
Protease-Triggered Release of Stabilized CXCL12 from Coated Scaffolds in an Ex Vivo Wound Model

 , , , and
, , , and
Abstract
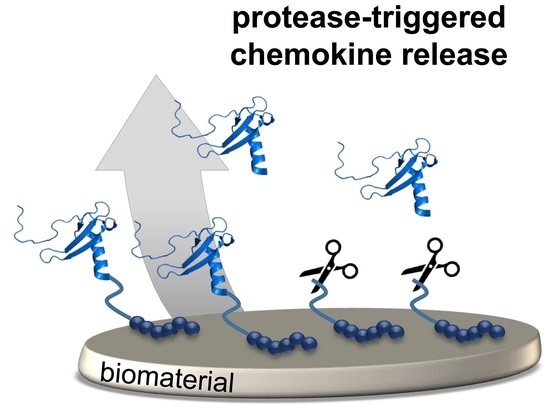
1. Introduction
2. Materials and Methods
2.1. Peptide Synthesis
2.2. Protein Expression and Purification
2.3. Expressed Protein Ligation and Refolding
2.4. PCL-co-LC Scaffold Preparation and Coating
2.5. MMP9 and MMP2 Activation and Digestion in Solution
2.6. Binding and Release Assay
2.7. Isolation and Culture of Murine Mesenchymal Stem Cells (MSCs)
2.8. Flow Cytometry Analysis of Murine MSCs
2.9. Immunofluorescence Staining of Murine MSCs
2.10. Transwell Migration Assays
2.10.1. Jurkat Cell Migration towards Coated or Uncoated PCL-co-LC
2.10.2. Concentration-Dependent Jurkat Cell Migration
2.10.3. Concentration-Dependent Migration of Murine MSCs
2.11. Inositol Phosphate Accumulation Assay
2.12. Analysis of HaCaT Cell Migration
2.13. Analysis of CXCL12 Signaling
2.14. Organ Culture of Porcine Skin
2.15. Histological Analysis
2.16. Statistical Methods
3. Results
3.1. Generation and Signal Transduction of CXCL12 Variants with Stable or Protease-Mediated Release Linker
3.2. Protease-Induced Surface Release of Compound 4 and Cellular Response to Gradient Formation
3.3. Ex Vivo Wound Closure Model Using Porcine Organ Culture
4. Discussion
4.1. CXC12 for Treatment of Disturbed Wound Healing
4.2. Immobilization of CXCL12 on Biomaterials with Gradient Formation
4.3. MMPs of the Wound Milieu May Be Used for Controlled Release of CXCL12 from Engineered Biomaterials
4.4. Functional Testing of CXCL12-Modified Biomaterials in an Ex Vivo Wound Healing Model
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rentsch, B.; Bernhardt, R.; Scharnweber, D.; Schneiders, W.; Rammelt, S.; Rentsch, C. Embroidered and surface coated polycaprolactone-co-lactide scaffolds: A potential graft for bone tissue engineering. Biomatter 2012, 2, 158–165. [Google Scholar] [CrossRef]
- Rentsch, C.; Schneiders, W.; Hess, R.; Rentsch, B.; Bernhardt, R.; Spekl, K.; Schneider, K.; Scharnweber, D.; Biewener, A.; Rammelt, S. Healing properties of surface-coated polycaprolactone-co-lactide scaffolds: A pilot study in sheep. J. Biomater. Appl. 2013, 28, 654–666. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Abbasi, N.; Lee, R.S.B.; Ivanovski, S.; Love, R.M.; Hamlet, S. In vivo bone regeneration assessment of offset and gradient melt electrowritten (MEW) PCL scaffolds. Biomater. Res. 2020, 24, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Mondal, D.; Griffith, M.; Venkatraman, S.S. Polycaprolactone-based biomaterials for tissue engineering and drug delivery: Current scenario and challenges. Int. J. Polym. Mater. 2016, 65, 255–265. [Google Scholar] [CrossRef]
- Woodruff, M.A.; Hutmacher, D.W. The return of a forgotten polymer—Polycaprolactone in the 21st century. Prog. Polym. Sci. 2010, 35, 1217–1256. [Google Scholar] [CrossRef]
- Joseph, B.; Augustine, R.; Kalarikkal, N.; Thomas, S.; Seantier, B.; Grohens, Y. Recent advances in electrospun polycaprolactone based scaffolds for wound healing and skin bioengineering applications. Mater. Today Commun. 2019, 19, 319–335. [Google Scholar] [CrossRef]
- Levengood, S.L.; Erickson, A.E.; Chang, F.-C.; Zhang, M. Chitosan-poly(caprolactone) nanofibers for skin repair. J. Mater. Chem. B 2017, 5, 1822–1833. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, B.K.; Park, S.H.; Kim, M.G.; Lee, J.W.; Lee, H.Y.; Lee, H.B.; Kim, J.H.; Kim, M.S. Preparation of biodegradable and elastic poly(ε-caprolactone-co-lactide) copolymers and evaluation as a localized and sustained drug delivery carrier. Int. J. Mol. Sci. 2017, 18, 671. [Google Scholar] [CrossRef] [PubMed]
- Sadeghianmaryan, A.; Karimi, Y.; Naghieh, S.; Alizadeh Sardroud, H.; Gorji, M.; Chen, X. Electrospinning of scaffolds from the polycaprolactone/polyurethane composite with graphene oxide for skin tissue engineering. Appl. Biochem. Biotechnol. 2020, 191, 567–578. [Google Scholar] [CrossRef]
