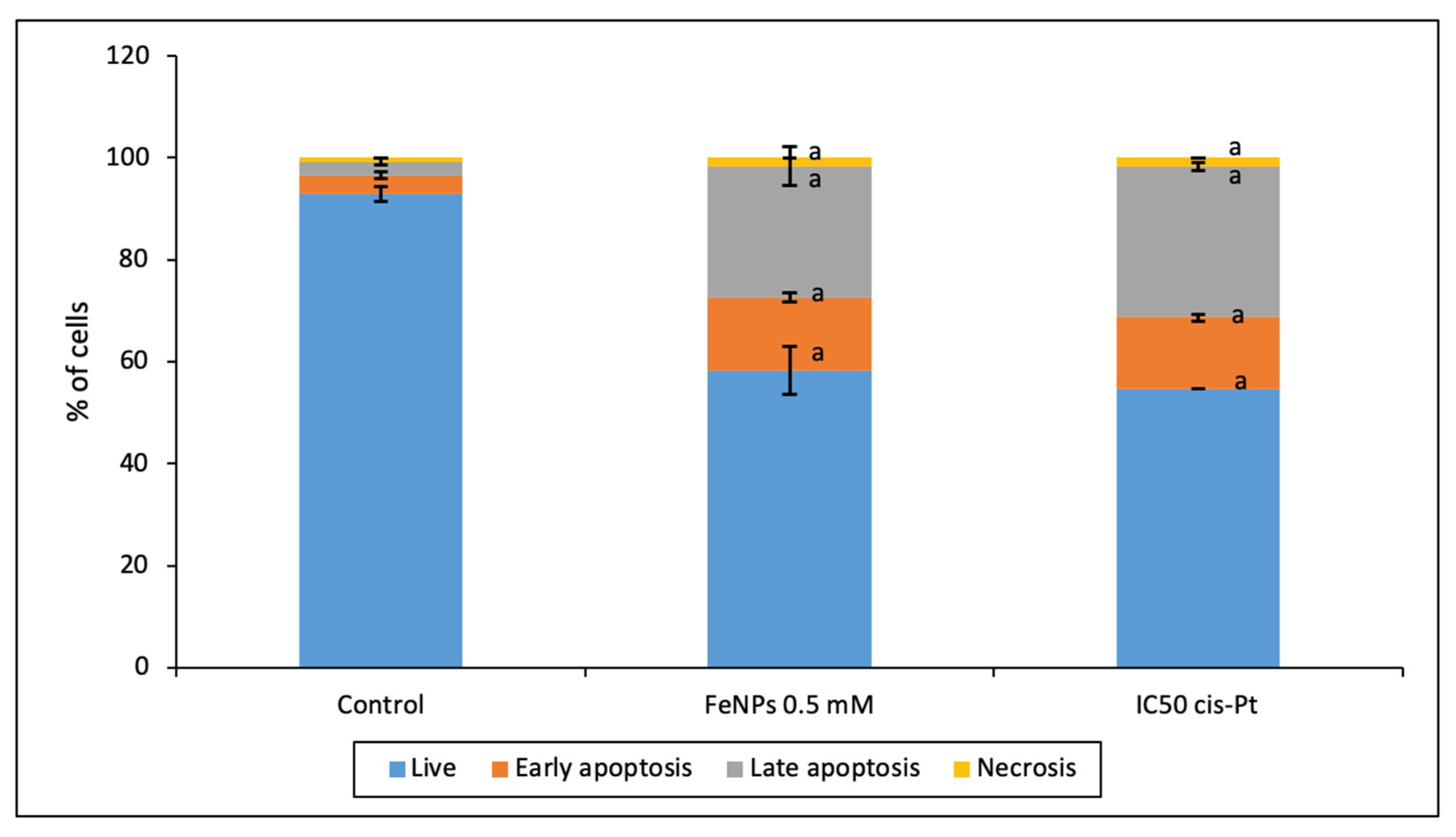
Furthermore, the ability of ultra-small iron nanoparticles to induce apoptosis/necrosis in colorectal tumoral cells was evaluated.
Figure 10 shows that at the present conditions FeNPs induced mainly apoptosis and low necrosis levels at similar conditions that IC
50 cis-platinum promoted. The role of iron nanoparticles in the cell death mechanism has been previously investigated by other authors that described that the rate of apoptosis induction depends on the size, form, and shape of nanomaterials [
7]. Indeed, this effect could be mediated by the capacity of Fe to conjugate with ROS and affect cell membranes, proteins, or mitochondria [
38].
