
Preparation and In Vitro/In Vivo Evaluation of Orally Disintegrating/Modified-Release Praziquantel Tablets


Abstract
:1. Introduction
2. Materials and methods
2.1. Materials
2.2. Extrudate Preparation
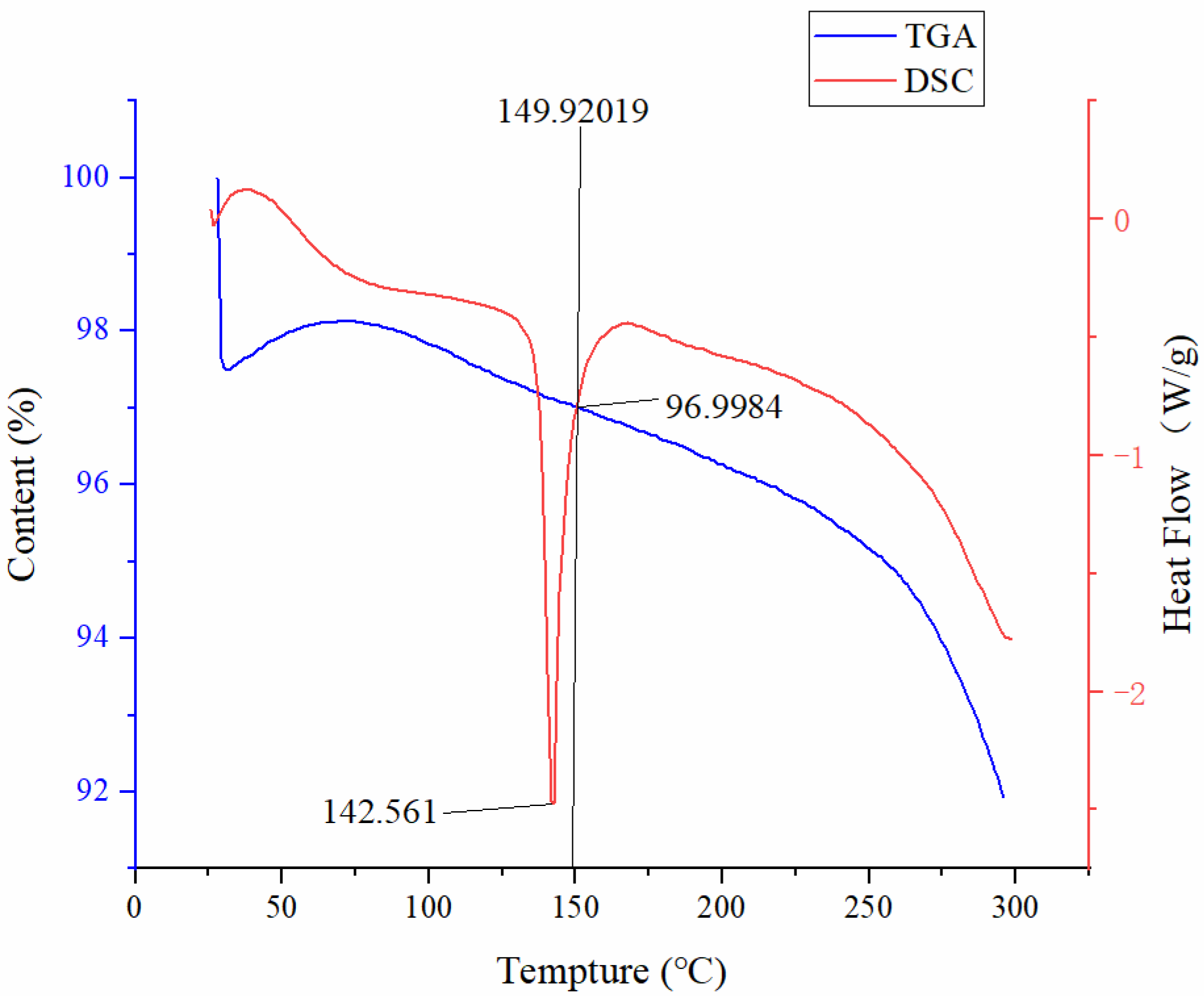
2.3. Thermogravimetric Analysis (TGA)
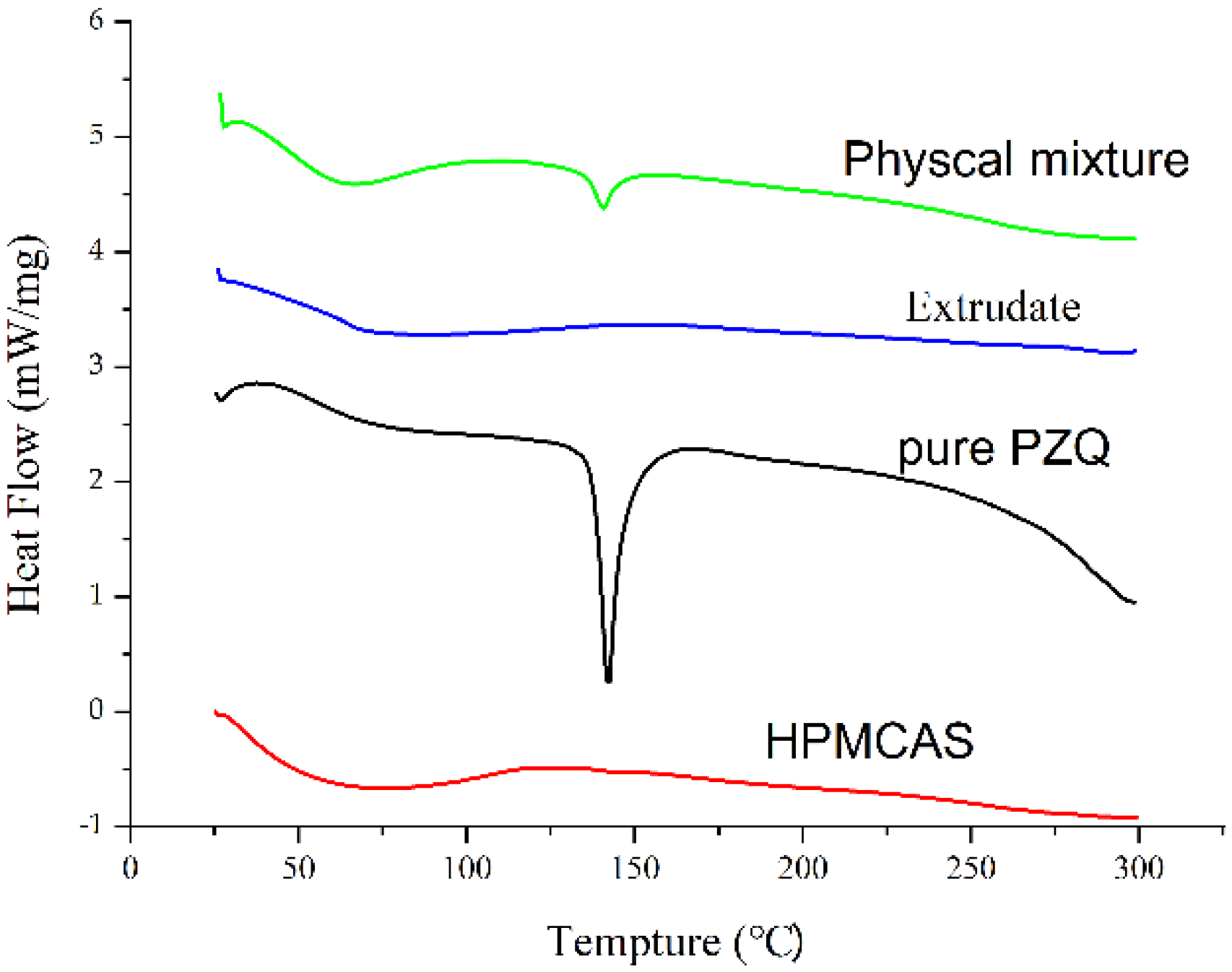
2.4. Differential Scanning Calorimetry (DSC) Analysis
2.5. Powder X-Ray Diffraction (PXRD)
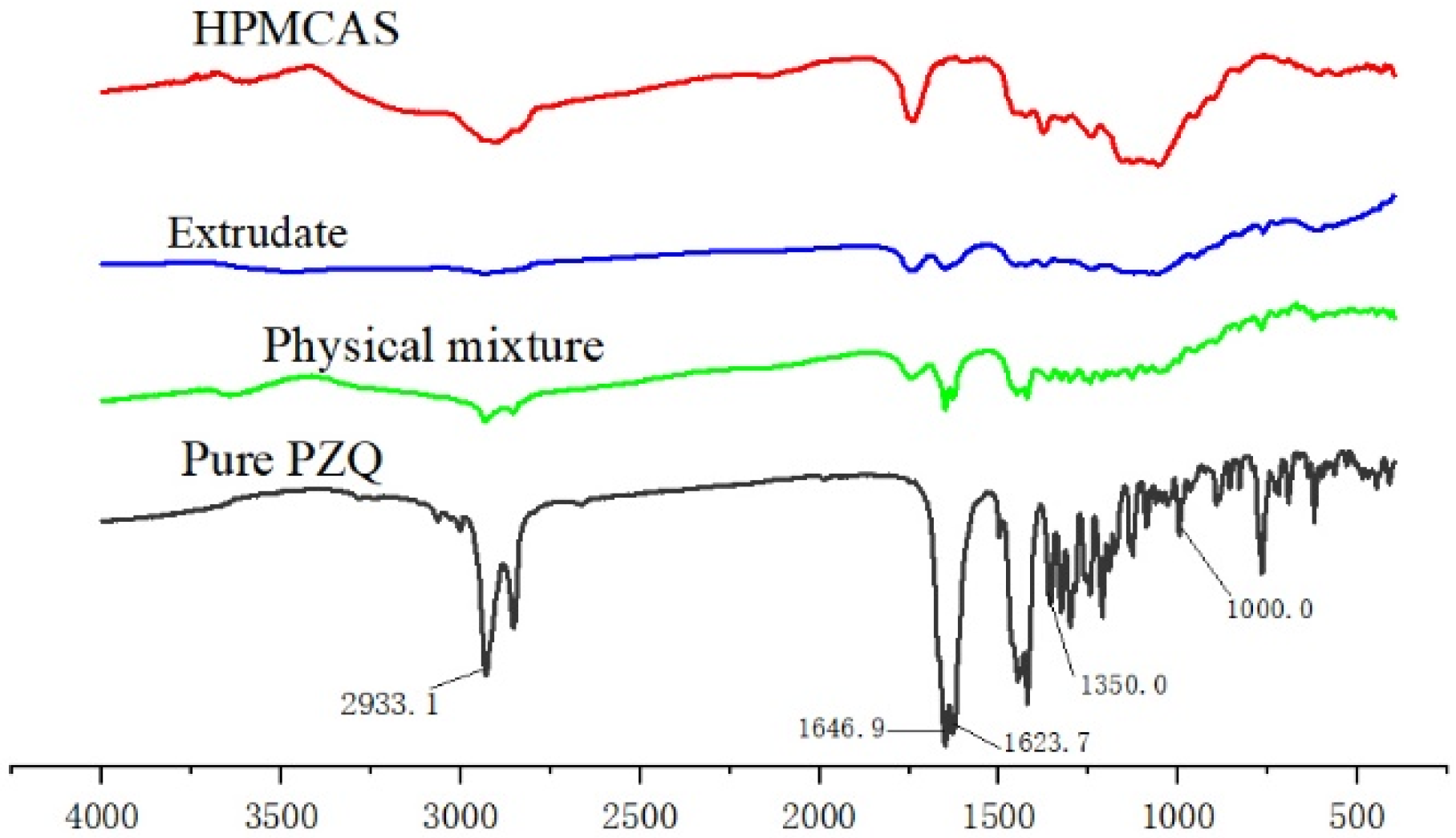
2.6. Fourier-Transform Infrared (FTIR) Spectroscopy
2.7. Drug Content Analysis
2.8. Preparation of PZQ ODSRTs
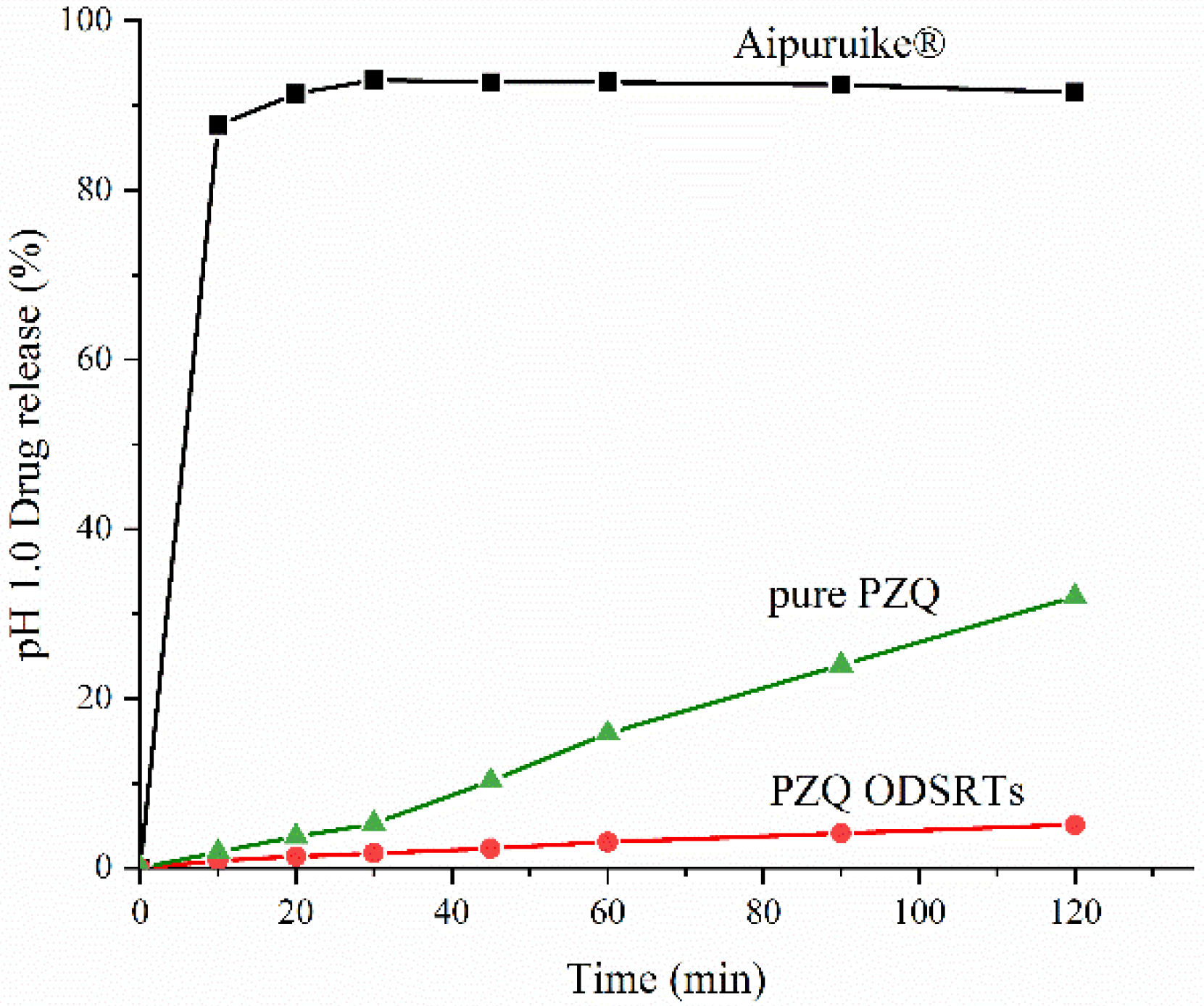
2.9. In Vitro Drug Release
2.10. Disintegration Time
2.11. Friability and Hardness Test
2.12. Stability Studies
2.13. Pharmacokinetic Analysis
2.13.1. Plasma Sample Preparation for Analysis
2.13.2. Statistical Analysis
3. Results
3.1. Preparation of the Hot-Melt Extrudate
3.2. Physical Characterization of the Extrudate
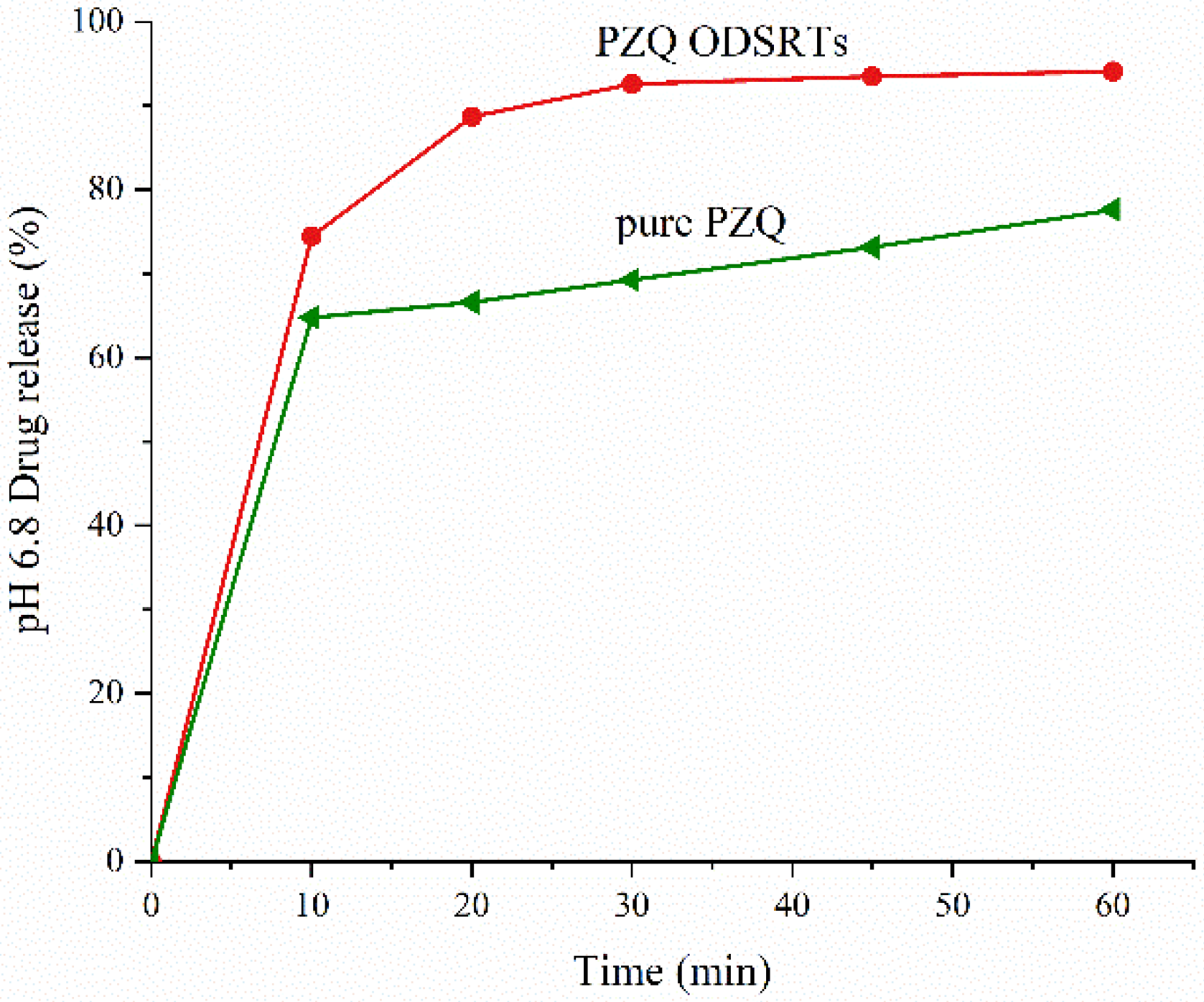
3.3. In Vitro Drug Release
3.4. Disintegration Time
3.5. Friability and Hardness Test
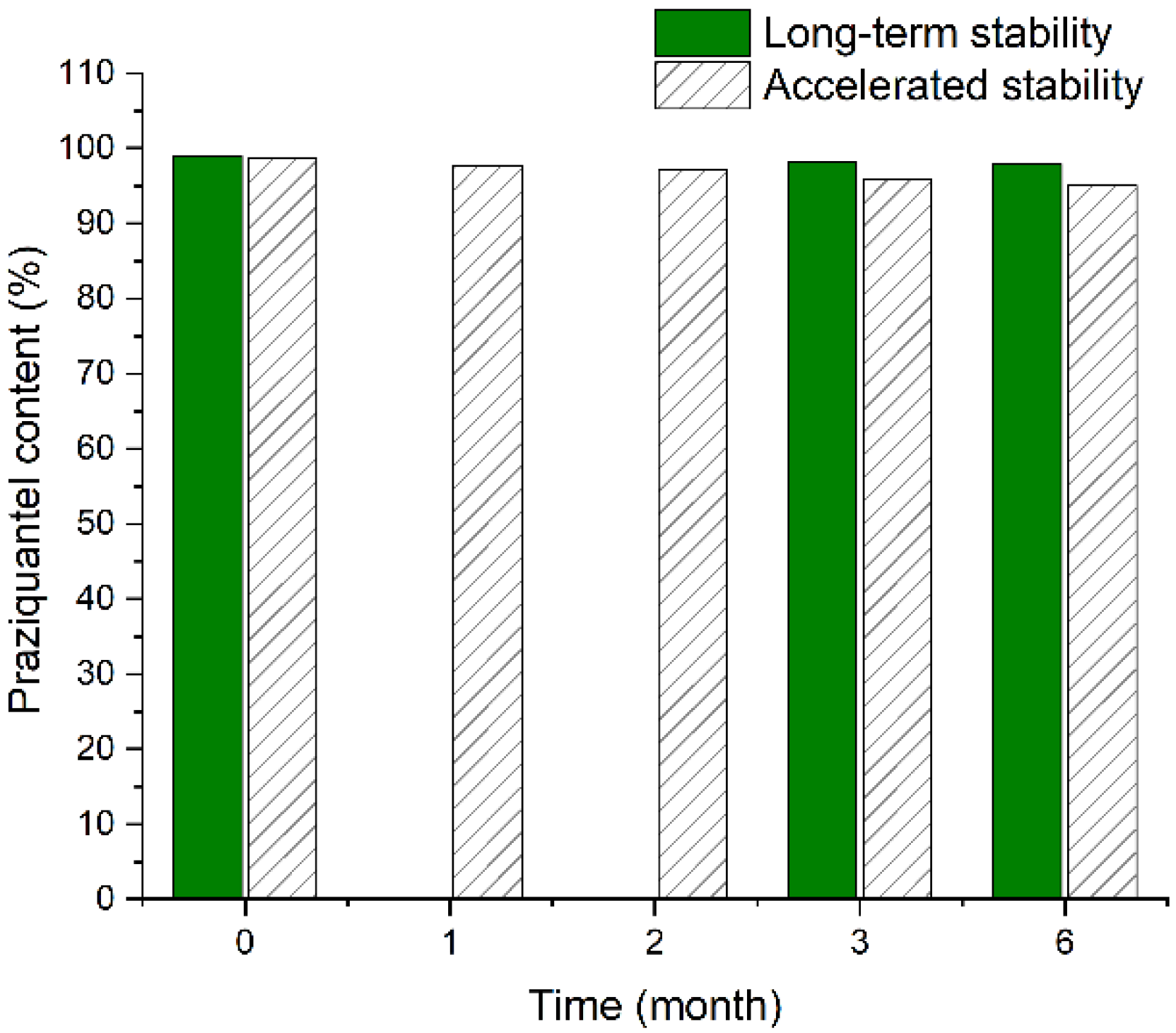
3.6. Stability
3.7. Pharmacokinetics of the PZQ ODSRTs in Dogs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jacob, J.; Lorber, B. Diseases transmitted by man’s best friend: The dog. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef] [Green Version]
- Shan, X.; Liu, S.; Liu, J.; Zhu, H.; Xiao, Y.; Chen, Y. Geographical survey of the intermediate host of Schistosoma japonicum: Toward precise management of Oncomelania hupensis. PLoS Negl. Trop. Dis. 2020, 14, e0008674. [Google Scholar] [CrossRef]
- Bergquist, R.; Utzinger, J.; Keiser, J. Controlling schistosomiasis with praziquantel: How much longer without a viable alternative? Infect. Dis. Poverty 2017, 6, 74. [Google Scholar] [CrossRef] [Green Version]