- Wei, L.-G.; Chang, H.-I.; Wang, Y.; Hsu, S.-H.; Dai, L.-G.; Fu, K.-Y.; Dai, N.-T. A gelatin/collagen/polycaprolactone scaffold for skin regeneration. PeerJ 2019, 7, e6358. [Google Scholar] [CrossRef]
- Anderson, J.M.; Rodriguez, A.; Chang, D.T. Foreign body reaction to biomaterials. Semin. Immunol. 2008, 20, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Klopfleisch, R.; Jung, F. The pathology of the foreign body reaction against biomaterials. J. Biomed. Mater. Res. A 2017, 105, 927–940. [Google Scholar] [CrossRef]
- Cencioni, C.; Capogrossi, M.C.; Napolitano, M. The SDF-1/CXCR4 axis in stem cell preconditioning. Cardiovasc. Res. 2012, 94, 400–407. [Google Scholar] [CrossRef]
- Hattori, K.; Heissig, B.; Rafii, S. The regulation of hematopoietic stem cell and progenitor mobilization by chemokine SDF-1. Leuk. Lymphoma 2003, 44, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Hocking, A.M. The role of chemokines in mesenchymal stem cell homing to wounds. Adv. Wound Care 2015, 4, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.K.; Tsui, J.; Xu, S.; Leoni, P.; Abraham, D.J.; Baker, D.M. Angiogenic effects of stromal cell-derived factor-1 (SDF-1/CXCL12) variants in vitro and the in vivo expressions of CXCL12 variants and CXCR4 in human critical leg ischemia. J. Vasc. Surg. 2010, 51, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Maan, Z.N.; Hu, M.S.; Rennert, R.; Barrera, J.A.; Duscher, D.; Januszyk, M.; Henn, D.; Chen, K.; Sivaraj, D.; Whittam, A.; et al. Endothelial CXCL12 regulates neovascularization during tissue repair and tumor progression. Cell Rep. 2021. [Google Scholar] [CrossRef]
- Yamaguchi, J.; Kusano, K.F.; Masuo, O.; Kawamoto, A.; Silver, M.; Murasawa, S.; Bosch-Marce, M.; Masuda, H.; Losordo, D.W.; Isner, J.M.; et al. Stromal cell-derived factor-1 effects on ex vivo expanded endothelial progenitor cell recruitment for ischemic neovascularization. Circulation 2003, 107, 1322–1328. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Chai, L.; Chen, L.; Chen, W.; Ge, L.; Li, X.; Li, H.; Li, S.; Cao, C. Stromal cell-derived factor 1 (SDF-1) accelerated skin wound healing by promoting the migration and proliferation of epidermal stem cells. Vitr. Cell Dev. Biol. Anim. 2015, 51, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z.; Zuba-Surma, E.; Kucia, M.; Reca, R.; Wojakowski, W.; Ratajczak, J. The pleiotropic effects of the SDF-1-CXCR4 axis in organogenesis, regeneration and tumorigenesis. Leukemia 2006, 20, 1915–1924. [Google Scholar] [CrossRef]
- Böhme, D.; Beck-Sickinger, A.G. Controlling toxicity of peptide-drug conjugates by different chemical linker structures. ChemMedChem 2015, 10, 804–814. [Google Scholar] [CrossRef]
- Baumann, L.; Beck-Sickinger, A.G. Identification of a potential modification site in human stromal cell-derived factor-1. Biopolymers 2010, 94, 771–778. [Google Scholar] [CrossRef] [PubMed]
- David, R.; Richter, M.P.O.; Beck-Sickinger, A.G. Expressed protein ligation. Eur. J. Biochem. 2004, 271, 663–677. [Google Scholar] [CrossRef]
- Chong, S.; Mersha, F.B.; Comb, D.G.; Scott, M.E.; Landry, D.; Vence, L.M.; Perler, F.B.; Benner, J.; Kucera, R.B.; Hirvonen, C.A.; et al. Single-column purification of free recombinant proteins using a self-cleavable affinity tag derived from a protein splicing element. Gene 1997, 192, 271–281. [Google Scholar] [CrossRef]
- Hirel, P.H.; Schmitter, M.J.; Dessen, P.; Fayat, G.; Blanquet, S. Extent of N-terminal methionine excision from Escherichia coli proteins is governed by the side-chain length of the penultimate amino acid. Proc. Natl. Acad. Sci. USA 1989, 86, 8247–8251. [Google Scholar] [CrossRef] [PubMed]
