
Customisable Tablet Printing: The Development of Multimaterial Hot Melt Inkjet 3D Printing to Produce Complex and Personalised Dosage Forms


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Printable Ink Preparation
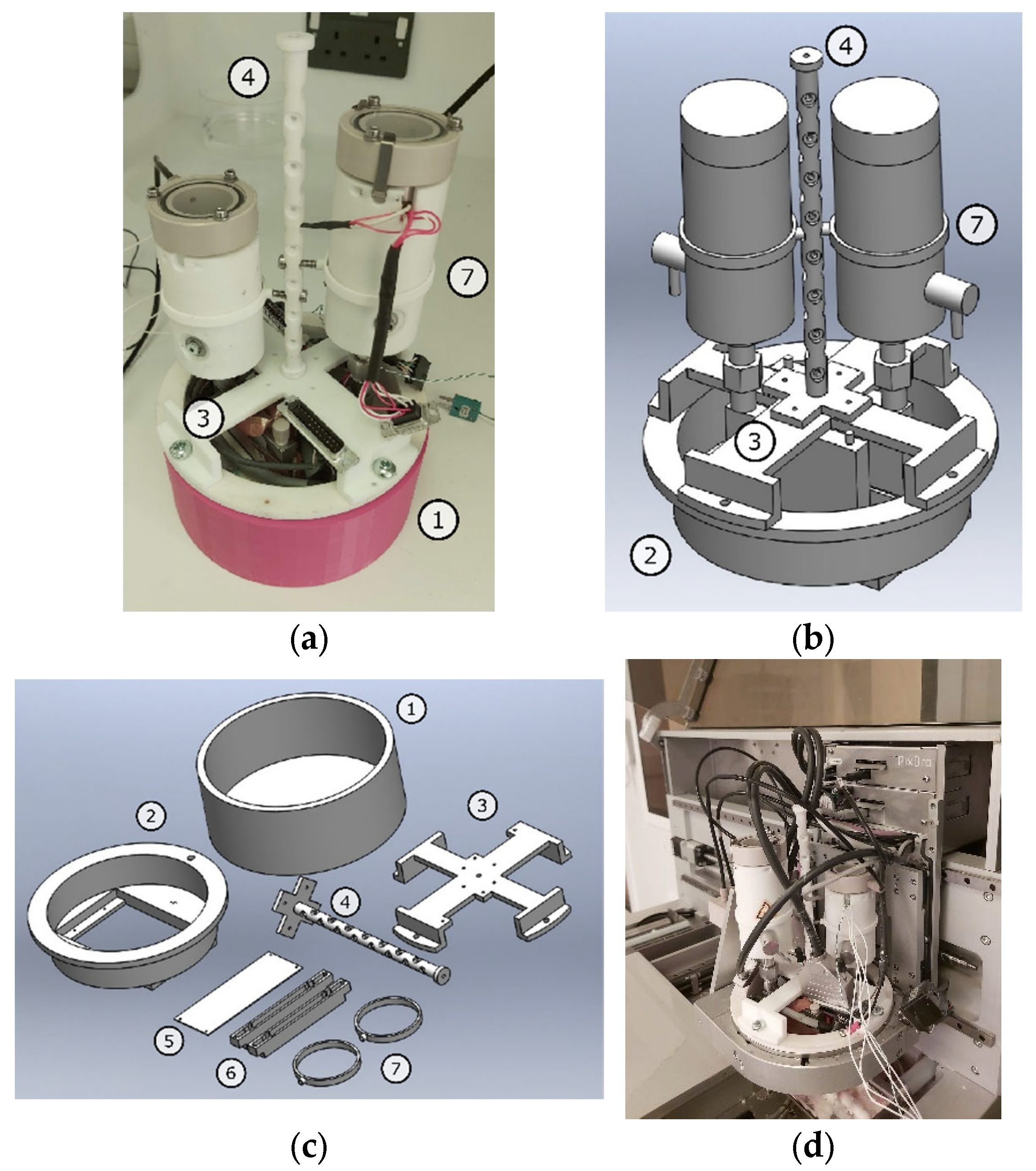
2.3. Tablets Fabrication and 3D Printing System
2.4. Tablet Designs
2.5. Differential Scanning Calorimetry (DSC)
2.6. Thermogravimetric Analysis (TGA)
2.7. Powder X-ray Diffraction (pXRD)
2.8. Scanning Electron Microscopy (SEM) and Energy-Dispersive X-ray (EDX)
2.9. Attenuated Total Reflection-Fourier Transform Infrared Spectroscopy (ATR-FTIR)
2.10. Raman Spectroscopy
2.11. In Vitro Release Study
3. Results and Discussion
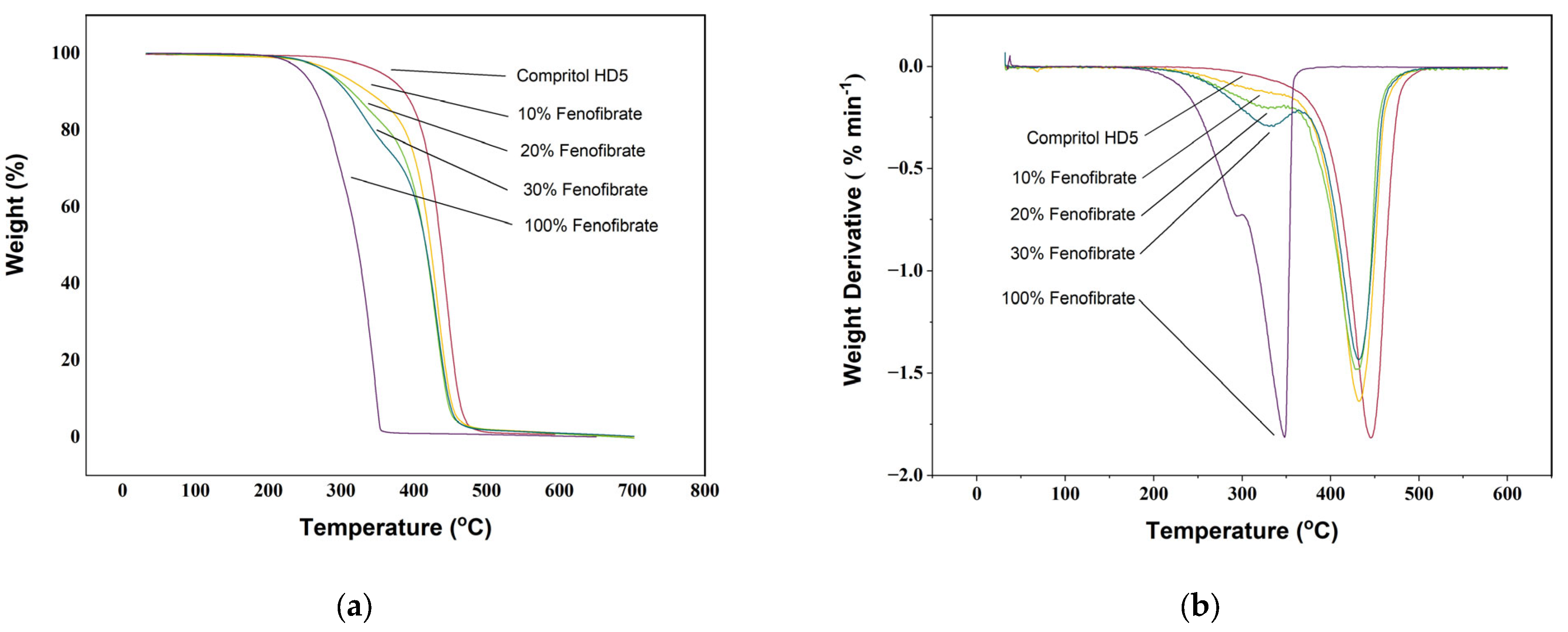
3.1. Thermogravimetric Analysis (TGA)
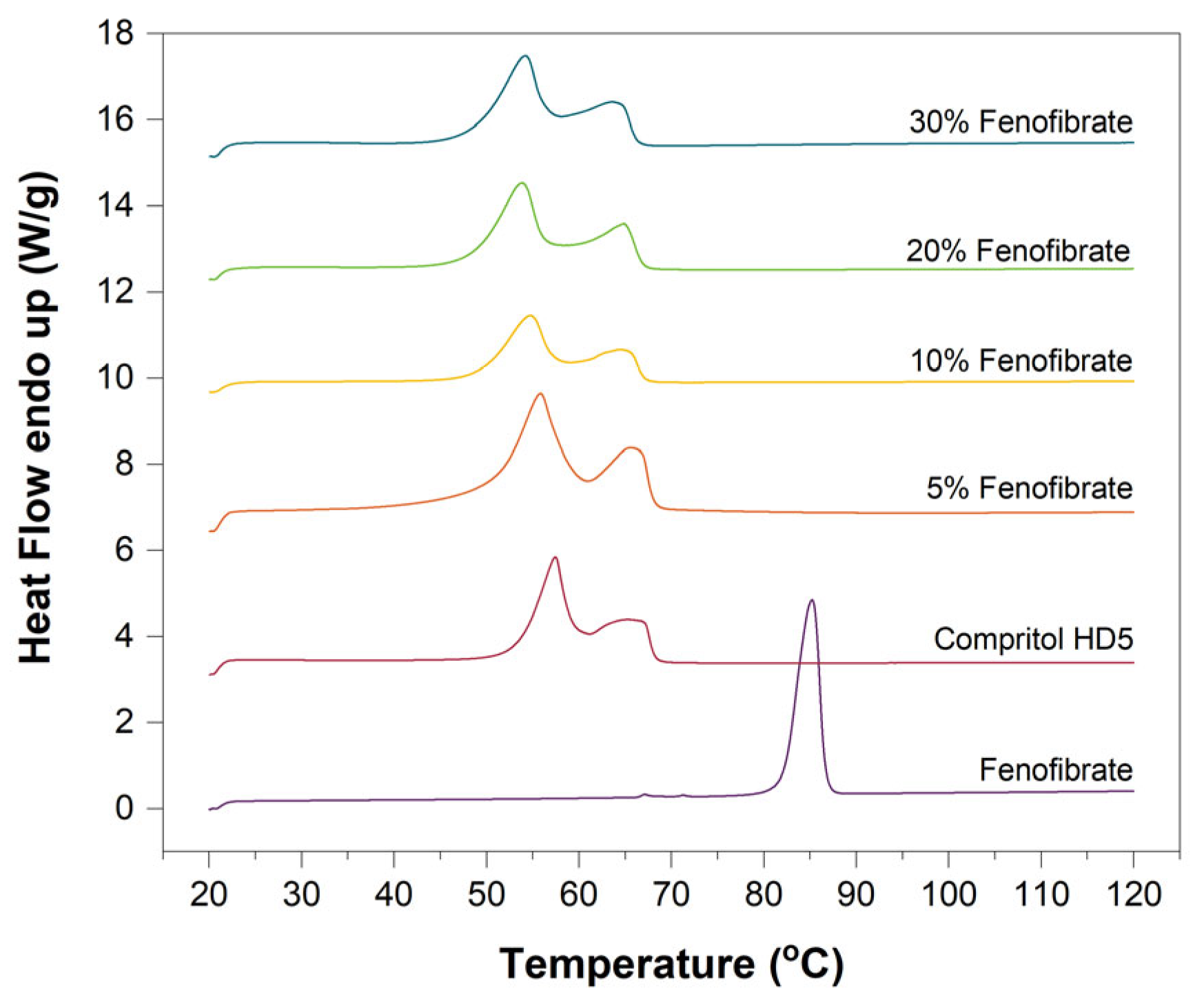
3.2. Differential Scanning Calorimetry (DSC)
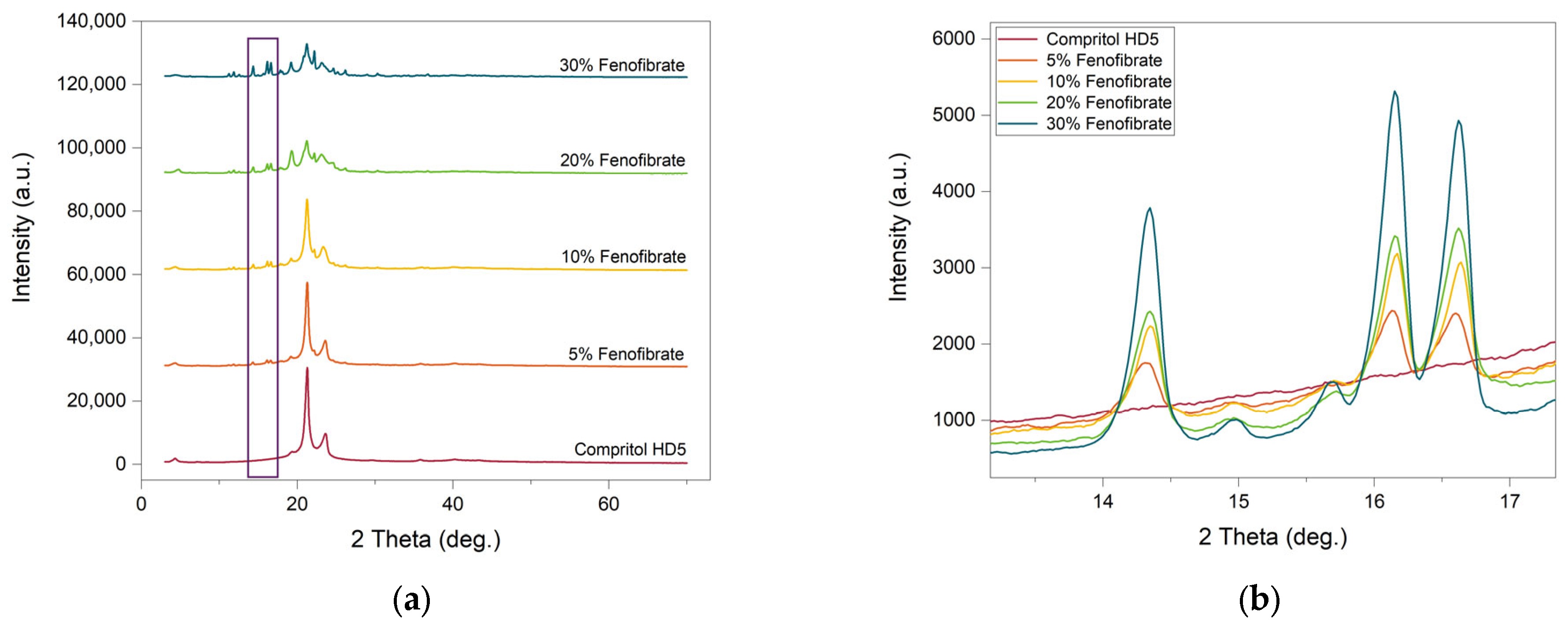
3.3. Powder X-ray Diffraction (pXRD)
3.4. Printed Tablets
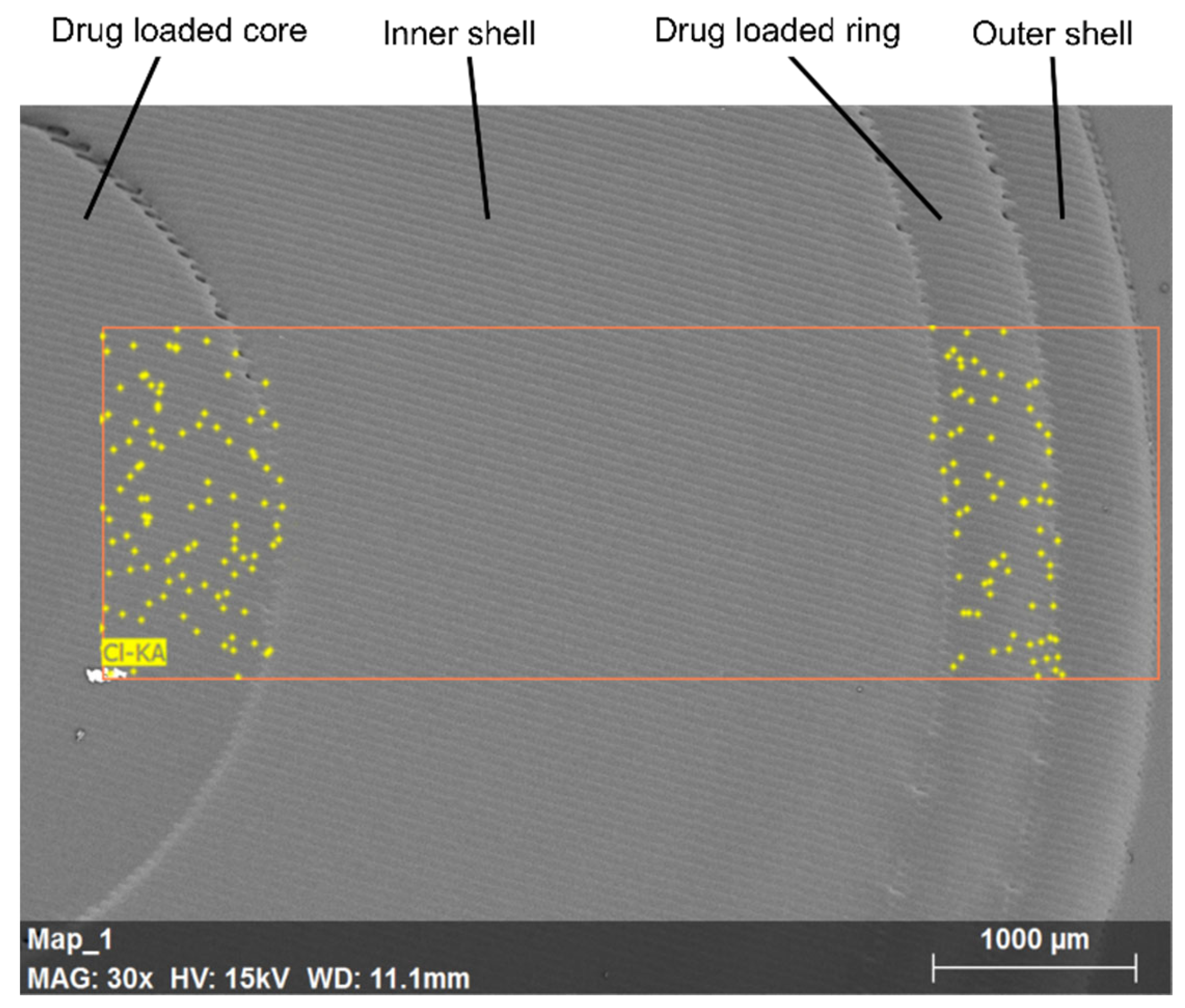
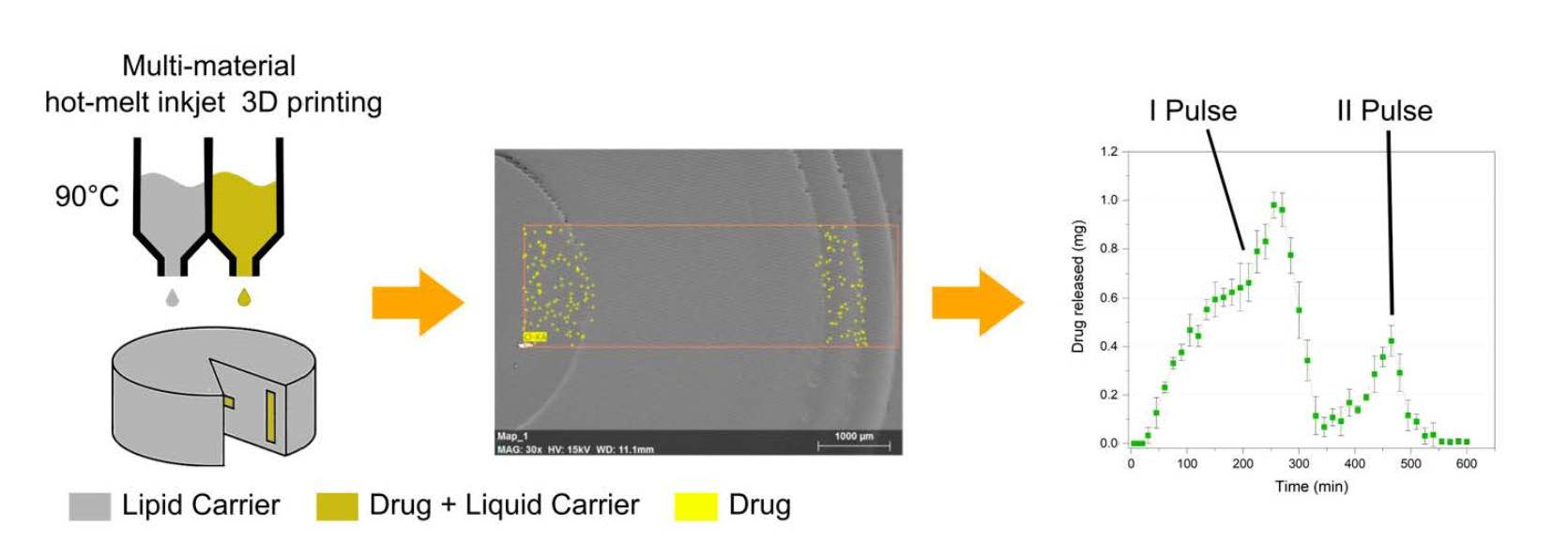
3.5. Scanning Electron Microscopy (SEM) and Energy-Dispersive X-ray (EDX)
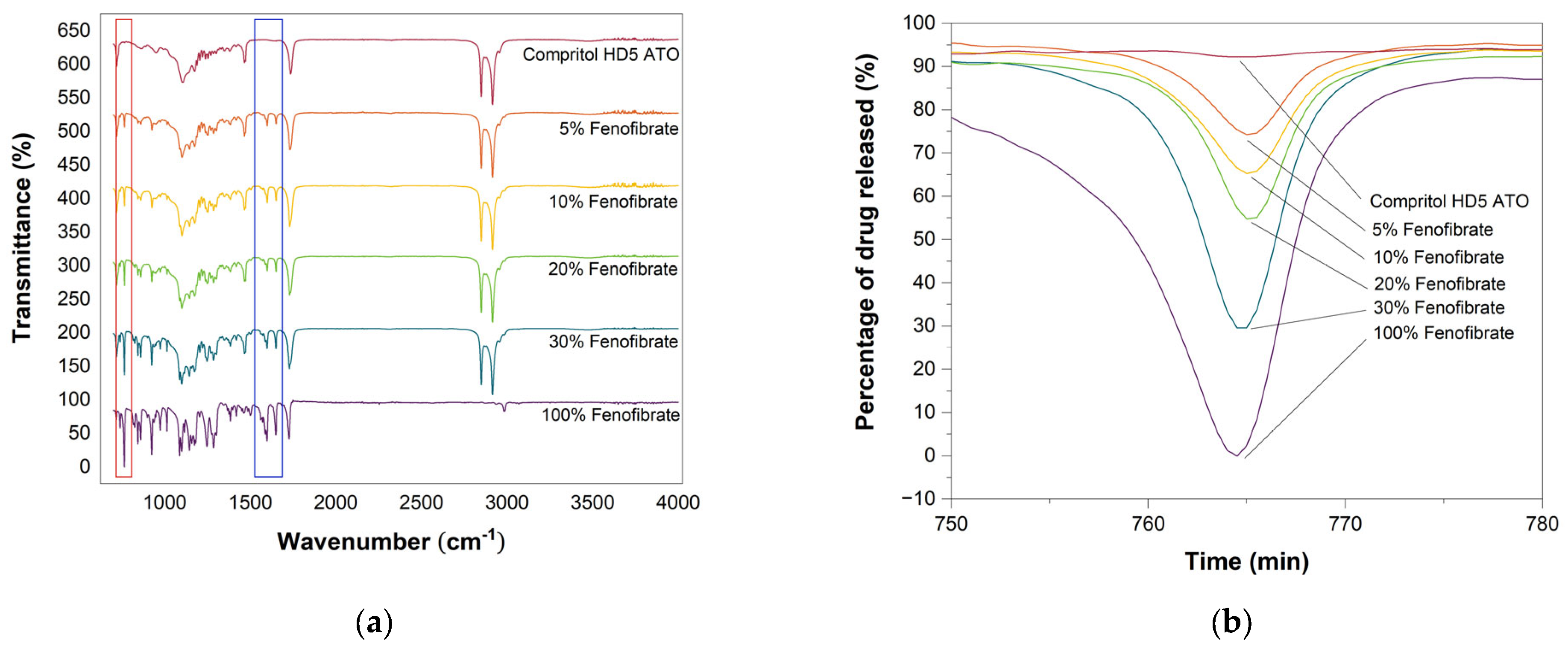
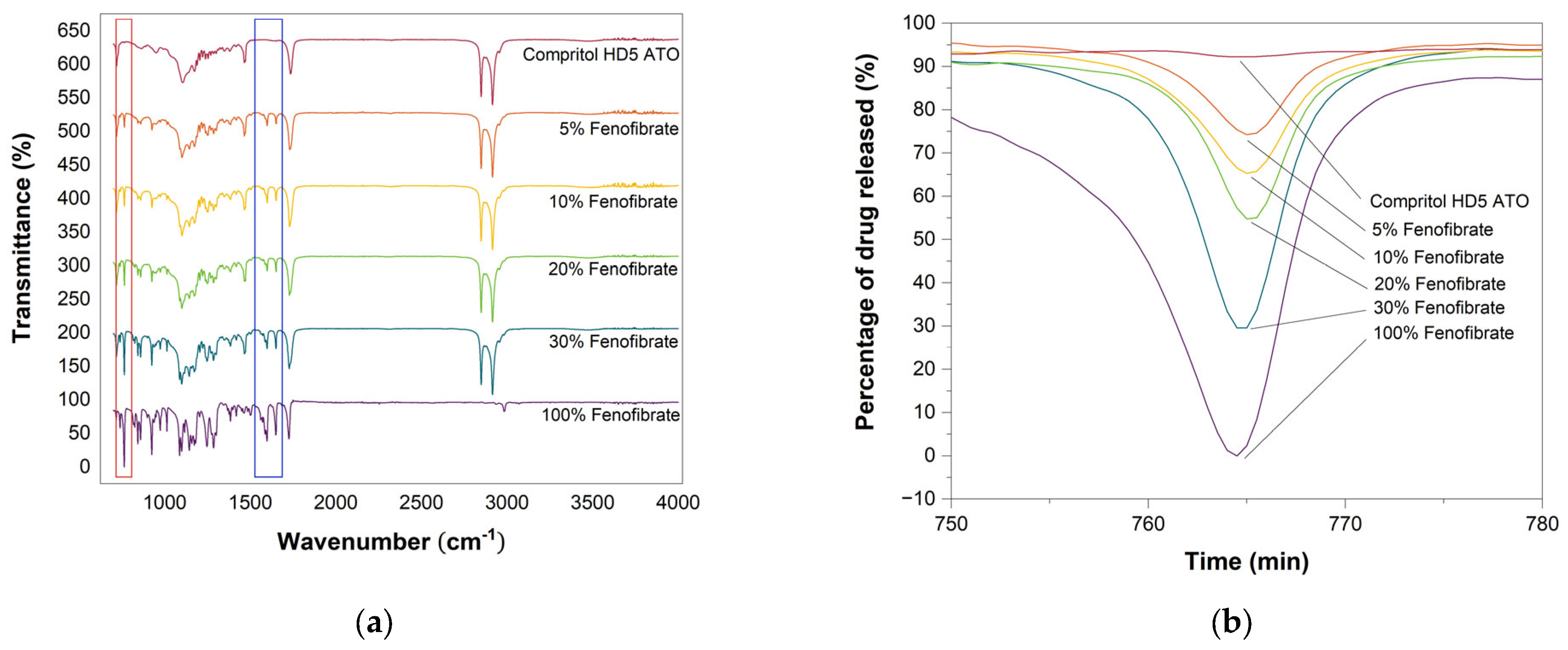
3.6. Attenuated Total Reflection-Fourier Transform Infrared Spectroscopy (ATR-FTIR)
3.7. Raman Spectroscopy
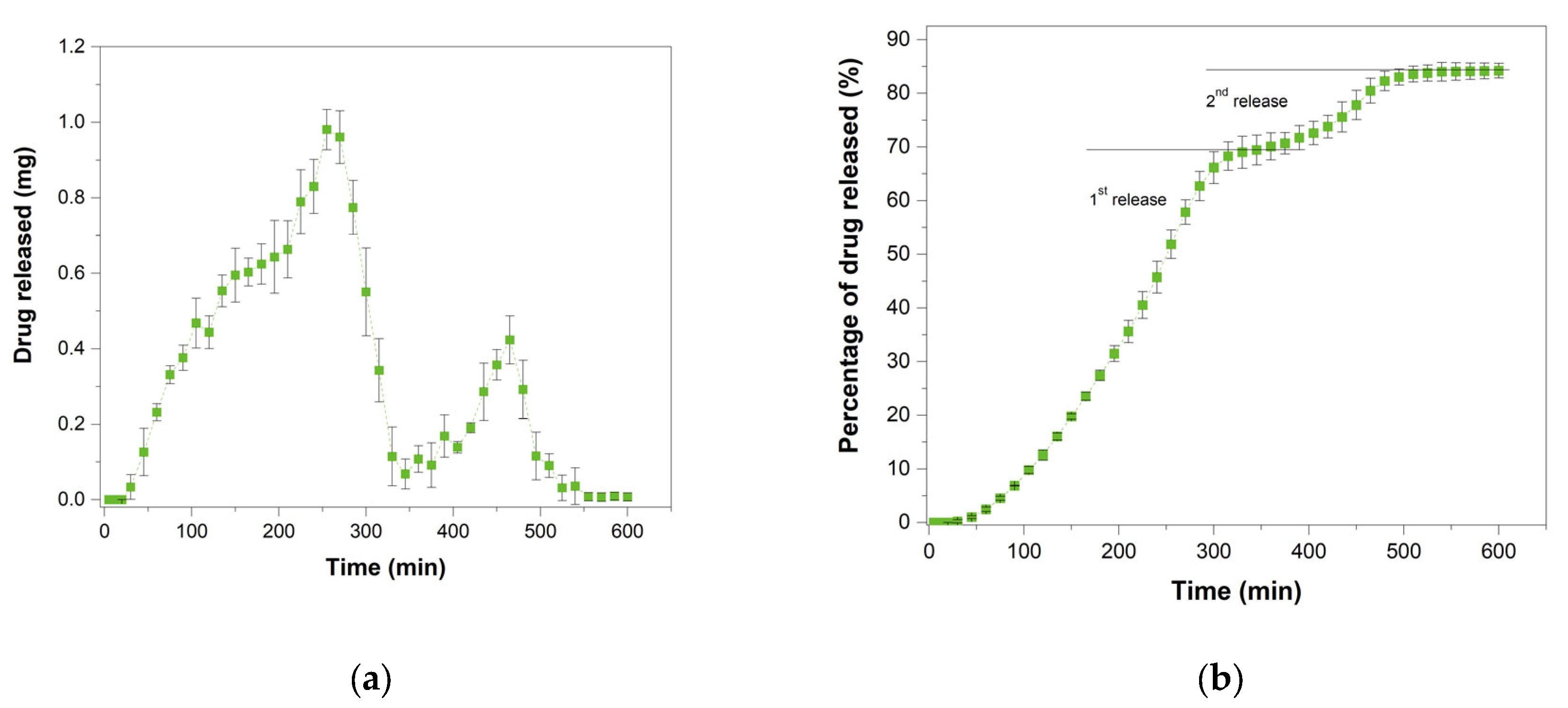
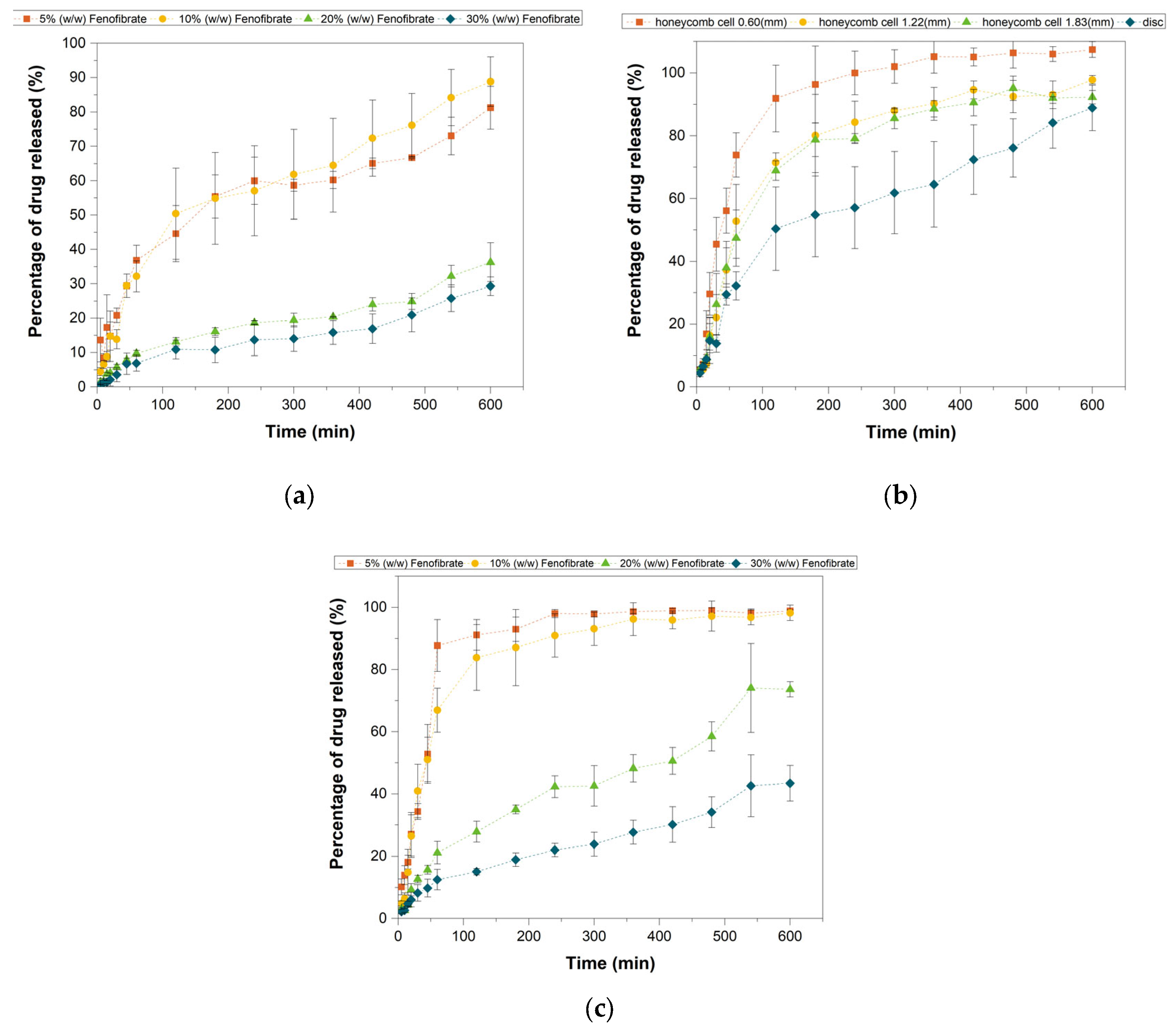
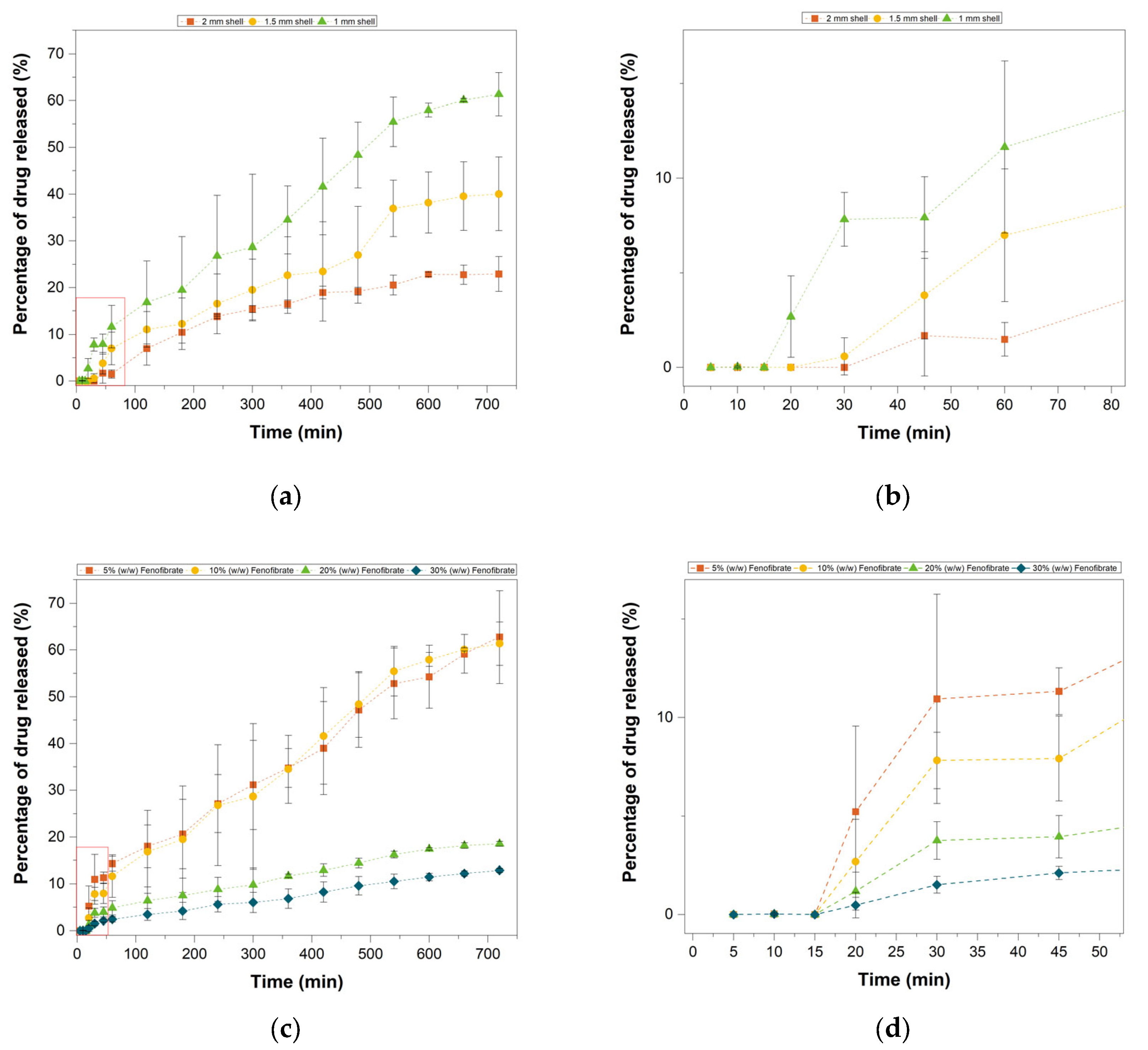
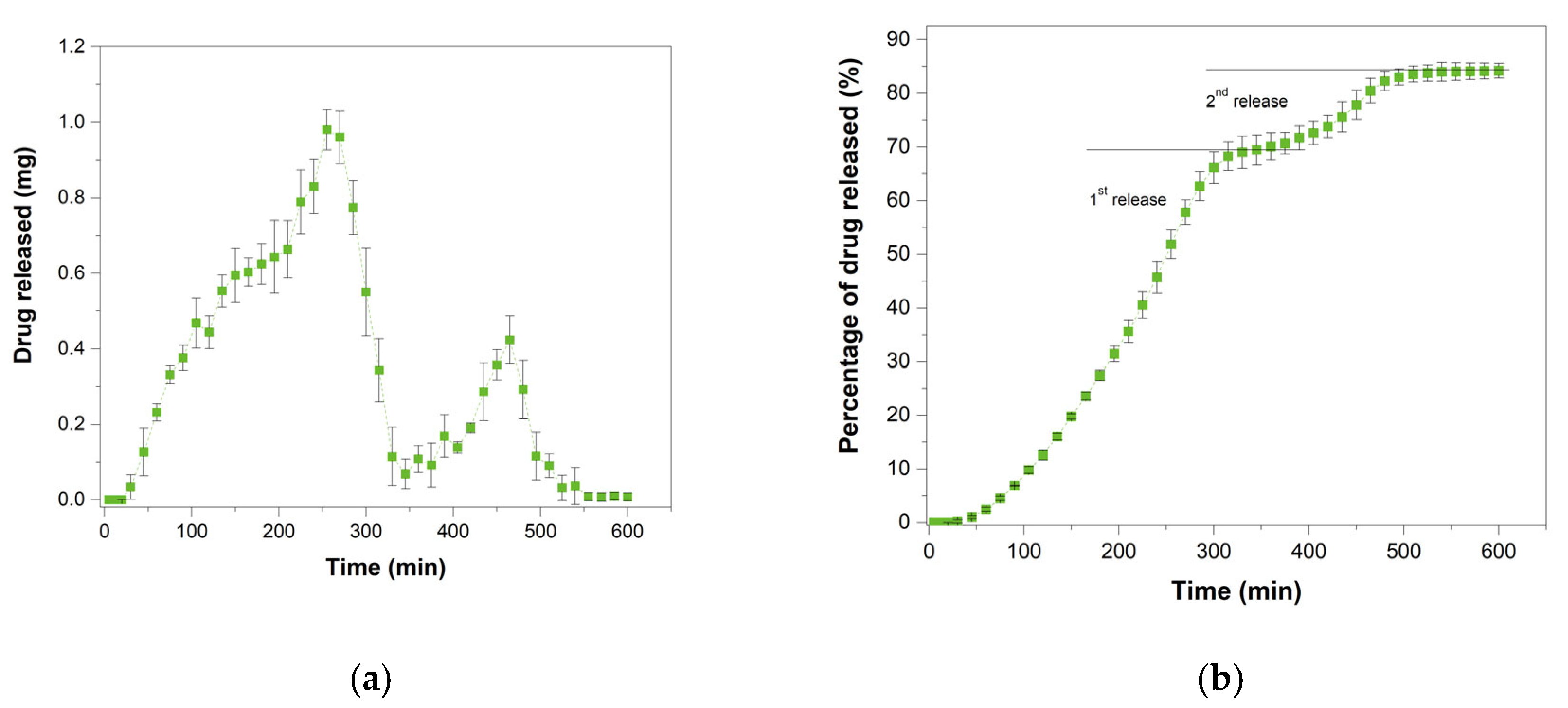
3.8. In Vitro Drug Release