- Faust, C.L.; Osakunor, D.N.M.; Downs, J.A.; Kayuni, S.; Stothard, J.R.; Lamberton, P.H.L.; Reinhard-Rupp, J.; Rollinson, D. Schistosomiasis control: Leave no age group behind. Trends Parasitol. 2020, 36, 582–591. [Google Scholar] [CrossRef]
- McManus, D.P.; Zhang, W.; Li, J.; Bartley, P.B. Echinococcosis. Lancet 2003, 362, 1295–1304. [Google Scholar] [CrossRef]
- Chapalamadugu, K.C.; Busboom, J.R.; Nelson, M.L.; Hancock, D.D.; Tang, J.; Jasmer, D.P. Taenia taeniaeformis: Effectiveness of staining oncospheres is related to both temperature of treatment and molecular weight of dyes utilized. Vet. Parasitol. 2008, 151, 203–211. [Google Scholar] [CrossRef]
- Yu, Q.F.; Zhang, J.Y.; Sun, M.T.; Gu, M.M.; Zou, H.Y.; Webster, J.P.; Lu, D.B. In vivo praziquantel efficacy of Schistosoma japonicum over time: A systematic review and meta-analysis. Acta Trop. 2021, 222, 106048. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, K.; Sakamoto, N.; Kobayashi, K.; Iwabuchi, S.; Nakamura-Uchiyama, F. Therapeutic effect of praziquantel against Taeniasis asiatica. Int. J. Infect. Dis. 2013, 17, e656–e657. [Google Scholar] [CrossRef] [Green Version]
- Pawlowski, Z.S. Role of chemotherapy of taeniasis in prevention of neurocysticercosis. Parasitol. Int. 2006, 55, S105–S109. [Google Scholar] [CrossRef] [PubMed]
- Cucchetto, G.; Buonfrate, D.; Marchese, V.; Rodari, P.; Ferrari, A.; Zanotti, P.; Bottieau, E.; Silva, R.; Bisoffi, Z.; Gobbi, F. High-dose or multi-day praziquantel for imported schistosomiasis? A systematic review. J. Travel Med. 2019, 26. [Google Scholar] [CrossRef] [PubMed]
- Gryseels, B. Schistosomiasis. Infect. Dis. Clin. N. Am. 2012, 26, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Zhou, X.N.; Zhang, H.B.; Tao, Y.; Huo, L.L.; Liu, N. Slow-release praziquantel for dogs: Presentation of a new formulation for echinococcosis control. Infect. Dis. Poverty 