- Panitz, N.; Theisgen, S.; Samsonov, S.A.; Gehrcke, J.-P.; Baumann, L.; Bellmann-Sickert, K.; Köhling, S.; Pisabarro, M.T.; Rademann, J.; Huster, D.; et al. The structural investigation of glycosaminoglycan binding to CXCL12 displays distinct interaction sites. Glycobiology 2016, 26, 1209–1221. [Google Scholar] [CrossRef]
- Schägger, H. Tricine-SDS-PAGE. Nat. Protoc. 2006, 1, 16–22. [Google Scholar] [CrossRef]
- Hassert, R.; Pagel, M.; Ming, Z.; Haupl, T.; Abel, B.; Braun, K.; Wießler, M.; Beck-Sickinger, A.G. Biocompatible silicon surfaces through orthogonal click chemistries and a high affinity silicon oxide binding peptide. Bioconjug. Chem. 2012, 23, 2129–2137. [Google Scholar] [CrossRef]
- Picke, A.-K.; Campbell, G.M.; Blüher, M.; Krügel, U.; Schmidt, F.N.; Tsourdi, E.; Winzer, M.; Rauner, M.; Vukicevic, V.; Busse, B.; et al. Thy-1 (CD90) promotes bone formation and protects against obesity. Sci. Transl. Med. 2018, 10, eaao6806. [Google Scholar] [CrossRef]
- Clauder, F.; Zitzmann, F.D.; Friebe, S.; Mayr, S.G.; Robitzki, A.A.; Beck-Sickinger, A.G. Multifunctional coatings combining bioactive peptides and affinity-based cytokine delivery for enhanced integration of degradable vascular grafts. Biomater. Sci. 2020, 8, 1734–1747. [Google Scholar] [CrossRef]
- Kostenis, E. Is Gα16 the optimal tool for fishing ligands of orphan G-protein-coupled receptors? Trends Pharmacol. Sci. 2001, 22, 560–564. [Google Scholar] [CrossRef]
- Kaiser, A.; Wanka, L.; Ziffert, I.; Beck-Sickinger, A.G. Biased agonists at the human Y1 receptor lead to prolonged membrane residency and extended receptor G protein interaction. Cell Mol. Life Sci. 2020, 77, 4675–4691. [Google Scholar] [CrossRef]
- Brandner, J.M.; Zacheja, S.; Houdek, P.; Moll, I.; Lobmann, R. Expression of matrix metalloproteinases, cytokines, and connexins in diabetic and nondiabetic human keratinocytes before and after transplantation into an ex vivo wound-healing model. Diabetes Care 2008, 31, 114–120. [Google Scholar] [CrossRef]
- Thönes, S.; Rother, S.; Wippold, T.; Blaszkiewicz, J.; Balamurugan, K.; Möller, S.; Ruiz-Gómez, G.; Schnabelrauch, M.; Scharnweber, D.; Saalbach, A.; et al. Hyaluronan/collagen hydrogels containing sulfated hyaluronan improve wound healing by sustained release of heparin-binding EGF-like growth factor. Acta Biomater. 2019, 86, 135–147. [Google Scholar] [CrossRef]
- Steinhagen, M.; Hoffmeister, P.-G.; Nordsieck, K.; Hötzel, R.; Baumann, L.; Hacker, M.C.; Schulz-Siegmund, M.; Beck-Sickinger, A.G. Matrix metalloproteinase 9 (MMP-9) mediated release of MMP-9 resistant stromal cell-derived factor 1α (SDF-1α) from surface modified polymer films. ACS Appl. Mater. Interfaces 2014, 6, 5891–5899. [Google Scholar] [CrossRef]
- Chong, S.; Montello, G.E.; Zhang, A.; Cantor, E.J.; Liao, W.; Xu, M.Q.; Benner, J. Utilizing the C-terminal cleavage activity of a protein splicing element to purify recombinant proteins in a single chromatographic step. Nucleic Acids Res. 1998, 26, 5109–5115. [Google Scholar] [CrossRef]
- Haase, C.; Rohde, H.; Seitz, O. Native chemical ligation at valine. Angew. Chem. Int. Ed. Engl. 2008, 47, 6807–6810. [Google Scholar] [CrossRef] [PubMed]
- Hackeng, T.M.; Griffin, J.H.; Dawson, P.E. Protein synthesis by native chemical ligation: Expanded scope by using straightforward methodology. Proc. Natl. Acad. Sci. USA 1999, 96, 10068–10073. [Google Scholar] [CrossRef] [PubMed]
- Avniel, S.; Arik, Z.; Maly, A.; Sagie, A.; Basst, H.B.; Yahana, M.D.; Weiss, I.D.; Pal, B.; Wald, O.; Ad-El, D.; et al. Involvement of the CXCL12/CXCR4 pathway in the recovery of skin following burns. J. Investig. Dermatol. 2006, 126, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhu, F.; Zhang, M.; Zeng, D.; Luo, D.; Liu, G.; Cui, W.; Wang, S.; Guo, W.; Xing, W.; et al. Stromal cell-derived factor-1 enhances wound healing through recruiting bone marrow-derived mesenchymal stem cells to the wound area and promoting neovascularization. Cells Tissues Organs 2013, 197, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Ågren, M.S. Gelatinase activity during wound healing. Br. J. Dermatol. 1994, 131, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Lobmann, R.; Ambrosch, A.; Schultz, G.; Waldmann, K.; Schiweck, S.; Lehnert, H. Expression of matrix-metalloproteinases and their inhibitors in the wounds of diabetic and non-diabetic patients. Diabetologia 2002, 45, 1011–1016. [Google Scholar] [CrossRef]
