
Progestins as Anticancer Drugs and Chemosensitizers, New Targets and Applications


Abstract
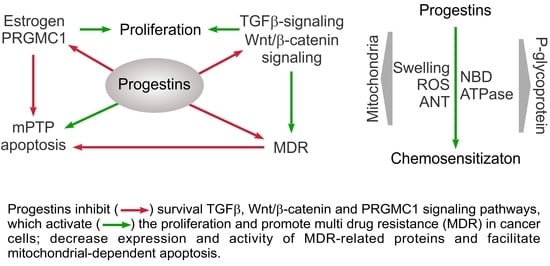
:
1. Introduction
2. Gestagens as Cytotoxic Drugs, Possible Mechanisms and Targets
2.1. Inhibition of ER Expression
2.2. Inhibition of Epithelial-to-Mesenchymal Transition (EMT)
2.3. Inhibition of PI3K/AKT, Ras/Raf/MEK/ERK, WNT/β-Catenin Cell Signaling Pathways
2.4. Inhibition of TGF-β Production and Signaling
2.5. Cell Cycle Inhibition in the G0/G1 Phase
2.6. Regulation of Mitochondrial-Dependent Apoptosis
3. Progestins as Chemosensitizers, Possible Targets
4. New MDR-Linked Targets of Progestin Action
5. Role of Wnt-Signaling in MDR and Cell Proliferation
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADR | adryamicin (doxorubicine) |
| ACC | adrenocortical carcinoma |
| ANT | adenylate translocase |
| BCRP | breast cancer resistance protein |
| CAT | carboxyatractyloside |
| CHOP | CCAAT/enhancer-binding protein homologous protein |
| CRC | colorectal cancer |
| EC | endometrial cancer |
| EMT | epithelial-to-mesenchymal transition |
| ER | estrogen receptor |
| ERK-1 and ERK-2 | extracellular signal-regulated kinase (ERK)1/2 |
| FDA | Food and Drug Administration |
| FOXO1 | forkhead box O1A |
| GB | pregnan progestogen, gestobutanoil |
| GPR30 | G protein-coupled receptor 30 |
| HER2 | human epidermal growth factor receptor-2 |
| HERPUD1 | homocysteine-responsive endoplasmic reticulum-resident ubiquitin-like domain member 1 protein |
| LBD | ligand binding domain |
| MA | megestrol acetate |
| MAP | kinase-mitogen-activated protein kinase |
| MDR | multidrug resistance |
| MGMT | O6-methylguanine-DNA-methyltransferase |
| MPA | medroxyprogesterone acetate |
| mPRα | membrane progesterone receptor alpha |
| MPTP | mitochondrial permeability transition pore |
| MMP9 | Matrix metallopeptidase 9 |
| MRP | multidrug resistance proteins |
| NBD | nucleotide binding domain |
| NBTI | S-[4-nitrobenzyl]-6-thioinosine |
| NEAT1 | NEAT1 Nuclear enriched abundant Transcript-1 |
| NEM | N-ethylmaleimide |
| NMGA | nomegestrol acetate |
| nPR | nuclear progesterone receptor |
| OSE | ovarian surface epithelium |
| P4 | progesterone |
| P-gp | P-glycoprotein |
| PR | progesterone receptor |
| PRGMC1 | progesterone receptor membrane component 1 |
| SERM | Selective estrogen receptor modulator |
| TGF-β | transforming growth factor β |
| TMD | transmembrane domain |
| TMZ | Temozolomide |
| TIL | tumor-infiltrating lymphocytes |
| TSPO | translocator protein or mitochondrial benzodiazepine receptor |
References
- Kim, J.J.; Kurita, T.; Bulun, S.E. Progesterone Action in Endometrial Cancer, Endometriosis, Uterine Fibroids, and Breast Cancer. Endocr. Rev. 2013, 34, 130–162. [Google Scholar] [CrossRef] [Green Version]
- Trojano, G.; Olivieri, C.; Tinelli, R.; Damiani, G.R.; Pellegrino, A.; Cicinelli, E. Conservative treatment in early stage endometrial cancer: A review. Acta Biomed. 2019, 90, 405–410. [Google Scholar] [CrossRef]
- Tamauchi, S.; Kajiyama, H.; Utsumi, F.; Suzuki, S.; Niimi, K.; Sakata, J.; Mizuno, M.; Shibata, K.; Kikkawa, F. Efficacy of medroxyprogesterone acetate treatment and retreatment for atypical endometrial hyperplasia and endometrial cancer. J. Obstet. Gynaecol. Res. 2017, 44, 151–156. [Google Scholar] [CrossRef] [Green Version]
- Modugno, F.; Laskey, R.; Smith, A.L.; Andersen, C.; Haluska, P.; Oesterreich, S. Hormone response in ovarian cancer: Time to reconsider as a clinical target? Endocr.-Relat. Cancer 2012, 19, R255–R279. [Google Scholar] [CrossRef] [Green Version]
- Fedotcheva, T. Clinical Use of Progestins and Their Mechanisms of Action: Present and Future (Review). Sovrem. Teh. v Med. 2021, 13, 93. [Google Scholar] [CrossRef]
- Nguyen, T.-T.; Duong, V.-A.; Maeng, H.-J. Pharmaceutical Formulations with P-Glycoprotein Inhibitory Effect as Promising Approaches for Enhancing Oral Drug Absorption and Bioavailability. Pharmaceutics 2021, 13, 1103. [Google Scholar] [CrossRef] [PubMed]
- Fürstenau, M.; Eichhorst, B. Novel Agents in Chronic Lymphocytic Leukemia: New Combination Therapies and Strategies to Overcome Resistance. Cancers 2021, 13, 1336. [Google Scholar] [CrossRef] [PubMed]
- Briston, T.; Selwood, D.; Szabadkai, G.; Duchen, M.R. Mitochondrial Permeability Transition: A Molecular Lesion with Multiple Drug Targets. Trends Pharmacol. Sci. 2019, 40, 50–70. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.; Can, A.S. Progestin; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK563211/ (accessed on 20 September 2021).
- Stanczyk, F.Z.; Hapgood, J.; Winer, S.; Mishell, D.R. Progestogens Used in Postmenopausal Hormone Therapy: Differences in Their Pharmacological Properties, Intracellular Actions, and Clinical Effects. Endocr. Rev. 2012, 34, 171–208. [Google Scholar] [CrossRef] [Green Version]
- Africander, D.; Verhoog, N.; Hapgood, J.P. Molecular mechanisms of steroid receptor-mediated actions by synthetic progestins used in HRT and contraception. Steroids 2011, 76, 636–652. [Google Scholar] [CrossRef]
- Kuhl, H.J. Pharmacology of Progestogens. J. Reprod. Med. Endocrinol. 2011, 8, 157–177. [Google Scholar]
- Zheng, Z.-Y.; Bay, B.-H.; Aw, S.-E.; Lin, V.C.-L. A Novel Antiestrogenic Mechanism in Progesterone Receptor-transfected Breast Cancer Cells. J. Biol. Chem. 2005, 280, 17480–17487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MedlinePlus. Megestrol. Available online: https://medlineplus.gov/druginfo/meds/a682003.html (accessed on 22 August 2021).