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hajjar, E.R.; Cafiero, A.C.; Hanlon, J.T. Polypharmacy in elderly patients. Am. J. Geriatr. Pharmacother. 2007, 5, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.I.; Young, A.; Maher, R.; Rodriguez, K.L.; Appelt, C.J.; Perera, S.; Hajjar, E.R.; Hanlon, J.T. Polypharmacy and health beliefs in older outpatients. Am. J. Geriatr. Pharmacother. 2007, 5, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Hanlon, J.T.; Weinberger, M.; Samsa, G.P.; Schmader, K.E.; Uttech, K.M.; Lewis, I.K.; Cowper, P.A.; Landsman, P.B.; Cohen, H.J.; Feussner, J.R. A randomized, controlled trial of a clinical pharmacist intervention to improve inappropriate prescribing in elderly outpatients with polypharmacy. Am. J. Med. 1996, 100, 428–437. [Google Scholar] [CrossRef]
- Montamat, S.C.; Cusack, B. Overcoming problems with polyphar-macy and drug misuse in the elderly. Clin. Geriatr. Med. 1992, 8, 143–158. [Google Scholar] [CrossRef]
- Maher, R.L.; Hanlon, J.; Hajjar, E.R. Clinical consequences of polypharmacy in elderly. Expert Opin. Drug Saf. 2014, 13, 57–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, X.; Zhu, H.; Huo, X.; Li, Z. Polypharmacy in the oldest old (≥80 years of age) patients in China: A cross-sectional study. BMC Geriatr. 2018, 18, 64. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.-G.; Frueh, F.W. Pharmacogenomics steps toward personalized medicine. Pers. Med. 2005, 2, 325–337. [Google Scholar] [CrossRef]
- Yang, L.; Fassihi, R. Zero-order release kinetics from a self-correcting floatable asymmetric configuration drug de-livery systemxd. J. Pharm. Sci. 1996, 85, 170–173. [Google Scholar] [CrossRef]