- Falanga, V. Wound healing and its impairment in the diabetic foot. Lancet 2005, 366, 1736–1743. [Google Scholar] [CrossRef]
- Tavakoli, S.; Klar, A.S. Advanced hydrogels as wound dressings. Biomolecules 2020, 10, 1169. [Google Scholar] [CrossRef] [PubMed]
- Ridiandries, A.; Tan, J.T.M.; Bursill, C.A. The role of chemokines in wound healing. Int. J. Mol. Sci. 2018, 19, 3217. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; DiPietro, L.A. Factors affecting wound healing. J. Dent. Res. 2010, 89, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, L.P.; Reis, R.L.; Correlo, V.M.; Marques, A.P. Hydrogel-based strategies to advance therapies for chronic skin wounds. Annu. Rev. Biomed. Eng. 2019, 21, 145–169. [Google Scholar] [CrossRef]
- Mulder, G.; Tenenhaus, M.; D’Souza, G.F. Reduction of diabetic foot ulcer healing times through use of advanced treatment modalities. Int. J. Low Extrem. Wounds 2014, 13, 335–346. [Google Scholar] [CrossRef]
- Steed, D.L. Clinical evaluation of recombinant human platelet-derived growth factor for the treatment of lower extremity ulcers. Plast. Reconstr. Surg. 2006, 117. [Google Scholar] [CrossRef]
- Zhao, X.-H.; Gu, H.-F.; Xu, Z.-R.; Zhang, Q.; Lv, X.-Y.; Zheng, X.-J.; Yang, Y.-M. Efficacy of topical recombinant human platelet-derived growth factor for treatment of diabetic lower-extremity ulcers: Systematic review and meta-analysis. Metabolism 2014, 63, 1304–1313. [Google Scholar] [CrossRef]
- Papanas, D.; Maltezos, E. Benefit-risk assessment of becaplermin in the treatment of diabetic foot ulcers. Drug Saf. 2010, 33, 455–461. [Google Scholar] [CrossRef]
- Lau, T.T.; Wang, D.-A. Stromal cell-derived factor-1 (SDF-1): Homing factor for engineered regenerative medicine. Expert Opin. Biol. Ther. 2011, 11, 189–197. [Google Scholar] [CrossRef]
- Fox, J.M.; Chamberlain, G.; Ashton, B.A.; Middleton, J. Recent advances into the understanding of mesenchymal stem cell trafficking. Br. J. Haematol. 2007, 137, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Hopman, R.K.; DiPersio, J.F. Advances in stem cell mobilization. Blood Rev. 2014, 28, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Quan, C.; Cho, M.K.; Shao, Y.; Mianecki, L.E.; Liao, E.; Perry, D.; Quan, T. Dermal fibroblast expression of stromal cell-derived factor-1 (SDF-1) promotes epidermal keratinocyte proliferation in normal and diseased skin. Protein Cell 2015, 6, 890–903. [Google Scholar] [CrossRef] [PubMed]
- Florin, L.; Maas-Szabowski, N.; Werner, S.; Szabowski, A.; Angel, P. Increased keratinocyte proliferation by JUN-dependent expression of PTN and SDF-1 in fibroblasts. J. Cell Sci. 2005, 118, 1981–1989. [Google Scholar] [CrossRef] [PubMed]
- Chow, K.Y.C.; Brotin, É.; Ben Khalifa, Y.; Carthagena, L.; Teissier, S.; Danckaert, A.; Galzi, J.-L.; Arenzana-Seisdedos, F.; Thierry, F.; Bachelerie, F. A pivotal role for CXCL12 signaling in HPV-mediated transformation of keratinocytes: Clues to understanding HPV-pathogenesis in WHIM syndrome. Cell Host Microbe 2010, 8, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Yellowley, C.E.; Toupadakis, C.A.; Vapniarsky, N.; Wong, A. Circulating progenitor cells and the expression of Cxcl12, Cxcr4 and angiopoietin-like 4 during wound healing in the murine ear. PLoS ONE 2019, 14, e0222462. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Hao, D.; Chai, J. Processing of CXCL12 impedes the recruitment of endothelial progenitor cells in diabetic wound healing. FEBS J. 2014, 281, 5054–5062. [Google Scholar] [CrossRef]