- Mittermeier, T.; Farrant, C.; Wise, M.R. Levonorgestrel-releasing intrauterine system for endometrial hyperplasia. Cochrane Database Syst. Rev. 2020, 2020. [Google Scholar] [CrossRef]
- Westin, S.N.; Fellman, B.; Sun, C.C.; Broaddus, R.R.; Woodall, M.L.; Pal, N.; Urbauer, D.L.; Ramondetta, L.M.; Schmeler, K.M.; Soliman, P.T.; et al. Prospective phase II trial of levonorgestrel intrauterine device: Nonsurgical approach for complex atypical hyperplasia and early-stage endometrial cancer. Am. J. Obstet. Gynecol. 2020, 224, 191.e1–191.e15. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.W.; Green, S.J.; Moinpour, C.M.; Bearden, J.D.; Giguere, J.K.; Jiang, C.S.; Lippman, S.M.; Martino, S.; Albain, K.S. Phase III Randomized Placebo-Controlled Trial of Two Doses of Megestrol Acetate as Treatment for Menopausal Symptoms in Women With Breast Cancer: Southwest Oncology Group Study 9626. J. Clin. Oncol. 2008, 26, 1650–1656. [Google Scholar] [CrossRef] [PubMed]
- Badwe, R.; Hawaldar, R.; Parmar, V.; Nadkarni, M.; Shet, T.; Desai, S.; Gupta, S.; Jalali, R.; Vanmali, V.; Dikshit, R.; et al. Single-Injection Depot Progesterone Before Surgery and Survival in Women With Operable Breast Cancer: A Randomized Controlled Trial. J. Clin. Oncol. 2011, 29, 2845–2851. [Google Scholar] [CrossRef] [PubMed]
- Sergeyev, P.V.; Atroshkin, K.A.; Semeikin, A.V.; Shimanovsky, N.L.; Fedotcheva, T.A.; Sekirina, M.A. Gestagen regulation of target cell proliferation activity. Bull. RONC 2008, 19, 22–28. [Google Scholar]
- Anderson, E. Cellular homeostasis and the breast. Maturitas 2004, 48, 13–17. [Google Scholar] [CrossRef]
- Chung, S.-H. Targeting female hormone receptors as cervical cancer therapy. Trends Endocrinol. Metab. 2015, 26, 399–401. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Gao, W.; Zheng, P.; Sun, X.; Wang, L. Medroxyprogesterone acetate causes the alterations of endoplasmic reticulum related mRNAs and lncRNAs in endometrial cancer cells. BMC Med. Genom. 2019, 12, 1–11. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.-J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Van Der Horst, P.H.; Wang, Y.; Vandenput, I.; Kühne, L.C.; Ewing, P.C.; Van Ijcken, W.F.J.; Van Der Zee, M.; Amant, F.; Burger, C.W.; Blok, L.J. Progesterone Inhibits Epithelial-to-Mesenchymal Transition in Endometrial Cancer. PLoS ONE 2012, 7, e30840. [Google Scholar] [CrossRef] [Green Version]
- Mehdinejadiani, S.; Amidi, F.; Mehdizadeh, M.; Barati, M.; Pazhohan, A.; Alyasin, A.; Mehdinejadiani, K.; Sobhani, A. Effects of letrozole and clomiphene citrate on Wnt signaling pathway in endometrium of polycystic ovarian syndrome and healthy women†. Biol. Reprod. 2018, 100, 641–648. [Google Scholar] [CrossRef]
- Kareva, E.N.; Solomatina, A.A.; Behbudova, L.K.; Kotsyubinskaya, N.A.; Gorenkova, O.S.; Tikhonov, D.A.; Ivanovskaya, T.N.; Bulatova, L.S. Molecular Mechanisms of Action of Progesterone in Endometry. Mol. Med. 2016, 14, 9–16. [Google Scholar]
- Shimanovskii, N.L.; Semeikin, A.V.; Fedotcheva, T.A.; Fedosov, A.V.; Kamernitskii, A.V.; Levina, I.S. 6α-Methyl-16α,17α-Cyclohexane Progesterone and Progesterone Inhibit Growth of Doxorubicin-Sensitive MCF-7 and HeLa Tumor Cells. Bull. Exp. Biol. Med. 2002, 134, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Thuneke, I.; Schulte, H.M.; Bamberger, A.-M. Biphasic effect of medroxyprogesterone-acetate (MPA) treatment on proliferation and cyclin D1 gene transcription in T47D breast cancer cells. Breast Cancer Res. Treat. 2000, 63, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Fjelldal, R.; Moe, B.T.; Ørbo, A.; Sager, G. MCF-7 cell apoptosis and cell cycle arrest: Non-genomic effects of progesterone and mifepristone (RU-486). Anticancer. Res. 2010, 30, 4835–4840. [Google Scholar]
- Ahola, T.M.; Alkio, N.; Manninen, T.; Ylikomi, T. Progestin and G Protein-Coupled Receptor 30 Inhibit Mitogen-Activated Protein Kinase Activity in MCF-7 Breast Cancer Cells. Endocrinology 2002, 143, 4620–4626. [Google Scholar] [CrossRef] [Green Version]
- Santen, R.J.; Song, R.X.; McPherson, R.; Kumar, R.; Adam, L.; Jeng, M.-H.; Yue, W. The role of mitogen-activated protein (MAP) kinase in breast cancer. J. Steroid Biochem. Mol. Biol. 2002, 80, 239–256. [Google Scholar] [CrossRef]
- Medina, M.A.; Oza, G.; Sharma, A.; Arriaga, L.; Hernández, J.M.H.; Rotello, V.M.; Ramirez, J.T. Triple-Negative Breast Cancer: A Review of Conventional and Advanced Therapeutic Strategies. Int. J. Environ. Res. Public Heal. 2020, 17, 2078. [Google Scholar] [CrossRef] [Green Version]
- Fabi, F.; Grenier, K.; Parent, S.; Adam, P.; Tardif, L.; Leblanc, V.; Asselin, E. Regulation of the PI3K/Akt pathway during decidualization of endometrial stromal cells. PLoS ONE 2017, 12, e0177387. [Google Scholar] [CrossRef]
- Gu, C.; Zhang, Z.; Yu, Y.; Liu, Y.; Zhao, F.; Yin, L.; Feng, Y.; Chen, X. Inhibiting the PI3K/Akt pathway reversed progestin resistance in endometrial cancer. Cancer Sci. 2011, 102, 557–564. [Google Scholar] [CrossRef]