2017, 6, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schimmel, A.; Altreuther, G.; Schroeder, I.; Charles, S.; Cruthers, L.; Kok, D.J.; Kraemer, F.; Krieger, K.J. Efficacy of emodepside plus praziquantel tablets (Profender tablets for dogs) against mature and immature adult Trichuris vulpis infections in dogs. Parasitol. Res. 2009, 105 (Suppl. 1), S17–S22. [Google Scholar] [CrossRef]
- Gutiérrez, M.L.; Di Federico, G.; Dale, J.A.; Minoia, J.M.; Corrales, C.D.; Schaiquevich, P.; Wikinski, S. Pharmacokinetics of a novel spot-on formulation of praziquantel for dogs. Vet. Parasitol. 2017, 239, 46–49. [Google Scholar] [CrossRef]
- Lindenberg, M.; Kopp, S.; Dressman, J.B. Classification of orally administered drugs on the World Health Organization model list of essential medicines according to the biopharmaceutics classification system. Eur. J. Pharm. Biopharm. 2004, 58, 265–278. [Google Scholar] [CrossRef]
- Yamasaki, K.; Taguchi, K.; Nishi, K.; Otagiri, M.; Seo, H. Enhanced dissolution and oral bioavailability of praziquantel by emulsification with human serum albumin followed by spray drying. Eur. J. Pharm. Sci. 2019, 139, 105064. [Google Scholar] [CrossRef]
- Tawfik, E.A.; Scarpa, M.; Abdelhakim, H.E.; Bukhary, H.A.; Craig, D.Q.M.; Barker, S.A.; Orlu, M. A potential alternative orodispersible formulation to prednisolone sodium phosphate orally disintegrating tablets. Pharmaceutics 2021, 13, 120. [Google Scholar] [CrossRef]
- Freitag, F.; Taylor, F.R.; Hamid, M.A.; Rodgers, A.; Hustad, C.M.; Ramsey, K.E.; Skobieranda, F. Elimination of migraine-associated nausea in patients treated with rizatriptan orally disintegrating tablet (ODT): A randomized, double-blind, placebo-controlled study. Headache 2008, 48, 368–377. [Google Scholar] [CrossRef]