- Gallagher, K.A.; Liu, Z.-J.; Xiao, M.; Chen, H.; Goldstein, L.J.; Buerk, D.G.; Nedeau, A.; Thom, S.R.; Velazquez, O.C. Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1 alpha. J. Clin. Investig. 2007, 117, 1249–1259. [Google Scholar] [CrossRef]
- Badillo, A.T.; Chung, S.; Zhang, L.; Zoltick, P.; Liechty, K.W. Lentiviral gene transfer of SDF-1α to wounds improves diabetic wound healing. J. Surg. Res. 2007, 143, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Restivo, T.E.; Mace, K.A.; Harken, A.H.; Young, D.M. Application of the chemokine CXCL12 expression plasmid restores wound healing to near normal in a diabetic mouse model. J. Trauma Acute Care Surg. 2010, 69, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Vågesjö, E.; Öhnstedt, E.; Mortier, A.; Lofton, H.; Huss, F.; Proost, P.; Roos, S.; Phillipson, M. Accelerated wound healing in mice by on-site production and delivery of CXCL12 by transformed lactic acid bacteria. Proc. Natl. Acad. Sci. USA 2018, 115, 1895. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Hoshi, R.; Chen, S.; Yi, J.; Duan, C.; Galiano, R.D.; Zhang, H.F.; Ameer, G.A. Sustained release of stromal cell derived factor-1 from an antioxidant thermoresponsive hydrogel enhances dermal wound healing in diabetes. J. Control. Release 2016, 238, 114–122. [Google Scholar] [CrossRef]
- Kojima, Y.; Acar, A.; Eaton, E.N.; Mellody, K.T.; Scheel, C.; Ben-Porath, I.; Onder, T.T.; Wang, Z.C.; Richardson, A.L.; Weinberg, R.A.; et al. Autocrine TGF-β and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 20009–20014. [Google Scholar] [CrossRef]
- Duo, J.; Stenken, J.A. Heparin-immobilized microspheres for the capture of cytokines. Anal. Bioanal. Chem. 2011, 399, 773–782. [Google Scholar] [CrossRef]
- Dinoro, J.; Maher, M.; Talebian, S.; Jafarkhani, M.; Mehrali, M.; Orive, G.; Foroughi, J.; Lord, M.S.; Dolatshahi-Pirouz, A. Sulfated polysaccharide-based scaffolds for orthopaedic tissue engineering. Biomaterials 2019, 214, 119214. [Google Scholar] [CrossRef]
- Hintze, V.; Miron, A.; Möller, S.; Schnabelrauch, M.; Wiesmann, H.P.; Worch, H.; Scharnweber, D. Sulfated hyaluronan and chondroitin sulfate derivatives interact differently with human transforming growth factor-β1 (TGF-β1). Acta Biomater. 2012, 8, 2144–2152. [Google Scholar] [CrossRef]
- Budiraharjo, R.; Neoh, K.G.; Kang, E.-T. Enhancing bioactivity of chitosan film for osteogenesis and wound healing by covalent immobilization of BMP-2 or FGF-2. J. Biomater. Sci. Polym. Ed. 2013, 24, 645–662. [Google Scholar] [CrossRef]
- Chiu, L.L.Y.; Radisic, M. Scaffolds with covalently immobilized VEGF and angiopoietin-1 for vascularization of engineered tissues. Biomaterials 2010, 31, 226–241. [Google Scholar] [CrossRef]
- Ren, J.; Tian, K.; Jia, L.; Han, X.; Zhao, M. Rapid covalent immobilization of proteins by phenol-based photochemical cross-linking. Bioconjug. Chem. 2016, 27, 2266–2270. [Google Scholar] [CrossRef]
- Choi, J.S.; Leong, K.W.; Yoo, H.S. In vivo wound healing of diabetic ulcers using electrospun nanofibers immobilized with human epidermal growth factor (EGF). Biomaterials 2008, 29, 587–596. [Google Scholar] [CrossRef]
- Recker, T.; Haamann, D.; Schmitt, A.; Küster, A.; Klee, D.; Barth, S.; Müller-Newen, G. Directed covalent immobilization of fluorescently labeled cytokines. Bioconjug. Chem. 2011, 22, 1210–1220. [Google Scholar] [CrossRef]
- Masters, K.S. Covalent growth factor immobilization strategies for tissue repair and regeneration. Macromol. Biosci. 2011, 11, 1149–1163. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Z.; Lu, W.W.; Zhen, W.; Yang, D.; Peng, S. Novel biomaterial strategies for controlled growth factor delivery for biomedical applications. NPG Asia Mater. 2017, 9, e435. [Google Scholar] [CrossRef]