- Giunchedi, P.; Conte, U.; Maggi, L.; La Manna, A. Hydrophilic matrices for the extended release of a model drug exhibiting pH-dependent solubility. Int. J. Pharm. 1992, 85, 141–147. [Google Scholar] [CrossRef]
- Shenouda, L.; Adams, K.; Zoglio, M. A controlled release delivery system using two hydrophilic polymers. Int. J. Pharm. 1990, 61, 127–134. [Google Scholar] [CrossRef]
- Hardy, I.J.; Windberg-Baarup, A.; Neri, C.; Byway, P.V.; Booth, S.W.; Fitzpatrick, S. Modulation of drug release kinetics from hydroxypropyl methyl cellulose matrix tablets using polyvinyl pyrrolidone. Int. J. Pharm. 2007, 337, 246–253. [Google Scholar] [CrossRef]
- Katstra, W.; Palazzolo, R.; Rowe, C.; Giritlioglu, B.; Teung, P.; Cima, M. Oral dosage forms fabricated by Three Dimensional Printing™. J. Control. Release 2000, 66, 1–9. [Google Scholar] [CrossRef]
- Lee, K.J.; Kang, A.; Delfino, J.J.; West, T.G.; Chetty, D.; Monkhouse, D.C.; Yoo, J. Evaluation of critical formula-tion factors in the development of a rapidly dispersing captopril oral dosage form. Drug Dev. Ind. Pharm. 2003, 29, 967–979. [Google Scholar] [CrossRef]
- Chia, H.N.; Wu, B.M. Recent advances in 3D printing of biomaterials. J. Biol. Eng. 2015, 9, 4. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.M.; Borland, S.W.; Giordano, R.A.; Cima, L.G.; Sachs, E.M.; Cima, M.J. Solid free-form fabrication of drug delivery devices. J. Control. Release 1996, 40, 77–87. [Google Scholar] [CrossRef]
- He, Y.; Foralosso, R.; Trindade, G.F.; Ilchev, A.; Ruiz-Cantu, L.; Clark, E.A.; Khaled, S.; Hague, R.J.M.; Tuck, C.J.; Rose, F.R.A.J.; et al. A Reactive Pro-drug Ink Formulation Strategy for Inkjet 3D Printing of Controlled Release Dosage Forms and Implants. Adv. Ther. 2020, 3, 1900187. [Google Scholar] [CrossRef]