- Fullerton, L.; Weiss, S.J.; Froman, P.; Oglesbee, S.; Cheney, P. Ondansetron oral dissolving tablets are superior to normal saline alone for prehospital nausea. Prehosp. Emerg. Care 2012, 16, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Stark, J.G.; Engelking, D.; McMahen, R.; Sikes, C. Pharmacokinetics of a novel amphetamine extended-release orally disintegrating tablet in children with attention-deficit/hyperactivity disorder. J. Child Adolesc. Psychopharmacol. 2017, 27, 216–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulsun, T.; Ozturk, N.; Kaynak, M.S.; Vural, I.; Sahin, S. Preparation and evaluation of furosemide containing orally disintegrating tablets by direct compression. Pharmazie 2017, 72, 389–394. [Google Scholar] [CrossRef]
- Stange, U.; Führling, C.; Gieseler, H. Freeze drying of orally disintegrating tablets containing taste masked naproxen sodium granules in blisters. Pharm. Dev. Technol. 2015, 20, 1018–1024. [Google Scholar] [CrossRef]
- Gryczke, A.; Schminke, S.; Maniruzzaman, M.; Beck, J.; Douroumis, D. Development and evaluation of orally disintegrating tablets (ODTs) containing Ibuprofen granules prepared by hot melt extrusion. Colloids Surf. B Biointerfaces 2011, 86, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.H.; Min, J.H.; Hwang, K.M.; Park, E.S. Development of sustained-release microparticles containing tamsulosin HCl for orally disintegrating tablet using melt-adsorption method. Drug Deliv. Transl. Res. 2018, 8, 552–564. [Google Scholar] [CrossRef]
- Xu, T.; Nahar, K.; Dave, R.; Bates, S.; Morris, K. Polymorphic transformation of indomethacin during hot melt extrusion granulation: Process and dissolution control. Pharm. Res. 2018, 35, 140. [Google Scholar] [CrossRef]
- Yeung, C.W.; Rein, H. Hot-melt extrusion of sugar-starch-pellets. Int. J. Pharm. 2015, 493, 390–403. [Google Scholar] [CrossRef]