- Prokoph, S.; Chavakis, E.; Levental, K.R.; Zieris, A.; Freudenberg, U.; Dimmeler, S.; Werner, C. Sustained delivery of SDF-1α from heparin-based hydrogels to attract circulating pro-angiogenic cells. Biomaterials 2012, 33, 4792–4800. [Google Scholar] [CrossRef]
- Baumann, L.; Prokoph, S.; Gabriel, C.; Freudenberg, U.; Werner, C.; Beck-Sickinger, A.G. A novel, biased-like SDF-1 derivative acts synergistically with starPEG-based heparin hydrogels and improves eEPC migration in vitro. J. Control. Release 2012, 162, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Chwalek, K.; Tsurkan, M.V.; Freudenberg, U.; Werner, C. Glycosaminoglycan-based hydrogels to modulate heterocellular communication in in vitro angiogenesis models. Sci. Rep. 2014, 4, 1–8. [Google Scholar] [CrossRef]
- Spiller, S.; Panitz, N.; Limasale, Y.D.P.; Atallah, P.M.; Schirmer, L.; Bellmann-Sickert, K.; Blaszkiewicz, J.; Köhling, S.; Freudenberg, U.; Rademann, J.; et al. Modulation of human CXCL12 binding properties to glycosaminoglycans to enhance chemotactic gradients. ACS Biomater. Sci. Eng. 2019, 5, 5128–5138. [Google Scholar] [CrossRef]
- Clauder, F.; Möller, S.; Köhling, S.; Bellmann-Sickert, K.; Rademann, J.; Schnabelrauch, M.; Beck-Sickinger, A.G. Peptide-mediated surface coatings for the release of wound-healing cytokines. J. Tissue Eng. Regen. Med. 2020, 14, 1738–1748. [Google Scholar] [CrossRef]
- Pagel, M.; Hassert, R.; John, T.; Braun, K.; Wießler, M.; Abel, B.; Beck-Sickinger, A.G. Multifunctional coating improves cell adhesion on titanium by using cooperatively acting peptides. Angew. Chem. Int. Ed. Engl. 2016, 55, 4826–4830. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.S.; Sim, S.B.; Cha, H.J. Cell adhesion biomaterial based on mussel adhesive protein fused with RGD peptide. Biomaterials 2007, 28, 4039–4046. [Google Scholar] [CrossRef] [PubMed]
- Dalsin, J.L.; Hu, B.-H.; Lee, B.P.; Messersmith, P.B. Mussel adhesive protein mimetic polymers for the preparation of nonfouling surfaces. J. Am. Chem. Soc. 2003, 125, 4253–4258. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Dellatore, S.M.; Miller, W.M.; Messersmith, P.B. Mussel-inspired surface chemistry for multifunctional coatings. Science 2007, 318, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Ton, X.-A.; Zhao, S.; Paez, J.I.; Del Campo, A. Mechanically reinforced catechol-containing hydrogels with improved tissue gluing performance. Biomimetics 2017, 2, 23. [Google Scholar] [CrossRef] [PubMed]
- Kord Forooshani, P.; Lee, B.P. Recent approaches in designing bioadhesive materials inspired by mussel adhesive protein. J. Polym. Sci. A Polym. Chem. 2017, 55, 9–33. [Google Scholar] [CrossRef] [PubMed]
- Clauder, F.; Czerniak, A.S.; Friebe, S.; Mayr, S.G.; Scheinert, D.; Beck-Sickinger, A.G. Endothelialization of titanium surfaces by bio-inspired cell adhesion peptide coatings. Bioconjug. Chem. 2019, 30, 2664–2674. [Google Scholar] [CrossRef]
- Kohn, J.M.; Riedel, J.; Horsch, J.; Stephanowitz, H.; Börner, H.G. Mussel-inspired polymerization of peptides: The chemical activation route as key to broaden the sequential space of artificial mussel-glue proteins. Macromol. Rapid Commun. 2020, 41, 1900431. [Google Scholar] [CrossRef]
- Wall, S.J.; Sampson, M.J.; Levell, N.; Murphy, G. Elevated matrix metalloproteinase-2 and -3 production from human diabetic dermal fibroblasts. Br. J. Dermatol. 2003, 149, 13–16. [Google Scholar] [CrossRef]
- McQuibban, G.A.; Butler, G.S.; Gong, J.H.; Bendall, L.; Power, C.; Clark-Lewis, I.; Overall, C.M. Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J. Biol. Chem. 2001, 276, 43503–43508. [Google Scholar] [CrossRef]
- Hsu, J.-Y.C.; McKeon, R.; Goussev, S.; Werb, Z.; Lee, J.-U.; Trivedi, A.; Noble-Haeusslein, L.J. Matrix metalloproteinase-2 facilitates wound healing events that promote functional recovery after spinal cord injury. J. Neurosci. 2006, 26, 9841–9850. [Google Scholar] [CrossRef] [PubMed]