- Krishnamurthy, N.; Kurzrock, R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 2017, 62, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Xu, X.; Chen, D.; Zhao, F.; Wang, W. Therapeutic potential of targeting the Wnt/β-catenin signaling pathway in colorectal cancer. Biomed. Pharmacother. 2018, 110, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Fragni, M.; Fiorentini, C.; Rossini, E.; Fisogni, S.; Vezzoli, S.; Bonini, S.A.; Dalmiglio, C.; Grisanti, S.; Tiberio, G.A.M.; Claps, M.; et al. In vitro antitumor activity of progesterone in human adrenocortical carcinoma. Endocrine 2018, 63, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhong, R.; He, X.; Deng, Q.; Peng, X.; Li, J.; Luo, X. Investigations on the mechanism of progesterone in inhibiting endometrial cancer cell cycle and viability via regulation of long noncoding RNA NEAT1/microRNA-146b-5p mediated Wnt/β-catenin signaling. IUBMB Life 2018, 71, 223–234. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Van Der Zee, M.; Fodde, R.; Blok, L.J. Wnt/Β-Catenin and Sex Hormone Signaling in Endometrial Homeostasis and Cancer. Oncotarget 2010, 1, 674–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasquez, Y.; Mazur, E.C.; Lichun, J.; Kommagani, R.; Jiang, L.; Chen, R.; Lanz, R.B.; Kovanci, E.; Gibbons, W.E.; DeMayo, F.J. FOXO1 is Required for Binding of PR on IRF4, Novel Transcriptional Regulator of Endometrial Stromal Decidualization. Mol. Endocrinol. 2015, 29, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Tao, F.; Zhang, X.; Zhang, Y.; Sun, X.; Wu, D. Role of Wnt/β-Catenin Signaling in the Chemoresistance Modulation of Colorectal Cancer. BioMed Res. Int. 2020, 2020, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Pedroza, D.A.; Subramani, R.; Lakshmanaswamy, R. Classical and Non-Classical Progesterone Signaling in Breast Cancers. Cancers 2020, 12, 2440. [Google Scholar] [CrossRef]
- Cantonero, C.; Salido, G.M.; Rosado, J.A.; Redondo, P.C. PGRMC1 Inhibits Progesterone-Evoked Proliferation and Ca2+ Entry Via STIM2 in MDA-MB-231 Cells. Int. J. Mol. Sci. 2020, 21, 7641. [Google Scholar] [CrossRef]
- Scherbakov, A.M.; Levina, I.S.; Kulikova, L.E.; Fedyushkina, I.V.; Skvortsov, V.S.; Veselovsky, A.V.; Kuznetsov, Y.V.; Zavarzin, I.V. Cytotoxic activity and molecular modeling of progestins - pregna-D′-pentarans. Biomeditsinskaya Khimiya 2016, 62, 290–294. [Google Scholar] [CrossRef] [Green Version]
- Tzavlaki, K.; Moustakas, A. TGF-βSignaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, G.C.; Nagarsheth, N.P.; Lee, K.L.; Bentley, R.C.; Walmer, D.K.; Cline, M.; Whitaker, R.S.; Isner, P.; Berchuck, A.; Dodge, R.K.; et al. Progestin-Induced Apoptosis in the Macaque Ovarian Epithelium: Differential Regulation of Transforming Growth Factor-. J. Natl. Cancer Inst. 2002, 94, 50–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, S.-M. Estrogen, Progesterone and Epithelial Ovarian Cancer. Reprod. Biol. Endocrinol. 2003, 1, 73. [Google Scholar] [CrossRef] [Green Version]
- Syed, V.; Ulinski, G.; Mok, S.C.; Yiu, G.K.; Ho, S.M. Expression of gonadotropin receptor and growth responses to key reproductive hormones in normal and malignant human ovarian surface epithelial cells. Cancer Res. 2001, 61. [Google Scholar]
- Li, S.; Hao, J.; Hong, Y.; Mai, J.; Huang, W. Long Non-Coding RNA NEAT1 Promotes the Proliferation, Migration, and Metastasis of Human Breast-Cancer Cells by Inhibiting miR-146b-5p Expression. Cancer Manag. Res. 2020, 12, 6091–6101. [Google Scholar] [CrossRef]
- A Fedotcheva, T.; Odintsova, E.V.; Shimanovskiĭ, N.L. Molecular mechanisms of cytostatic and chemosensitizing action of gestagens. Ann. Russ. Acad. Med Sci. 2010. [Google Scholar]
- Tang, L.; Zhang, Y.; Pan, H.; Luo, Q.; Zhu, X.-M.; Dong, M.-Y.; Leung, P.C.; Sheng, J.-Z.; Huang, H.-F. Involvement of cyclin B1 in progesterone-mediated cell growth inhibition, G2/M cell cycle arrest, and apoptosis in human endometrial cell. Reprod. Biol. Endocrinol. 2009, 7, 144. [Google Scholar] [CrossRef] [Green Version]
- Fedotcheva, T.A.; Teplova, V.V.; Fedotcheva, N.I. Activation of the calcium-dependent cyclosporin-sensitive mitochondrial pore by doxorubicin in combination with iron ions. Biol. Memb. 2018, 35, 79–84. [Google Scholar] [CrossRef]
- Fedotcheva, T.A.; Fedotcheva, N.I. Protectors of the Mitochondrial Permeability Transition Pore Activated by Iron and Doxorubicin. Curr. Cancer Drug Targets 2021, 21, 514–525. [Google Scholar] [CrossRef]
- Fedotcheva, N.I.; Teplova, V.V.; Fedotcheva, T.A.; Rzheznikov, V.M.; Shimanovskii, N.L. Effect of progesterone and its synthetic analogues on the activity of mitochondrial permeability transition pore in isolated rat liver mitochondria. Biochem. Pharmacol. 2009, 78, 1060–1068. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, S.; Di Sante, M.; Sileikyte, J.; Deveraux, J.; Bauer, T.; Bround, M.J.; Menabò, R.; Paillard, M.; Alanova, P.; Carraro, M.; et al. A novel class of cardioprotective small-molecule PTP inhibitors. Pharmacol. Res. 2019, 151, 104548. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Tang, M.; Bode, A.M.; Liao, W.; Cao, Y. ANTs and cancer: Emerging pathogenesis, mechanisms, and perspectives. Biochim. et Biophys. Acta (BBA) - Bioenerg. 2020, 1875, 188485. [Google Scholar] [CrossRef]