- Speer, I.; Preis, M.; Breitkreutz, J. Prolonged drug release properties for orodispersible films by combining hot-melt extrusion and solvent casting methods. Eur. J. Pharm. Biopharm. 2018, 129, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Solanki, N.G.; Kathawala, M.; Serajuddin, A.T.M. Effects of surfactants on itraconazole-hydroxypropyl methylcellulose acetate succinate solid dispersion prepared by hot melt extrusion III: Tableting of extrudates and drug release from tablets. J. Pharm. Sci. 2019, 108, 3859–3869. [Google Scholar] [CrossRef]
- Li, D.; Guo, G.; Fan, R.; Liang, J.; Deng, X.; Luo, F.; Qian, Z. PLA/F68/dexamethasone implants prepared by hot-melt extrusion for controlled release of anti-inflammatory drug to implantable medical devices: I. Preparation, characterization and hydrolytic degradation study. Int. J. Pharm. 2013, 441, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Sarabu, S.; Bandari, S.; Kallakunta, V.R.; Tiwari, R.; Patil, H.; Repka, M.A. An update on the contribution of hot-melt extrusion technology to novel drug delivery in the twenty-first century: Part II. Expert Opin. Drug Deliv. 2019, 16, 567–582. [Google Scholar] [CrossRef] [PubMed]
- Pimparade, M.B.; Morott, J.T.; Park, J.B.; Kulkarni, V.I.; Majumdar, S.; Murthy, S.N.; Lian, Z.; Pinto, E.; Bi, V.; Durig, T.; et al. Development of taste masked caffeine citrate formulations utilizing hot melt extrusion technology and in vitro-in vivo evaluations. Int. J. Pharm. 2015, 487, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Xu, N.; Dong, J.; Yang, Y.; Ai, X. Pharmacokinetics and residue depletion of praziquantel in rice field eels Monopterus albus. Dis. Aquat. Org. 2016, 119, 67. [Google Scholar] [CrossRef]
- Tang, S.; Chen, L.; Guo, Z.; Hu, X.; He, J.; Wang, G.; Zhao, T.; Xiao, X. Pharmacokinetics of a new ivermectin/praziquantel oil suspension after intramuscular administration in pigs. Vet. Parasitol. 2012, 185, 229–235. [Google Scholar] [CrossRef]