- Leppert, D.; Waubant, E.; Galardy, R.; Bunnett, N.W.; Hauser, S.L. T cell gelatinases mediate basement membrane transmigration in vitro. J. Immunol. 1995, 154, 4379–4389. [Google Scholar]
- Liu, Y.; Min, D.; Bolton, T.; Nubé, V.; Twigg, S.M.; Yue, D.K.; McLennan, S.V. Increased matrix metalloproteinase-9 predicts poor wound healing in diabetic foot ulcers. Diabetes Care 2009, 32, 117–119. [Google Scholar] [CrossRef]
- Soo, C.; Shaw, W.W.; Zhang, X.; Longaker, M.T.; Howard, E.W.; Ting, K. Differential expression of matrix metalloproteinases and their tissue-derived inhibitors in cutaneous wound repair. Plast. Reconstr. Surg. 2000, 105, 638–647. [Google Scholar] [CrossRef]
- Caley, M.P.; Martins, V.L.C.; O’Toole, E.A. Metalloproteinases and wound healing. Adv. Wound Care 2015, 4, 225–234. [Google Scholar] [CrossRef]
- Mott, J.D.; Werb, Z. Regulation of matrix biology by matrix metalloproteinases. Curr. Opin. Cell Biol. 2004, 16, 558–564. [Google Scholar] [CrossRef]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef]
- Löffek, S.; Schilling, O.; Franzke, C.W. Biological role of matrix metalloproteinases: A critical balance. Eur. Respir. J. 2011, 38, 191–208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; McQuibban, G.A.; Silva, C.; Butler, G.S.; Johnston, J.B.; Holden, J.; Clark-Lewis, I.; Overall, C.M.; Power, C. HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nat. Neurosci. 2003, 6, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Segers, V.F.M.; Revin, V.; Wu, W.; Qiu, H.; Yan, Z.; Lee, R.T.; Sandrasagra, A. Protease-resistant stromal cell-derived factor-1 for the treatment of experimental peripheral artery disease. Circulation 2011, 123, 1306–1315. [Google Scholar] [CrossRef]
- Segers, V.F.M.; Tokunou, T.; Higgins, L.J.; MacGillivray, C.; Gannon, J.; Lee, R.T. Local delivery of protease-resistant stromal cell derived factor-1 for stem cell recruitment after myocardial infarction. Circulation 2007, 116, 1683–1692. [Google Scholar] [CrossRef]
- Hesse, E.; Freudenberg, U.; Niemietz, T.; Greth, C.; Weisser, M.; Hagmann, S.; Binner, M.; Werner, C.; Richter, W. Peptide-functionalized starPEG/heparin hydrogels direct mitogenicity, cell morphology and cartilage matrix distribution in vitro and in vivo. J. Tissue Eng. Regen. Med. 2018, 12, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Vessillier, S.; Adams, G.; Chernajovsky, Y. Latent cytokines: Development of novel cleavage sites and kinetic analysis of their differential sensitivity to MMP-1 and MMP-3. Protein Eng. Des. Sel. 2004, 17, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, K.J.; Jensen, M.M.; Subrahmanyam, N.B.; Ghandehari, H. Matrix-metalloproteinases as targets for controlled delivery in cancer: An analysis of upregulation and expression. J. Control. Release 2017, 259, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Kou, L.; Tu, Y.; Zhu, L. MMP-responsive ‘smart’ drug delivery and tumor targeting. Trends Pharmacol. Sci. 2018, 39, 766–781. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.B.; Lin, C.-C.; Kuntzler, D.V.; Anseth, K.S. The performance of human mesenchymal stem cells encapsulated in cell-degradable polymer-peptide hydrogels. Biomaterials 2011, 32, 3564–3574. [Google Scholar] [CrossRef]
- Holloway, J.L.; Ma, H.; Rai, R.; Hankenson, K.D.; Burdick, J.A. Synergistic effects of SDF-1α and BMP-2 delivery from proteolytically degradable hyaluronic acid hydrogels for bone repair. Macromol. Biosci. 2015, 15, 1218–1223. [Google Scholar] [CrossRef]
- Jian, W.-H.; Wang, H.-C.; Kuan, C.-H.; Chen, M.-H.; Wu, H.-C.; Sun, J.-S.; Wang, T.-W. Glycosaminoglycan-based hybrid hydrogel encapsulated with polyelectrolyte complex nanoparticles for endogenous stem cell regulation in central nervous system regeneration. Biomaterials 2018, 174, 17–30. [Google Scholar] [CrossRef]