- Lytovchenko, O.; Kunji, E.R. Expression and putative role of mitochondrial transport proteins in cancer. Biochim. et Biophys. Acta (BBA)-Bioenerg. 2017, 1858, 641–654. [Google Scholar] [CrossRef]
- Jang, J.-Y.; Choi, Y.; Jeon, Y.-K.; Aung, K.C.A.; Kim, C.-W. Over-expression of Adenine Nucleotide Translocase 1 (ANT1) Induces Apoptosis and Tumor Regression in vivo. BMC Cancer 2008, 8, 160. [Google Scholar] [CrossRef] [Green Version]
- Gallerne, C.; Touat, Z.; Chen, Z.X.; Martel, C.; Mayola, E.; El Dein, O.S.; Buron, N.; Le Bras, M.; Jacotot, E.; Borgne-Sanchez, A. The fourth isoform of the adenine nucleotide translocator inhibits mitochondrial apoptosis in cancer cells. Int. J. Biochem. Cell Biol. 2010, 42, 623–629. [Google Scholar] [CrossRef]
- Jang, J.-Y.; Kim, Y.-G.; Nam, S.J.; Keam, B.; Kim, T.M.; Jeon, Y.K.; Kim, C.W. Targeting Adenine Nucleotide Translocase-2 (ANT2) to Overcome Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor in Non–Small Cell Lung Cancer. Mol. Cancer Ther. 2016, 15, 1387–1396. [Google Scholar] [CrossRef] [Green Version]
- Stepien, G.; Torroni, A.; Chung, A.B.; Hodge, J.A.; Wallace, D.C. Differential expression of adenine nucleotide translocator isoforms in mammalian tissues and during muscle сell differentiation. J. Biol. Chem. 1992, 267, 14592–14597. [Google Scholar] [CrossRef]
- Wang, J.-Q.; Yang, Y.; Cai, C.-Y.; Teng, Q.-X.; Cui, Q.; Lin, J.; Assaraf, Y.G.; Chen, Z.-S. Multidrug resistance proteins (MRPs): Structure, function and the overcoming of cancer multidrug resistance. Drug Resist. Updat. 2021, 54, 100743. [Google Scholar] [CrossRef]
- Nanayakkara, A.K.; Follit, C.A.; Chen, G.; Williams, N.S.; Vogel, P.D.; Wise, J.G. Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells. Sci. Rep. 2018, 8, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Lai, J.-I.; Tseng, Y.-J.; Chen, M.-H.; Huang, C.-Y.F.; Chang, P.M.-H. Clinical Perspective of FDA Approved Drugs With P-Glycoprotein Inhibition Activities for Potential Cancer Therapeutics. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Yang, C.P.H.; DePinho, S.G.; Greenberger, L.M.; Arceci, R.J.; Horwitz, S.B. Progesterone Interacts with P-Glycoprotein in Multidrug-resistant Cells and in the Endometrium of Gravid Uterus. J. Biol. Chem. 1989, 264, 782–788. [Google Scholar] [CrossRef]
- Barnes, K.M.; Dickstein, B.; Cutler, G.B.; Fojo, T.; Bates, S.E. Steroid Transport, Accumulation, and Antagonism of P-Glycoprotein in Multidrug-Resistant Cells. Biochemistry 1996, 35, 4820–4827. [Google Scholar] [CrossRef] [PubMed]
- Gruol, D.J.; Zee, M.C.; Trotter, J.; Bourgeois, S. Reversal of multidrug resistance by RU 486. Cancer Res. 1994, 54. [Google Scholar]
- Leonessa, F.; Kim, J.-H.; Ghiorghis, A.; Kulawiec, R.J.; Hammer, C.; Talebian, A.; Clarke, R. C-7 Analogues of Progesterone as Potent Inhibitors of the P-Glycoprotein Efflux Pump. J. Med. Chem. 2001, 45, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Victoria, J.M.; Conseil, G.; Munoz-Martinez, F.; Dayan, G.; Marsaud, V.; Castanys, S.; Gamarro, F.; Renoir, J.M.; Di Pietro, A. RU49953: A non-hormonal steroid derivative that potently inhibits P-glycoprotein and reverts cellular multidrug resistance. Cell. Mol. Life Sci. 2003, 60, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Alameh, G.; Emptoz-Bonneton, A.; De Ravel, M.R.; Matera, E.L.; Mappus, E.; Balaguer, P.; Rocheblave, L.; Lomberget, T.; Dumontet, C.; Le Borgne, M.; et al. In vitro modulation of multidrug resistance by pregnane steroids and in vivo inhibition of tumour development by 7α-OBz-11α(R)-OTHP-5β-pregnanedione in K562/R7 and H295R cell xenografts. J. Enzym. Inhib. Med. Chem. 2019, 34, 684–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, C.-H. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int. 2005, 5, 30. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, P.M.; Jackman, J.; Bae, I.; Myers, T.G.; Fan, S.; Mutoh, M.; A Scudiero, D.; Monks, A.; A Sausville, E.; Weinstein, J.N.; et al. Characterization of the p53 tumor suppressor pathway in cell lines of the National Cancer Institute anticancer drug screen and correlations with the growth-inhibitory potency of 123 anticancer agents. Cancer Res. 1997, 57. [Google Scholar]
- Rao, U.; Fine, R.L.; Scarborough, G.A. Antiestrogens and steroid hormones: Substrates of the human P-glycoprotein. Biochem. Pharmacol. 1994, 48, 287–292. [Google Scholar] [CrossRef]