- Kallakunta, V.R.; Sarabu, S.; Bandari, S.; Tiwari, R.; Patil, H.; Repka, M.A. An update on the contribution of hot-melt extrusion technology to novel drug delivery in the twenty-first century: Part I. Expert Opin. Drug Deliv. 2019, 16, 539–550. [Google Scholar] [CrossRef]
- Sarode, A.L.; Obara, S.; Tanno, F.K.; Sandhu, H.; Iyer, R.; Shah, N. Stability assessment of hypromellose acetate succinate (HPMCAS) NF for application in hot melt extrusion (HME). Carbohydr. Polym. 2014, 101, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, Y.; Zhao, Y.; Ding, Z.; Fan, Z.; Zhang, H.; Liu, M.; Wang, Z.; Han, J. Effect of HPMCAS on recrystallization inhibition of nimodipine solid dispersions prepared by hot-melt extrusion and dissolution enhancement of nimodipine tablets. Colloids Surf. B Biointerfaces 2018, 172, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Murdande, S.B.; Pikal, M.J.; Shanker, R.M.; Bogner, R.H. Solubility advantage of amorphous pharmaceuticals, part 3: Is maximum solubility advantage experimentally attainable and sustainable? J. Pharm. Sci. 2011, 100, 4349–4356. [Google Scholar] [CrossRef]
- Guo, Z.; Lu, M.; Li, Y.; Pang, H.; Lin, L.; Liu, X.; Wu, C. The utilization of drug-polymer interactions for improving the chemical stability of hot-melt extruded solid dispersions. J. Pharm. Pharmacol. 2014, 66, 285–296. [Google Scholar] [CrossRef]
- Xi, L.; Song, H.; Wang, Y.; Gao, H.; Fu, Q. Lacidipine amorphous solid dispersion based on hot melt extrusion: Good miscibility, enhanced dissolution, and favorable stability. AAPS PharmSciTech 2018, 19, 3076–3084. [Google Scholar] [CrossRef]