Indeed, the results obtained showed that ultra-small nanoparticles could be used to promote cell death using the mitochondria as a target, when necessary, although the impact on healthy neighbor cells remains to be determined.
In a previous investigation from our research group [
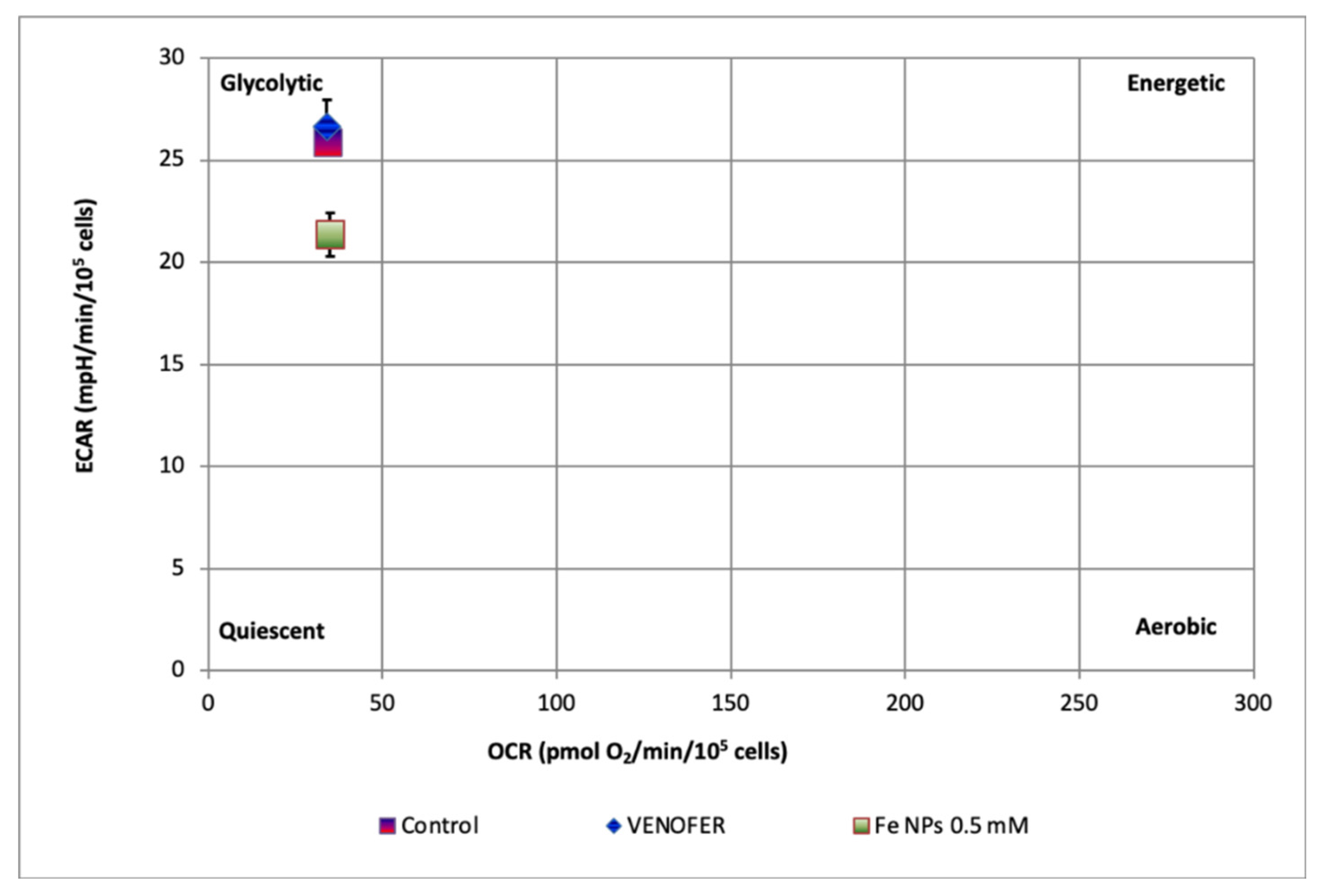
15], it was observed that the same FeNPs tested in this study increased ROS generation, which could lead to an increased mitochondrial autophagy as consequence of the increased oxidative damage at this organelle. This situation would cause the reported fall in mitochondrial respiration, as well as in glycolytic capacity. This sequence of events would be responsible for the entry of the cells into the quiescence state.

 ,
, 

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}