- Wang, E.-J.; Casciano, C.N.; Clement, R.P.; Johnson, W.W. Cholesterol Interaction with the Daunorubicin Binding Site of P-Glycoprotein. Biochem. Biophys. Res. Commun. 2000, 276, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, X.; Sun, L.; Zhang, H.; Li, L.; Wang, X.; Li, W.; Su, P.; Hu, J.; Gao, P.; et al. Progesterone Negatively Regulates BCRP in Progesterone Receptor-Positive Human Breast Cancer Cells. Cell. Physiol. Biochem. 2013, 32, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xu, L.; He, K. Modulation of multiple drug resistance by nomegestrol acetate and droloxifene in K562/A02. Zhonghua Xue Ye Xue Za Zhi 1999, 20, 288–291. [Google Scholar] [PubMed]
- Matin, K.; Egorin, M.J.; Ballesteros, M.F.; Smith, D.; Lembersky, B.; Day, R.S.; Johnson, C.S.; Trump, D.L. Phase I and pharmacokinetic study of vinblastine and high-dose megestrol acetate. Cancer Chemother. Pharmacol. 2002, 50, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xu, L.-Z.; He, K.-L.; Guo, W.-J.; Zheng, Y.-H.; Xia, P.; Chen, Y. Reversal effects of nomegestrol acetate on multidrug resistance in adriamycin-resistant MCF7 breast cancer cell line. Breast Cancer Res. 2001, 3, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, C.-P.H.; Horwitz, S.B.; Trail, P.A.; Casazza, A.M. Reversal of the human and murine multidrug-resistance phenotype with megestrol acetate. Cancer Chemother. Pharmacol. 1994, 34, 96–102. [Google Scholar] [CrossRef]
- Fleming, G.F.; Amato, J.M.; Agresti, M.; Safa, A.R. Megestrol acetate reverses multidrug resistance and interacts with P-glycoprotein. Cancer Chemother. Pharmacol. 1992, 29, 445–449. [Google Scholar] [CrossRef]
- Paucarmayta, A.; Taitz, H.; McGlorthan, L.; Casablanca, Y.; Maxwell, G.L.; Darcy, K.M.; Syed, V. Progesterone-Calcitriol Combination Enhanced Cytotoxicity of Cisplatin in Ovarian and Endometrial Cancer Cells In Vitro. Biomedicines 2020, 8, 73. [Google Scholar] [CrossRef] [Green Version]
- McGlorthan, L.; Paucarmayta, A.; Casablanca, Y.; Maxwell, G.L.; Syed, V. Progesterone induces apoptosis by activation of caspase-8 and calcitriol via activation of caspase-9 pathways in ovarian and endometrial cancer cells in vitro. Apoptosis 2021, 26, 184–194. [Google Scholar] [CrossRef]
- Tansan, S.; Aydin, H.; Urbano, G.; McCaffrey, R. Augmentation of vincristine cytotoxicity by megestrol acetate. Cancer Chemother. Pharmacol. 1997, 39, 333–340. [Google Scholar] [CrossRef]
- Brayboy, L.M.; O Knapik, L.; Long, S.; Westrick, M.; Wessel, G.M. Ovarian hormones modulate multidrug resistance transporters in the ovary. Contracept. Reprod. Med. 2018, 3, 26. [Google Scholar] [CrossRef]
- Laberge, R.-M.; Ambadipudi, R.; Georges, E. P-glycoprotein mediates the collateral sensitivity of multidrug resistant cells to steroid hormones. Biochem. Biophys. Res. Commun. 2014, 447, 574–579. [Google Scholar] [CrossRef]
- Argov, M.; Bod, T.; Batra, S.; Margalit, R. Novel steroid carbamates reverse multidrug-resistance in cancer therapy and show linkage among efficacy, loci of drug action and P-glycoprotein’s cellular localization. Eur. J. Pharm. Sci. 2010, 41, 53–59. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, L.; Gupta, A.; Vethanayagam, R.R.; Zhang, Y.; Unadkat, J.D.; Mao, Q. Regulation of BCRP/ABCG2 expression by progesterone and 17β-estradiol in human placental BeWo cells. Am. J. Physiol. Metab. 2006, 290, E798–E807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claudio, J.A.; Emerman, J.T. The effects of cyclosporin A, tamoxifen, and medroxyprogesterone acetate on the enhancement of Adriamycin cytotoxicity in primary cultures of human breast epithelial cells. Breast Cancer Res. Treat. 1996, 41, 111–122. [Google Scholar] [CrossRef]
- Orlowski, S.; Valente, D.; Garrigos, M.; Ezan, E. Bromocriptine Modulates P-Glycoprotein Function. Biochem. Biophys. Res. Commun. 1998, 244, 481–488. [Google Scholar] [CrossRef]
- Orlowski, S.; Mir, L.M.; Belehradek, J.; Garrigos, M. Effects of steroids and verapamil on P-glycoprotein ATPase activity: Progesterone, desoxycorticosterone, corticosterone and verapamil are mutually non-exclusive modulators. Biochem. J. 1996, 317, 515–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buxbaum, E. Co-operative binding sites for transported substrates in the multiple drug resistance transporter Mdr1. JBIC J. Biol. Inorg. Chem. 1999, 265, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Atif, F.; Patel, N.R.; Yousuf, S.; Stein, N.G. The Synergistic Effect of Combination Progesterone and Temozolomide on Human Glioblastoma Cells. PLoS ONE 2015, 10, e0131441. [Google Scholar] [CrossRef] [PubMed]
- Odintsova, E.V.; Fedotcheva, T.A.; Banin, V.V.; Shimanovsky, N.L. Investigation of the mechanisms of sensitization of tumor cells with a new synthetic gestagenbuterol. Bull. RSMU 2012, 4, 69–74. [Google Scholar]
- Shchulkin, A.; Chernykh, I.; Popova, N.; Slepnev, A.; Yakusheva, E. Evaluation of female sex hormones influence on the protein-transporter p-glycoprotein functioning in vitro. Biomeditsinskaya Khimiya 2020, 66, 444–449. [Google Scholar] [CrossRef]