- Liaskoni, A.; Wildman, R.D.; Roberts, C.J. 3D printed polymeric drug-eluting implants. Int. J. Pharm. 2021, 597, 120330. [Google Scholar] [CrossRef]
- Cader, H.K.; Rance, G.; Alexander, M.; Gonçalves, A.D.; Roberts, C.; Tuck, C.; Wildman, R. Water-based 3D inkjet printing of an oral pharmaceutical dosage form. Int. J. Pharm. 2019, 564, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Cantu, L.; Trindade, G.; Taresco, V.; Zhou, Z.; Burroughs, L.; Clark, E.; Rose, F.R.A.J.; Alexander, M.; Tuck, C.; Hague, R.; et al. Microstructural Control of Polymers Achieved Using Controlled Phase Separation during 3D Printing with Oligomer Libraries: Dictating Drug Release for Personalized Subdermal Implants; Cambridge Open Engage: Cambridge, UK, 2020. [Google Scholar]
- Louzao, I.; Koch, B.; Taresco, V.; Ruiz-Cantu, L.; Irvine, D.J.; Roberts, C.J.; Tuck, C.; Alexander, C.; Hague, R.; Wildman, R.; et al. Identification of novel “Inks” for 3D printing using high-throughput screening: Bioresorbable photocurable polymers for controlled drug de-livery. ACS Appl. Mater. Interfaces 2018, 10, 6841–6848. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-C.; Motwani, M.R.T.; Roach, W.J.; Kay, J.L.; Yoo, J.; Surprenant, H.L.; Monkhouse, D.C.; Pryor, T.J. Development of Near Zero-Order Release Dosage Forms Using Three-Dimensional Printing (3-DP™) Technology. Drug Dev. Ind. Pharm. 2006, 32, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.G.; Yang, X.L.; Huang, W.D.; Liu, J.; Wang, Y.G.; Xu, H. Tablets With Material Gradients Fabricated by Three-Dimensional Printing. J. Pharm. Sci. 2007, 96, 2446–2456. [Google Scholar] [CrossRef]