- Andrade, L.N.; Oliveira, D.M.L.; Chaud, M.V.; Alves, T.F.R.; Nery, M.; da Silva, C.F.; Gonsalves, J.K.C.; Nunes, R.S.; Corrêa, C.B.; Amaral, R.G.; et al. Praziquantel-solid lipid nanoparticles produced by supercritical carbon dioxide extraction: Physicochemical characterization, release profile, and cytotoxicity. Molecules 2019, 24, 3881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrego-Sánchez, A.; Carazo, E.; Albertini, B.; Passerini, N.; Perissutti, B.; Cerezo, P.; Viseras, C.; Hernández-Laguna, A.; Aguzzi, C.; Sainz-Díaz, C.I. Conformational polymorphic changes in the crystal structure of the chiral antiparasitic drug praziquantel and interactions with calcium carbonate. Eur. J. Pharm. Biopharm. 2018, 132, 180–191. [Google Scholar] [CrossRef]
- Ponnammal, P.; Kanaujia, P.; Yani, Y.; Ng, W.K.; Tan, R.B.H. Orally disintegrating tablets containing melt extruded amorphous solid dispersion of tacrolimus for dissolution enhancement. Pharmaceutics 2018, 10, 35. [Google Scholar] [CrossRef] [Green Version]
- Dengale, S.J.; Grohganz, H.; Rades, T.; Löbmann, K. Recent advances in co-amorphous drug formulations. Adv. Drug Deliv. Rev. 2016, 100, 116–125. [Google Scholar] [CrossRef]
- Al-Khattawi, A.; Mohammed, A.R. Challenges and emerging solutions in the development of compressed orally disintegrating tablets. Expert Opin. Drug Discov. 2014, 9, 1109–1120. [Google Scholar] [CrossRef]
- Cioli, D.; Pica-Mattoccia, L. Praziquantel. Parasitol. Res. 2003, 90, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Zhang, T.; Yu, J.; Liu, Y.; Wang, Y.; He, Z. In vitro/vivo assessment of praziquantel nanocrystals: Formulation, characterization, and pharmacokinetics in beagle dogs. Asian J. Pharm. Sci. 2019, 14, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, T.; Ding, W.; Dong, C.; Wang, X.; Chen, J.; Li, Y. Dissolution and oral bioavailability enhancement of praziquantel by solid dispersions. Drug Deliv. Transl. Res. 2018, 8, 580–590. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredients | Quantity (mg/Tablet) | Function |