- Safa, A.R. Identification and Characterization of the Binding Sites of P-Glycoprotein for Multidrug Resistance-Related Drugs and Modulators. Curr. Med. Chem. Agents 2004, 4, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, K.; Okamura, N.; Hirai, M.; Tanigawara, Y.; Saeki, T.; Kioka, N.; Komano, T.; Hori, R. Human P-glycoprotein transports cortisol, aldosterone, and dexamethasone, but not progesterone. J. Biol. Chem. 1992, 267, 24248–24252. [Google Scholar] [CrossRef]
- Fedotcheva, T. Molecular Mechanisms of Cytostatic and Chemosensitizing Action of Gestagens on Tumor Cells. Ph.D. Thesis, Pirogov Russian National Research Medical University, Ministry of Health of the Russian Federation, Moscow, Russia, 30 May 2012. Available online: http://medical-diss.com/medicina/molekulyarnye-mehanizmy-tsitostaticheskogo-i-himiosensibiliziruyuschego-deystviya-gestagenov-na-opuholevye-kletki (accessed on 20 August 2021).
- Vasconcelos, F.C.; de Souza, P.S.; Hancio, T.; de Faria, F.C.C.; Maia, R.C. Update on drug transporter proteins in acute myeloid leukemia: Pathological implication and clinical setting. Crit. Rev. Oncol. 2021, 160, 103281. [Google Scholar] [CrossRef]
- Liu, J.; Pandya, P.; Afshar, S. Therapeutic Advances in Oncology. Int. J. Mol. Sci. 2021, 22, 2008. [Google Scholar] [CrossRef]
- Yin, P.; Lin, Z.; Cheng, Y.-H.; Marsh, E.E.; Utsunomiya, H.; Ishikawa, H.; Xue, Q.; Reierstad, S.; Innes, J.; Thung, S.; et al. Progesterone Receptor Regulates Bcl-2 Gene Expression through Direct Binding to Its Promoter Region in Uterine Leiomyoma Cells. J. Clin. Endocrinol. Metab. 2007, 92, 4459–4466. [Google Scholar] [CrossRef] [Green Version]
- Ory, K.; Lebeau, J.; Levalois, C.; Bishay, K.; Fouchet, P.; Allemand, I.; Therwath, A.; Chevillard, S. Apoptosis inhibition mediated by medroxyprogesterone acetate treatment of breast cancer cell lines. Breast Cancer Res. Treat. 2001, 68, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Subhawong, A.P.; Nassar, H.; Halushka, M.K.; Illei, P.B.; Vang, R.; Argani, P. Heterogeneity of Bcl-2 expression in metastatic breast carcinoma. Mod. Pathol. 2010, 23, 1089–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, M.; Yamashita, J.; Ogawa, M. Medroxyprogesterone acetate inhibits human pancreatic carcinoma cell growth by inducing apoptosis in association with Bcl-2 phosphorylation. Cancer 2000, 88, 2000–2009. [Google Scholar] [CrossRef]
- Zhang, Y.-L.; Wen, X.-D.; Guo, X.; Huang, S.-Q.; Wang, T.-T.; Zhou, P.-T.; Li, W.; Zhou, L.-F.; Hu, Y.-H. Progesterone suppresses the progression of colonic carcinoma by increasing the activity of the GADD45α/JNK/c-Jun signalling pathway. Oncol. Rep. 2021, 45, 1–13. [Google Scholar] [CrossRef]
- Tocchetti, G.N.; Domínguez, C.J.; Zecchinati, F.; Arana, M.R.; Ruiz, M.L.; Villanueva, S.S.M.; Weiss, J.; Mottino, A.D.; Rigalli, J.P. Biphasic modulation of cAMP levels by the contraceptive nomegestrol acetate. Impact on P-glycoprotein expression and activity in hepatic cells. Biochem. Pharmacol. 2018, 154, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, M.; Hakuno, F.; Kamei, H.; Yamanaka, D.; Chida, K.; Minami, S.; Coe, I.R.; Takahashi, S.-I. Steroid hormones are novel nucleoside transport inhibitors by competition with nucleosides for their transporters. Biochem. Biophys. Res. Commun. 2014, 443, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-T.; May, E.W.S.; Chang, J.-F.; Hu, R.-Y.; Wang, L.H.-C.; Chan, H.-L. PGRMC1 contributes to doxorubicin-induced chemoresistance in MES-SA uterine sarcoma. Cell. Mol. Life Sci. 2015, 72, 2395–2409. [Google Scholar] [CrossRef] [PubMed]
- Rohe, H.J.; Ahmed, I.S.; Twist, K.E.; Craven, R.J. PGRMC1 (progesterone receptor membrane component 1): A targetable protein with multiple functions in steroid signaling, P450 activation and drug binding. Pharmacol. Ther. 2009, 121, 14–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, A.C.W.; Roche, S.L.; Byrne, A.M.; Ruiz-Lopez, A.M.; Cotter, T.G. Progesterone receptor signalling in retinal photoreceptor neuroprotection. J. Neurochem. 2015, 136, 63–77. [Google Scholar] [CrossRef] [Green Version]
- Friel, A.; Zhang, L.; Pru, C.A.; Clark, N.C.; McCallum, M.L.; Blok, L.J.; Shioda, T.; Peluso, J.J.; Rueda, B.R.; Pru, J.K. Progesterone receptor membrane component 1 deficiency attenuates growth while promoting chemosensitivity of human endometrial xenograft tumors. Cancer Lett. 2014, 356, 434–442. [Google Scholar] [CrossRef] [Green Version]
- Clark, N.C.; Friel, A.; Pru, C.A.; Zhang, L.; Shioda, T.; Rueda, B.R.; Peluso, J.J.; Pru, J.K. Progesterone receptor membrane component 1 promotes survival of human breast cancer cells and the growth of xenograft tumors. Cancer Biol. Ther. 2016, 17, 262–271. [Google Scholar] [CrossRef]
- Zhu, X.; Han, Y.; Fang, Z.; Wu, W.; Ji, M.; Teng, F.; Zhu, W.; Yang, X.; Jia, X.; Zhang, C. Progesterone protects ovarian cancer cells from cisplatin-induced inhibitory effects through progesterone receptor membrane component 1/2 as well as AKT signaling. Oncol. Rep. 2013, 30, 2488–2494. [Google Scholar] [CrossRef] [Green Version]