- Yu, D.G.; Branford-White, C.; Ma, Z.H.; Zhu, L.M.; Li, X.Y.; Yang, X.L. Novel drug delivery devices for providing linear release profiles fabricated by 3DP. Int. J. Pharm. 2009, 370, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Khaled, S.A.; Burley, J.C.; Alexander, M.R.; Roberts, C.J. Desktop 3D printing of controlled release pharmaceutical bilayer tablets. Int. J. Pharm. 2014, 461, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Khaled, S.A.; Burley, J.; Alexander, M.; Yang, J.; Roberts, C.J. 3D printing of tablets containing multiple drugs with defined release profiles. Int. J. Pharm. 2015, 494, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Khaled, S.A.; Burley, J.C.; Alexander, M.; Yang, J.; Roberts, C.J. 3D printing of five-in-one dose combination polypill with defined immediate and sustained release profiles. J. Control. Release 2015, 217, 308–314. [Google Scholar] [CrossRef]
- Goyanes, A.; Martinez, P.R.; Buanz, A.; Basit, A.W.; Gaisford, S. Effect of geometry on drug release from 3D printed tablets. Int. J. Pharm. 2015, 494, 657–663. [Google Scholar] [CrossRef]
- Goyanes, A.; Buanz, A.; Hatton, G.B.; Gaisford, S.; Basit, A.W. 3D printing of modified-release aminosalicylate (4-ASA and 5-ASA) tablets. Eur. J. Pharm. Biopharm. 2015, 89, 157–162. [Google Scholar] [CrossRef]
- Goyanes, A.; Buanz, A.B.; Basit, A.W.; Gaisford, S. Fused-filament 3D printing (3DP) for fabrication of tablets. Int. J. Pharm. 2014, 476, 88–92. [Google Scholar] [CrossRef]