- Kofuku, Y.; Yoshiura, C.; Ueda, T.; Terasawa, H.; Hirai, T.; Tominaga, S.; Hirose, M.; Maeda, Y.; Takahashi, H.; Terashima, Y.; et al. Structural basis of the interaction between chemokine stromal cell-derived factor-1/CXCL12 and its G-protein-coupled receptor CXCR4. J. Biol. Chem. 2009, 284, 35240–35250. [Google Scholar] [CrossRef]
- Tamamis, P.; Floudas, C.A. Elucidating a key component of cancer metastasis: CXCL12 (SDF-1α) binding to CXCR4. J. Chem. Inf. Model. 2014, 54, 1174–1188. [Google Scholar] [CrossRef]
- Legler, D.F.; Thelen, M. New insights in chemokine signaling. F1000Research 2018, 7, 1–8. [Google Scholar] [CrossRef]
- Welf, E.S.; Haugh, J.M. Signaling pathways that control cell migration: Models and analysis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 231–240. [Google Scholar] [CrossRef]
- Eiger, D.S.; Boldizsar, N.; Honeycutt, C.C.; Gardner, J.; Rajagopal, S. Biased agonism at chemokine receptors. Cell Signal. 2021, 78, 109862. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Yong, X.; Li, C.; Lü, M.; Liu, D.; Chen, L.; Hu, J.; Teng, M.; Zhang, D.; Fan, Y.; et al. CXCL12/CXCR4 axis promotes mesenchymal stem cell mobilization to burn wounds and contributes to wound repair. J. Surg. Res. 2013, 183, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Byrne, S.N.; Sarchio, S.N. AMD3100 protects from UV-induced skin cancer. Oncoimmunology 2014, 3, e27562. [Google Scholar] [CrossRef][Green Version]
- Wescott, M.P.; Kufareva, I.; Paes, C.; Goodman, J.R.; Thaker, Y.; Puffer, B.A.; Berdougo, E.; Rucker, J.B.; Handel, T.M.; Doranz, B.J. Signal transmission through the CXC chemokine receptor 4 (CXCR4) transmembrane helices. Proc. Natl. Acad. Sci. USA 2016, 113, 9928–9933. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | B | C | D | E | F | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | CXCL12 Variant | Stabilized | Adhesive | Releasable | Monoisotopic Mass [Da] | Elution [%ACN] | Purity [%] | EC50 [nM] | pEC50 ± SEM | Emax [%] ± SEM |
| wt | CXCL12 wild type | − | − | − | 8085.30 | 37.3 | >95 | 5.4 | 8.27 ± 0.03 | 100.0 |
| 1 | [V49A]-CXCL12-SBP(Biotin) | − | + | − | 9505.01 | 37.6 | >95 | 6.2 | 8.21 ± 0.05 | 91.5 ± 2.4 |
| 2 | [V49A]-CXCL12-MMPCS-SBP(Biotin) | − | + | + | 12,005.05 | 39.1 | >95 | 126.6 | 6.90 ± 0.05 | 93.7 ± 2.8 |
| 3 | [S4V]-[V49A]-CXCL12-SBP | + | + | − | 9064.80 | 37.6 | >95 | 43.3 | 7.36 ± 0.06 | 91.1 ± 3.0 |
| 4 | [S4V]-[V49A]-CXCL12-MMPCS-SBP | + | + | + | 11,565.29 | 39.0 | >95 | 54.0 | 7.27 ± 0.17 | 84.0 ± 7.5 |
| 5 | [S4V]-[V49A]-CXCL12-GPLS | + | − | − | 8427.53 | 39.0 | >95 | 55.5 | 7.25 ± 0.07 | 87.4 ± 2.9 |
| 6 | [S4V]-[V49A]-CXCL12 | + | − | − | 8073.34 | 38.3 | >95 | 53.7 | 7.27 ± 0.06 | 89.6 ± 2.5 |
| SBP | SBP(Biotin) | − | + | − | 1465.75 | 24.6 | >95 | nd | nd | 0 ± 2.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spiller, S.; Wippold, T.; Bellmann-Sickert, K.; Franz, S.; Saalbach, A.; Anderegg, U.; Beck-Sickinger, A.G. Protease-Triggered Release of Stabilized CXCL12 from Coated Scaffolds in an Ex Vivo Wound Model. Pharmaceutics 2021, 13, 1597. https://doi.org/10.3390/pharmaceutics13101597
Spiller S, Wippold T, Bellmann-Sickert K, Franz S, Saalbach A, Anderegg U, Beck-Sickinger AG. Protease-Triggered Release of Stabilized CXCL12 from Coated Scaffolds in an Ex Vivo Wound Model. Pharmaceutics. 2021; 13(10):1597. https://doi.org/10.3390/pharmaceutics13101597
Chicago/Turabian StyleSpiller, Sabrina, Tom Wippold, Kathrin Bellmann-Sickert, Sandra Franz, Anja Saalbach, Ulf Anderegg, and Annette G. Beck-Sickinger. 2021. "Protease-Triggered Release of Stabilized CXCL12 from Coated Scaffolds in an Ex Vivo Wound Model" Pharmaceutics 13, no. 10: 1597. https://doi.org/10.3390/pharmaceutics13101597
APA StyleSpiller, S., Wippold, T., Bellmann-Sickert, K., Franz, S., Saalbach, A., Anderegg, U., & Beck-Sickinger, A. G. (2021). Protease-Triggered Release of Stabilized CXCL12 from Coated Scaffolds in an Ex Vivo Wound Model. Pharmaceutics, 13(10), 1597. https://doi.org/10.3390/pharmaceutics13101597