- Kareva, E.N.; Bulgakova, V.A.; Gutorova, D.S.; Oleinikova, O.M.; Kononova, I.N.; Gorbunov, A.A.; Breusenko, V.G.; Fisen-ko, V.P. Progesterone membrane receptor PGRMC1: Potential drug target. Eksp. Klin. Farmakol. 2020, 83, 19–29. [Google Scholar] [CrossRef]
- Lin, Y.; Higashisaka, K.; Shintani, T.; Maki, A.; Hanamuro, S.; Haga, Y.; Maeda, S.; Tsujino, H.; Nagano, K.; Fujio, Y.; et al. Progesterone receptor membrane component 1 leads to erlotinib resistance, initiating crosstalk of Wnt/β-catenin and NF-κB pathways, in lung adenocarcinoma cells. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Ahmed, I.S.; Rohe, H.J.; Twist, K.E.; Mattingly, M.N.; Craven, R.J. Progesterone Receptor Membrane Component 1 (Pgrmc1): A Heme-1 Domain Protein That Promotes Tumorigenesis and Is Inhibited by a Small Molecule. J. Pharmacol. Exp. Ther. 2010, 333, 564–573. [Google Scholar] [CrossRef] [Green Version]
- Nagendra, P.B.; Goad, J.; Nielsen, S.; Rassam, L.; Lombard, J.M.; Nahar, P.; Tanwar, P.S. Ovarian hormones through Wnt signalling regulate the growth of human and mouse ovarian cancer initiating lesions. Oncotarget 2016, 7, 64836–64853. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Lin, C.; Zhang, Y.; Zhang, X.; Zhang, C.; Zhang, P.; Xie, X.; Ren, Z. miR-506 enhances the sensitivity of human colorectal cancer cells to oxaliplatin by suppressing MDR1/P-gp expression. Cell Prolif. 2017, 50, e12341. [Google Scholar] [CrossRef]
- Ghandadi, M.; Valadan, R.; Mohammadi, H.; Akhtari, J.; Khodashenas, S.; Ashari, S. Wnt-β-catenin Signaling Pathway, the Achilles’ Heels of Cancer Multidrug Resistance. Curr. Pharm. Des. 2019, 25, 4192–4207. [Google Scholar] [CrossRef]
- Ciardiello, D.; Elez, E.; Tabernero, J.; Seoane, J. Clinical development of therapies targeting TGFβ: Current knowledge and future perspectives. Ann. Oncol. 2020, 31, 1336–1349. [Google Scholar] [CrossRef]
- Kim, B.-G.; Malek, E.; Choi, S.H.; Ignatz-Hoover, J.J.; Driscoll, J.J. Novel therapies emerging in oncology to target the TGF-β pathway. J. Hematol. Oncol. 2021, 14, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Huynh, L.K.; Hipolito, C.J.; Dijke, P.T. A Perspective on the Development of TGF-β Inhibitors for Cancer Treatment. Biomolecules 2019, 9, 743. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M.; Katoh, M. Molecular genetics and targeted therapy of WNT-related human diseases (Review). Int. J. Mol. Med. 2017, 40, 587–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
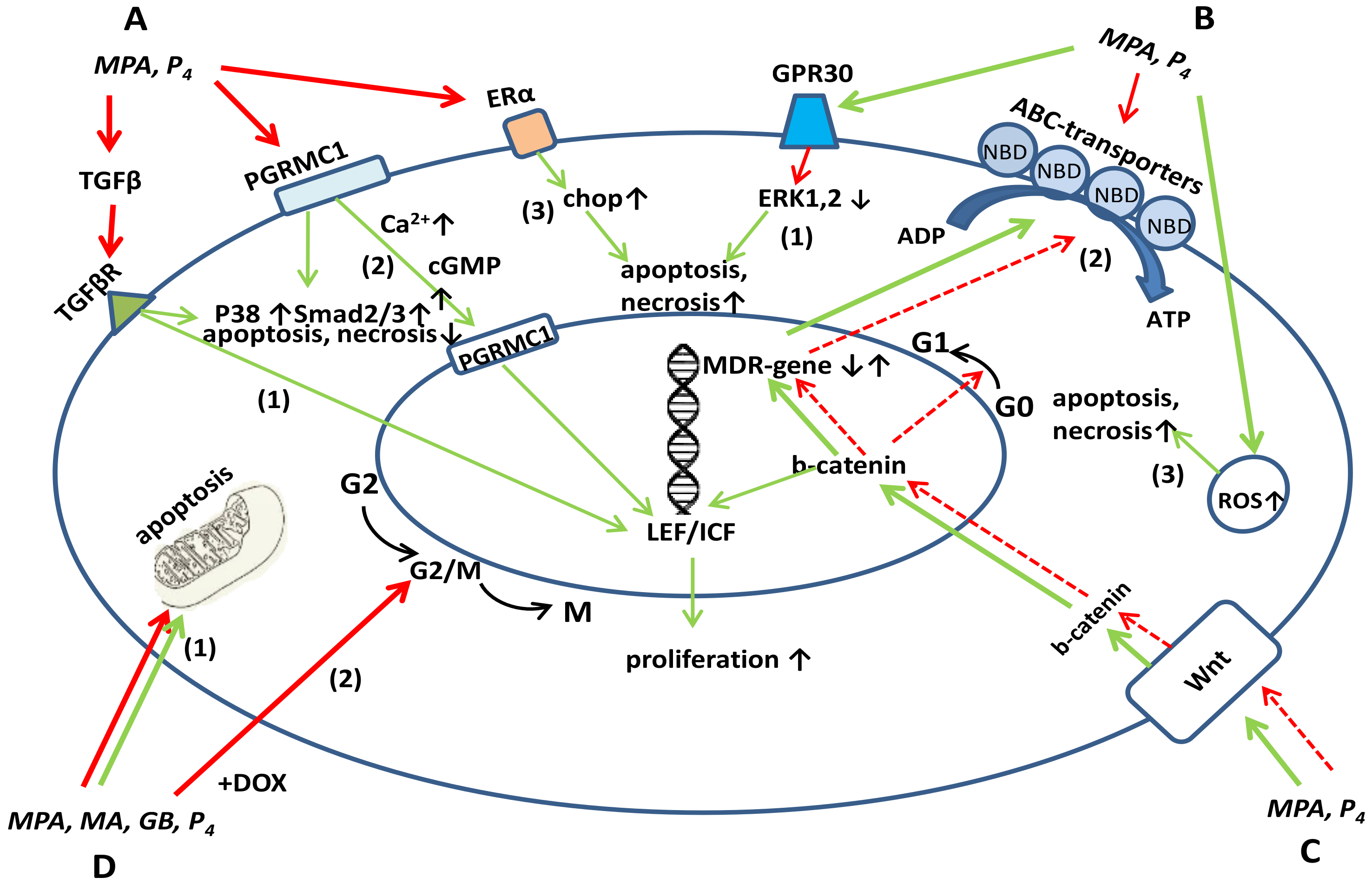
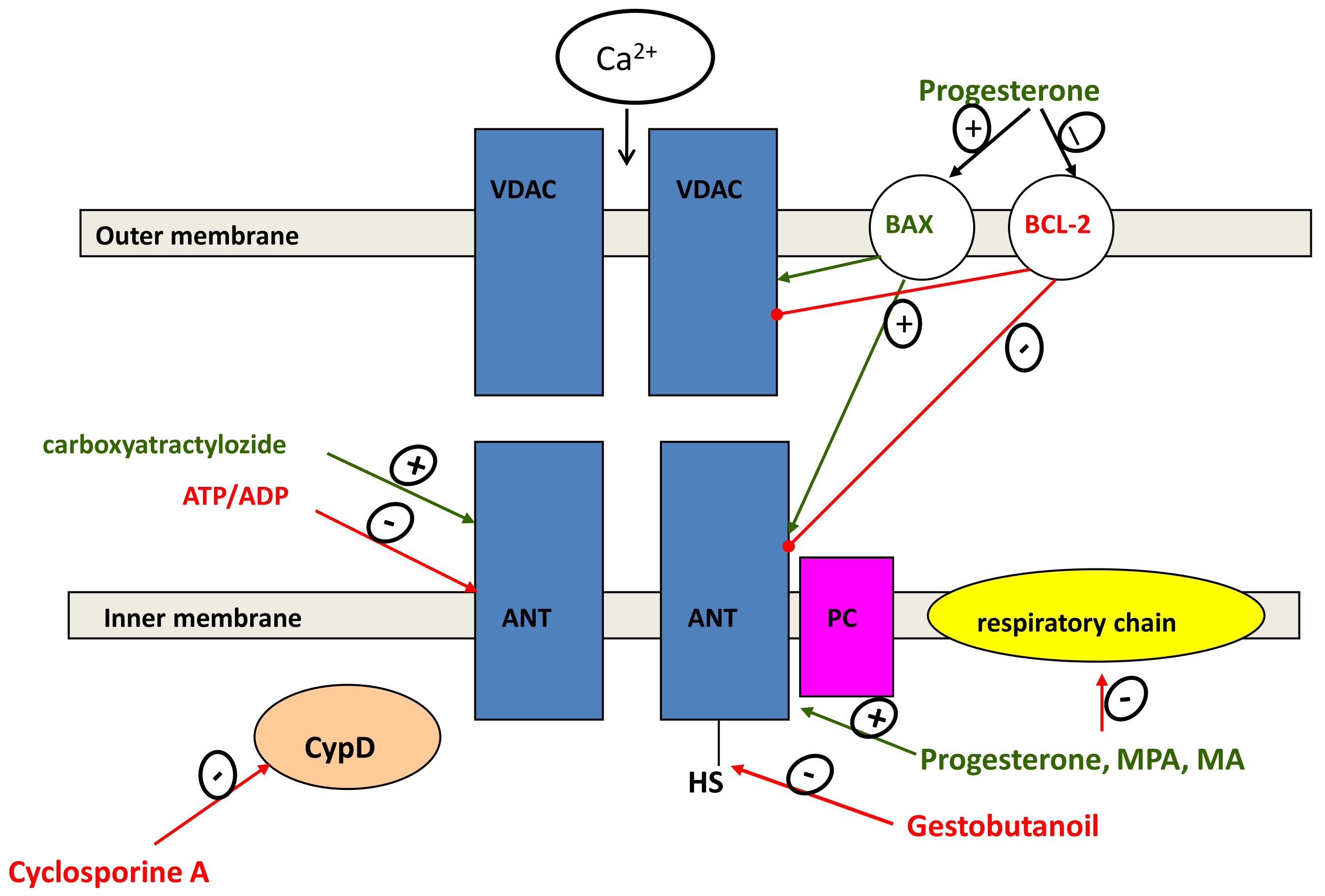

{kind=link}
{kind=link}
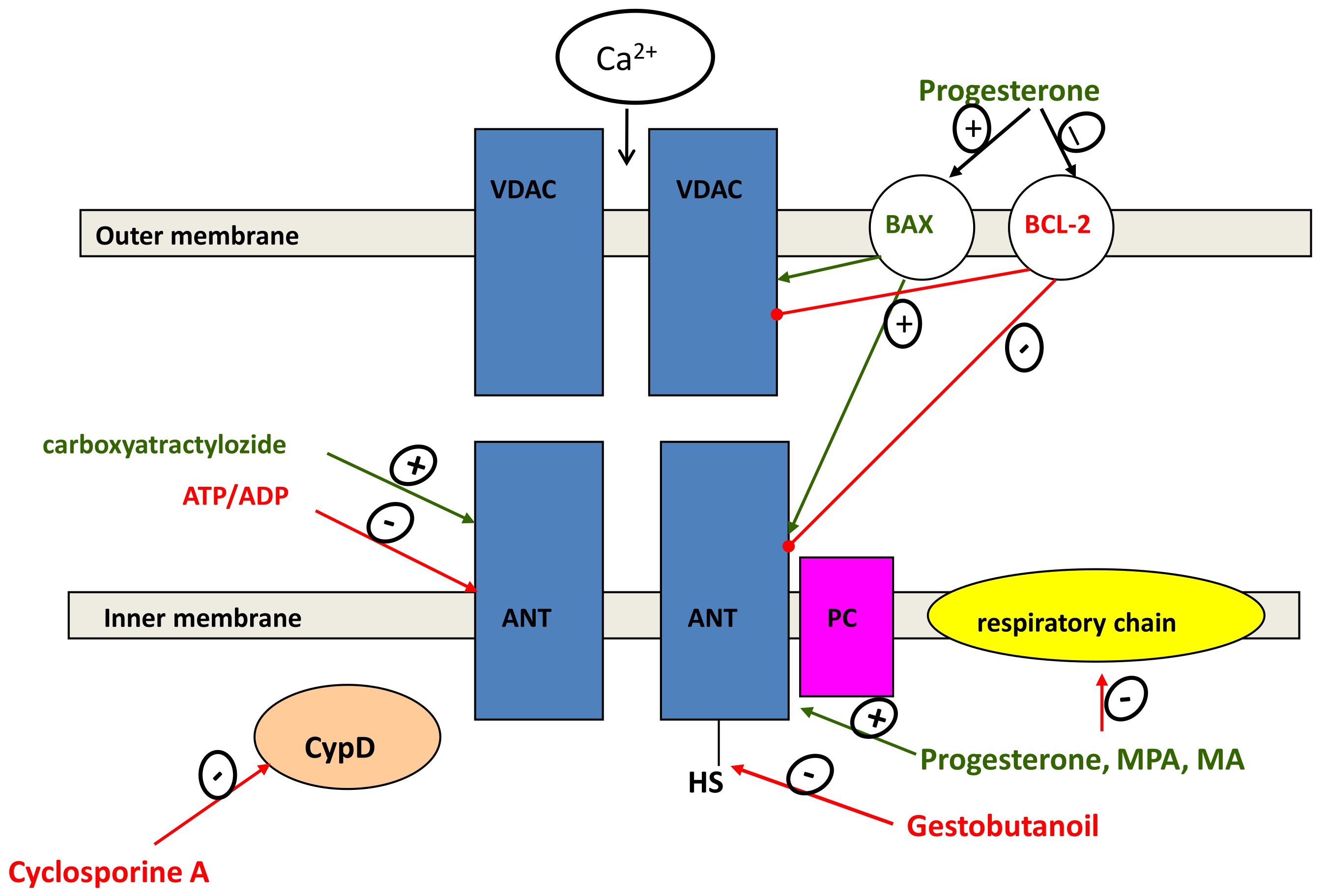{kind=link}
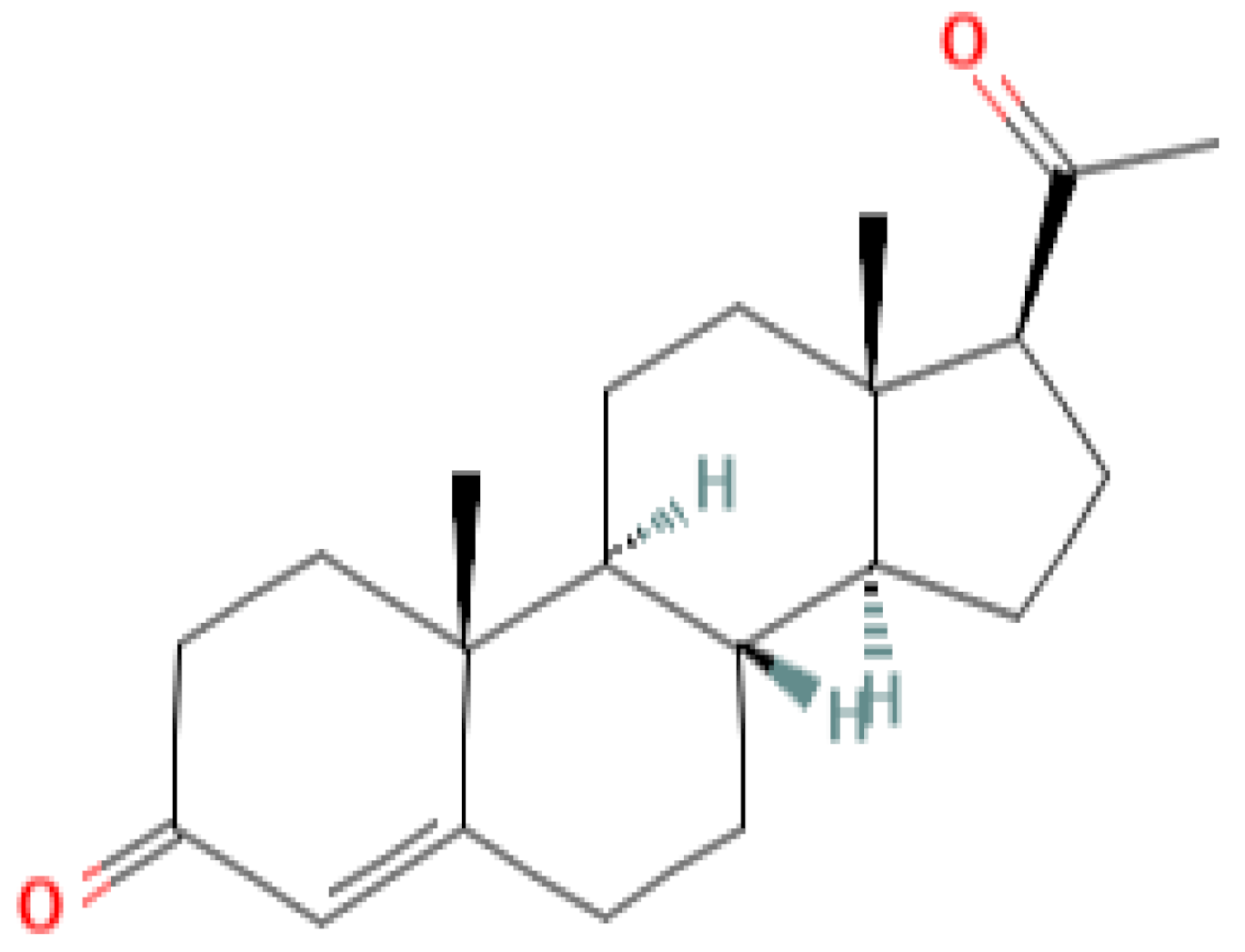 | 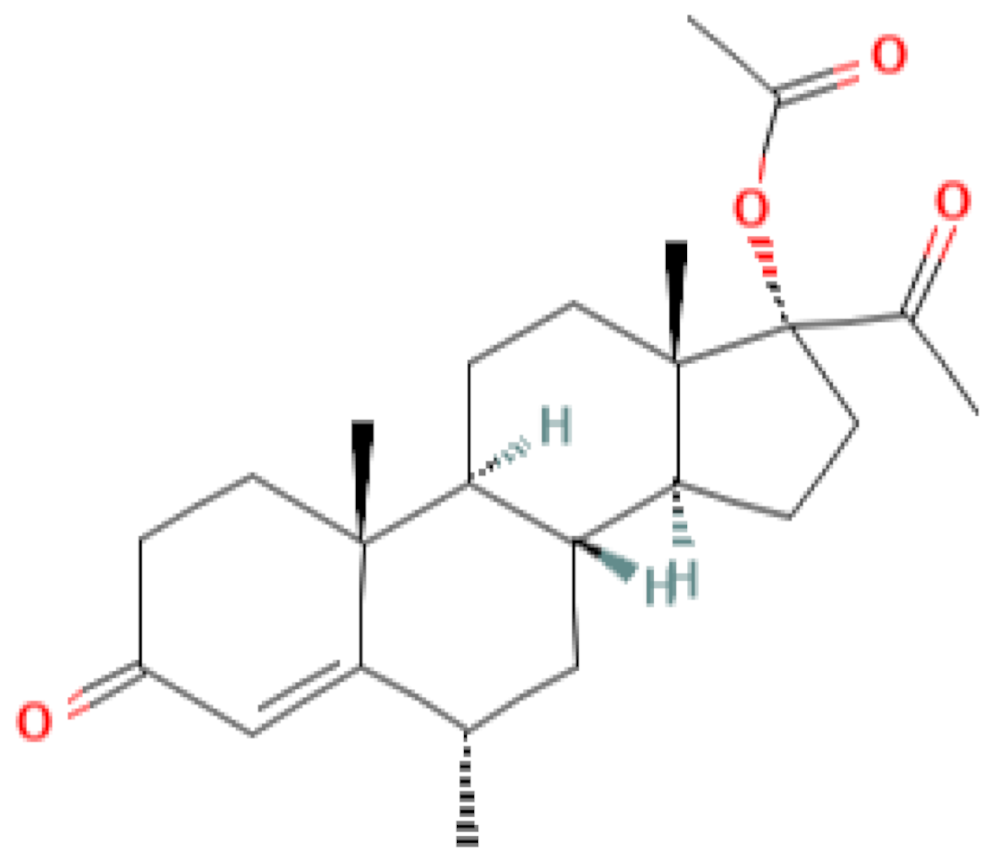 | 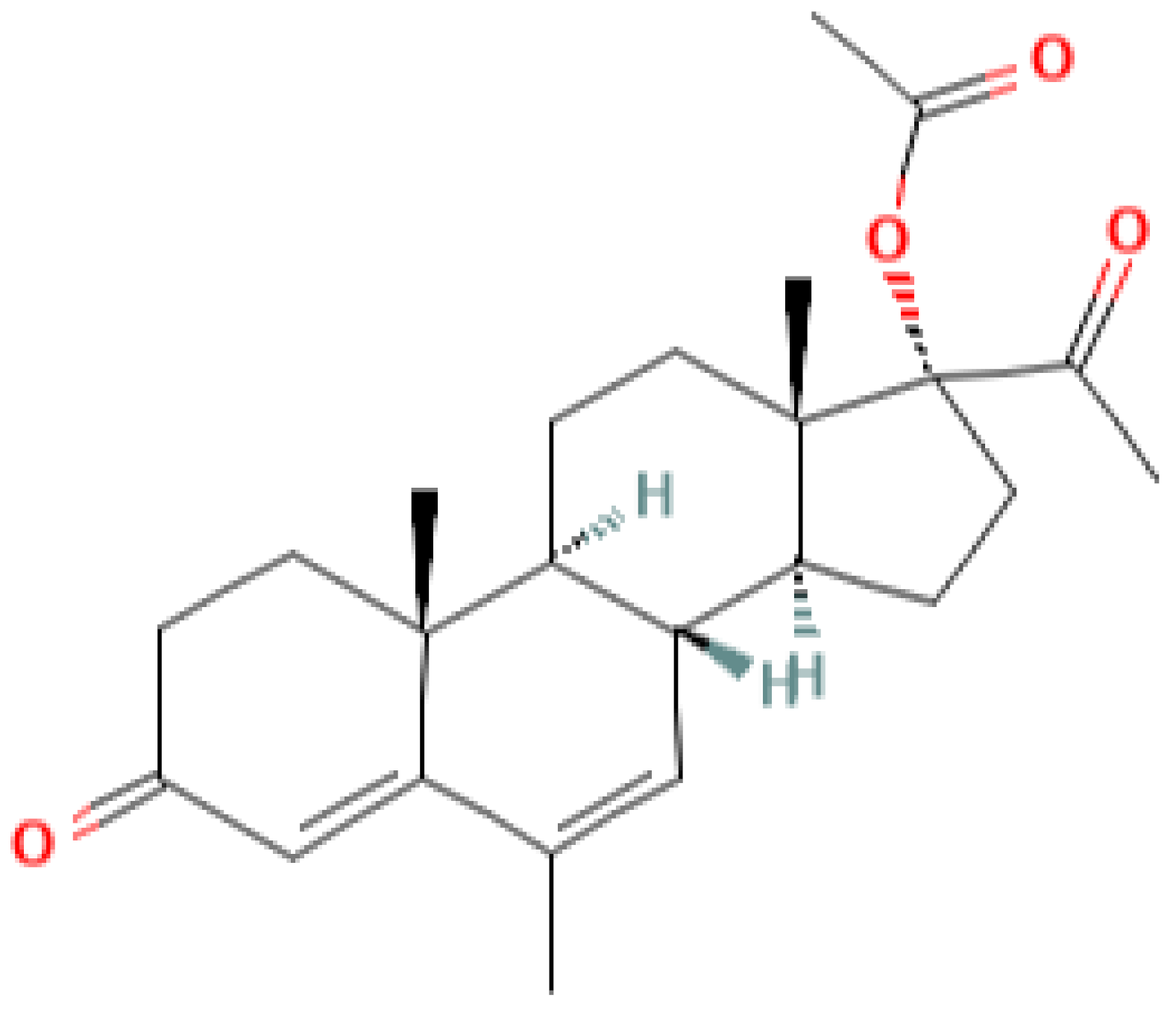 | 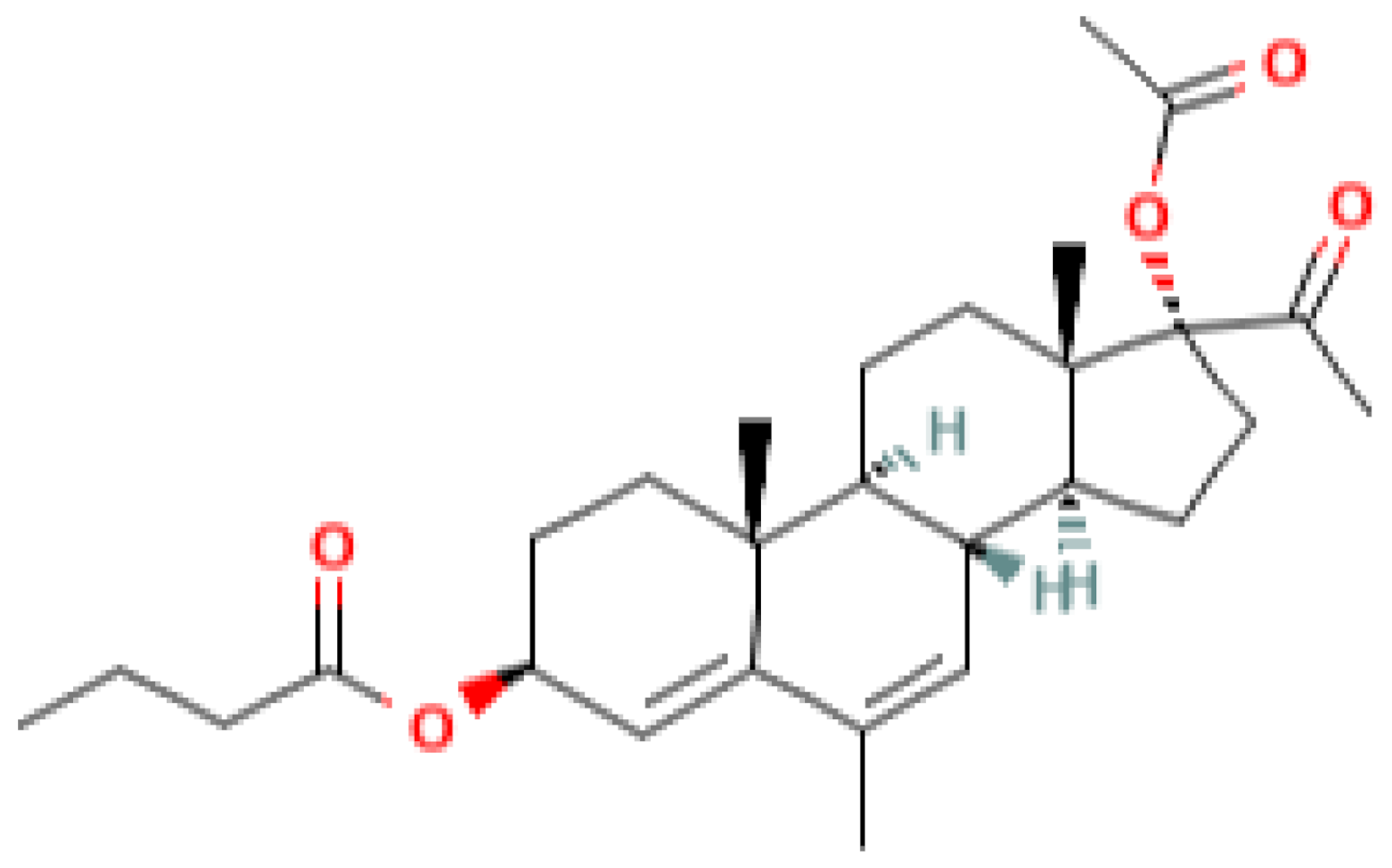 |
| progesterone | medroxyprogesterone acetate (MPA) | megestrol acetate (MA) | gestobutanoyl (butagest, buterol) (GB) |
| Type of Disease | Drugs | Estimated Performance Indicators | Clinical Trial Number |
|---|---|---|---|
| Endometrial hyperplasia | - MPA vs. Dydrogesterone; - Vaginal Micronized - Progesterone vs. LNG-IUS; - Megestrol acetate plus LNG-IUS; - LNG-IUS | - Complete response rate (CR) -Regression and remission rate of endometrial hyperplasia - Changes in the level of hormone receptor expression measured by immunohistochemistry; - Genes related to proliferation, estrogen signaling, and Wnt signaling | NCT03675139 NCT03992937 NCT03241888 NCT00788671 |
| Endometrial Cancer | - PD-1 inhibitor combined with progesterone; - MPA vs. MPA plus Entinostat; - LNG; - MPA; - LNG-IUS with or without everolimus; - MA with or without Temsirolimus and Tamoxifen; -MPA + Tamoxifen vs. Everolimus + Letrozole | - Expression of the PR; co-expression of ER; Ki67 and p21; - Hyperplastic or neoplastic lesion; - Fertility rate; - Frequency of the endometrial adenocarcinomas of the uterine corpus; - Life expectancy, number of relapses, side effects | NCT04046185 NCT03018249 NCT03463252 NCT00064025 NCT02397083 NCT03077698 NCT00729586 NCT02228681 |
| Cervical Intraepithelial Neoplasia | Progesterone | Regression rates of CIN I and II | NCT00247169 |
| Breast Cancer | - Megestrol Acetate + letrolzole vs. letrozole; - Exemestane vs. - Megestrol Acetate; - Megestrolacetate; - Megestrol Acetate vs. MPA; - MPA with or without Cyclophosphamide & Methotrexate; - Depot hydroxy-progesterone; - Letrozole vs. Letrozole plus Prometrium vs. Tamoxifen + Prometrium | - Determination of change in tumor proliferation; Geometric mean suppression of proliferation marker Ki67; - Definition of a gene set as a predictive biomarker for a reduction in Ki67; - Changes in the apoptotic markers Bcl-2 and Caspase 3 in the tumors following intervention; - Changes in ER, PR, AR, FoxA1, Cyclin D1 protein and mRNA expression in the tumors following intervention | NCT03306472 NCT01237327 NCT00005975 NCT03024580 NCT00002920 NCT00577122 NCT00123669 NCT03906669 |