- Krause, J.; Müller, L.; Sarwinska, D.; Seidlitz, A.; Sznitowska, M.; Weitschies, W. 3D Printing of Mini Tablets for Pediatric Use. Pharmaceuticals 2021, 14, 143. [Google Scholar] [CrossRef]
- Scoutaris, N.; Ross, S.A.; Douroumis, D. 3D printed “Starmix” drug loaded dosage forms for paediatric applications. Pharm. Res. 2018, 35, 34. [Google Scholar] [CrossRef]
- Kyobula, M.; Adedeji, A.; Alexander, M.; Saleh, E.; Wildman, R.; Ashcroft, I.; Gellert, P.R.; Roberts, C.J. 3D inkjet printing of tablets exploiting bespoke complex geometries for controlled and tuneable drug release. J. Control. Release 2017, 261, 207–215. [Google Scholar] [CrossRef] [PubMed]
- US Department of Health and Human Services, & Food and Drug Administration. Size, Shape, and Other Physical Attributes of Generic Tablets and Capsules Guidance for Industry; Office of Communications, Division of Drug Information Center for Drug Evaluation and Research, Food and Drug Administration: Silver Spring, MD, USA, 2015; Volume 80, pp. 35366–35367.
- Kadry, H.; Al-Hilal, T.A.; Keshavarz, A.; Alam, F.; Xu, C.; Joy, A.; Ahsan, F. Multi-purposable filaments of HPMC for 3D printing of medications with tailored drug release and timed-absorption. Int. J. Pharm. 2018, 544, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Punčochová, K.; Ewing, A.V.; Gajdošová, M.; Sarvašová, N.; Kazarian, S.G.; Beranek, J.; Štěpánek, F. Identifying the mechanisms of drug release from amorphous solid dispersions using MRI and ATR-FTIR spectroscopic imaging. Int. J. Pharm. 