|---|---|---|
| PZQ hot-melt extrudate | 250 | active ingredient |
| Microcrystalline cellulose | 187.5 | dry binder |
| mannitol | 515 | diluent, sweetening agent |
| Low-Substituted Hydroxypropyl Cellulose | 37.5 | disintegrant |
| Magnesium stearate | 10 | lubricant |
| Total | 1000 |
| PZQ ODSRTs (p.o.) | Aipuruike® | p-Value (ANOVA) | |||
|---|---|---|---|---|---|
| t1/2λ (h) | 4.28 ± 0.6701 | 1.78 ± 0.44 | 0.000 | ||
| AUC0–∞ (μg·h/mL) | 2.30 ± 0.28 | 1.24 ± 0.51 | 0.000 | ||
| AUMC0–∞ (μg·h2/mL) | 9.99 ± 1.87 | 3.13 ± 1.69 | 0.001 | ||
| MRT0–∞ (h) | 4.34 ± 1.221 | 2.42 ± 0.98 | 0.001 | ||
| Cmax (μg/mL) | 0.43 ± 0.0084 | 0.39 ± 0.28 | 0.252 | ||
| F (%) | 184.48 ± 54.90 | — | — | ||
| Tmax (h) | Median | Range | Median | Range | — |
| 3.00 | 2.00–2.00 | 1.00 | 0.25–1.50 | 0.000 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, X.; Deng, Z.; Xu, Y.; Yan, G.; Deng, X.; Wu, L.; Liang, Q.; Fang, F.; Feng, X.; Yu, M.; et al. Preparation and In Vitro/In Vivo Evaluation of Orally Disintegrating/Modified-Release Praziquantel Tablets. Pharmaceutics 2021, 13, 1567. https://doi.org/10.3390/pharmaceutics13101567
Wen X, Deng Z, Xu Y, Yan G, Deng X, Wu L, Liang Q, Fang F, Feng X, Yu M, et al. Preparation and In Vitro/In Vivo Evaluation of Orally Disintegrating/Modified-Release Praziquantel Tablets. Pharmaceutics. 2021; 13(10):1567. https://doi.org/10.3390/pharmaceutics13101567
Chicago/Turabian StyleWen, Xuemei, Zhaoyou Deng, Yangfeng Xu, Guoqing Yan, Xin Deng, Liqin Wu, Qiuling Liang, Fang Fang, Xin Feng, Meiling Yu, and et al. 2021. "Preparation and In Vitro/In Vivo Evaluation of Orally Disintegrating/Modified-Release Praziquantel Tablets" Pharmaceutics 13, no. 10: 1567. https://doi.org/10.3390/pharmaceutics13101567