| Cell Line/Object | Progestin or Combination Progestin + Modulator | Cytostatic Drug | Progestin Effect | Ref. |
|---|---|---|---|---|
| K562 | megestrole + droloxifene | adriamycin | significantly inhibit mdr1 and GST pi expression | [76] |
| In vivo, patients | high-dose oral megestrol acetate | vinblastine | in vivo decrease the general toxicity of vinblastine | [77] |
| Breast carcinoma MCF-7/ADR | nomegestrol acetate | adriamycin | Downregulating the mRNA and protein expression levels of MDR1 and GSTpi, increasing intracellular drug concentration and arresting cells at the G2M phase (NOM in combination with ADR) | [78] |
| Colon carcinoma HCT-116/VM46, breast carcinoma MCF-7/ADR, murine cell line, J774.2 | megestrol acetate | doxorubicin | increasing DOX accumulation measured by flow cytometry method | [79] |
| Human neuroblastic SH-SY5Y/VCR cells | megestrol acetate | vincristine | steroids inhibited the binding to P-glycoprotein | [80] |
| Human ovarian cancer cell lines, ES-2, TOV-21G, and UWB1.289; Endometrial cancer cell lines, HEC-1A and HEC-59 | progesterone-calcitriol | cisplatin | increased caspase-3, BAX, and decreased bcl-2, progesterone-calcitriol potentiated the anti-growth effects of cisplatin on cancer cells by attenuating the expression of SMAD2/3, multidrug resistance protein- 1 (MDR-1), and ABC transporters (ABCG1, and ABCG2), | [81] |
| Ovarian cancer cells, ES-2, and TOV-21, OV-90, TOV-112D, HEC-1A, and HEC-59 | progesterone-calcitriol | - | progesterone induced apoptosis through activation of caspase-8 | [82] |
| K562/R7 erythroleukaemiacells, steroidogenic NCI-H295R adrenocortical carcinoma cells | pregnane steroids | doxorubicine | interactions of pregnane modulators with human progesterone and pregnane X receptors | [70] |
| HL60 | pregnane steroids | mitoxantrone | specific P-gp, MRP1 and BCRPbinding and blocking | [70] |
| MCF7/MTX | pregnanesteroids | mitoxantrone | specific P-gp, MRP1 and BCRPbinding and blocking | [70] |
| P-gp-positive human colon cancer cell lines (HT-29 and CACO-2) and the human T-cell leukemia cell line (HUT-102) | megestrol acetate | vincristine | augmentation of vincristine cytotoxicity | [83] |
| Mice, Female | progesterone | influence MDR-1 transcript levels | [84] | |
| P-gp-positive Chinese hamster ovary cells (CHO)C5 cells | progesterone | deoxycorticosterone | P-gp, mitochondrial electron transport chain interaction | [85] |
| T47D, progesterone receptor positive (PR+) | progesterone | mitoxantrone | Progesterone negatively regulates BCRP | [75] |
| PR-negative MDA-MB-231 cells | progesterone | mitoxantrone | Progesterone negatively regulates BCRP | [75] |
| Human colon cancer line HCT-15 | progesterone-derived carbamates | paclitaxel and doxorubicin | increase in intracellular accumulation and reduced drug efflux | [86] |
| Human placental BeWo cells | progesterone | - | progesterone up-regulates BCRP via a mechanism other than PR, presumably via a nonclassical PR | [87] |
| MCF-7/ADR (adriamycin resistant) | medroxyprogesterone acetate + cyclosporine | adriamycin | enhancement of adriamycin cytotoxicity | [88] |
| CHO/ADR | progesterone + bromocriptine | vincristine | non-competitively inhibits the vinblastine-dependent P-gp ATPase activity | [89] |
| Chinese hamster DC-3F lung fibroblasts and its DC3F/ADX resistance variant | progesterone + verapamil | vincristine | progesterone and verapamil induce a synergistic modulation of P-gp ATPase activity | [90] |
| CHRB30 | progesterone + verapamil | adriamycin | inhibition of ATPase activity | [91] |
| U87MG Human Glioblastoma Cells | Progesterone alone and in combination with TMZ | TMZ | progesterone suppressed EGFR/PI3K/Akt/mTOR signaling pathway and MGMT expression; enhanced the cytotoxic effects of TMZ and reduced its toxic side effects in healthy primary cells. | [92] |
| MCF-7/dox | gestobutanoyl | etoposide, doxorubicin | MRP1, BCRP, BCL-2 mRNA expression inhibition | [93] |
| Caco-2 | progesterone | - | inhibition P-gp activity (100µM) | [94] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fedotcheva, T.A.; Fedotcheva, N.I.; Shimanovsky, N.L. Progestins as Anticancer Drugs and Chemosensitizers, New Targets and Applications. Pharmaceutics 2021, 13, 1616. https://doi.org/10.3390/pharmaceutics13101616
Fedotcheva TA, Fedotcheva NI, Shimanovsky NL. Progestins as Anticancer Drugs and Chemosensitizers, New Targets and Applications. Pharmaceutics. 2021; 13(10):1616. https://doi.org/10.3390/pharmaceutics13101616
Chicago/Turabian StyleFedotcheva, Tatiana A., Nadezhda I. Fedotcheva, and Nikolai L. Shimanovsky. 2021. "Progestins as Anticancer Drugs and Chemosensitizers, New Targets and Applications" Pharmaceutics 13, no. 10: 1616. https://doi.org/10.3390/pharmaceutics13101616
APA StyleFedotcheva, T. A., Fedotcheva, N. I., & Shimanovsky, N. L. (2021). Progestins as Anticancer Drugs and Chemosensitizers, New Targets and Applications. Pharmaceutics, 13(10), 1616. https://doi.org/10.3390/pharmaceutics13101616