2015, 483, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Hanh, B.D.; Neubert, R.H.; Wartewig, S. Investigation of drug release from suspension using FTIR-ATR technique: Part I. Determination of effective diffusion coefficient of drugs. Int. J. Pharm. 2000, 204, 145–150. [Google Scholar] [CrossRef]
- De Waard, H.; De Beer, T.; Hinrichs, W.L.; Vervaet, C.; Remon, J.P.; Frijlink, H.W. Controlled crystallization of the lipophilic drug fenofibrate during freeze-drying: Elucidation of the mechanism by in-line Raman spectroscopy. AAPS J. 2010, 12, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Okwuosa, T.C.; Pereira, B.C.; Arafat, B.; Cieszynska, M.; Isreb, A.; Alhnan, M.A. Fabricating a Shell-Core Delayed Release Tablet Using Dual FDM 3D Printing for Patient-Centred Therapy. Pharm. Res. 2016, 34, 427–437. [Google Scholar] [CrossRef]
- Fernández, J.M.; Plante, A.F.; Leifeld, J.; Rasmussen, C. Methodological considerations for using thermal analysis in the characterization of soil organic matter. J. Therm. Anal. Calorim. 2010, 104, 389–398. [Google Scholar] [CrossRef]
- Gaál, F.; Szöllösy, I.; Arnold, M.; Paulik, F. Determination of the organic matter, metal carbonate and mobile water in soils simultaneous TG, DTG, DTA and EGA techniques. J. Therm. Anal. Calorim. 1994, 42, 1007–1016. [Google Scholar] [CrossRef]
- Miyazawa, M.; Pavan, M.A.; De Oliveira, E.L.; Ionashiro, M.; Silva, A.K. Gravimetric determination of soil organic matter. Braz. Arch. Biol. Technol. 2000, 43, 475–478. [Google Scholar] [CrossRef]
- Salgado, J.; Mato, M.M.; Vázquez-Galiñanes, A.; Paz-Andrade, M.; Carballas, T. Comparison of two calorimetric methods to determine the loss of organic matter in Galician soils (NW Spain) due to forest wildfires. Thermochim. Acta 2004, 410, 141–148. [Google Scholar] [CrossRef]
- Siewert, C. Rapid Screening of Soil Properties using Thermogravimetry. Soil Sci. Soc. Am. J. 2004, 68, 1656–1661. [Google Scholar] [CrossRef]
- Duguy, B.; Rovira, P. Differential thermogravimetry and differential scanning calorimetry of soil organic matter in mineral horizons: Effect of wildfires and land use. Org. Geochem. 2010, 41, 742–752. [Google Scholar] [CrossRef]
- Coats, A.W.; Redfern, J.P. Thermogravimetric analysis: A review. Analyst 1963, 88, 906–924. [Google Scholar] [CrossRef]
- Gattefosse. Available online: https://www.gattefosse.com/pharmaceuticals-products/compritol-hd5-ato (accessed on 21 August 2021).
- N’Diaye, A.; Jannin, V.; Bérard, V.; Andrès, C.; Pourcelot, Y. Comparative study of the lubricant performance of Compritol® HD5 ATO and Compritol® 888 ATO: Effect of polyethylene glycol behenate on lubricant capacity. Int. J. Pharm. 2003, 254, 263–269. [Google Scholar] [CrossRef]
- Bruni, G.; Milanese, C.; Bellazzi, G.; Berbenni, V.; Cofrancesco, P.; Marini, A.; Villa, M. Quantification of drug amorphous fraction by DSC. J. Therm. Anal. Calorim. 2007, 89, 761–766. [Google Scholar] [CrossRef]
- Schwantes, J.M.; Marsden, O.; Pellegrini, K. State of practice and emerging application of analytical techniques of nuclear forensic analysis: Highlights from the 4th Collaborative Materials Exercise of the Nuclear Forensics International Technical Working Group (ITWG). J. Radioanal. Nucl. Chem. 2016, 311, 1441–1452. [Google Scholar] [CrossRef]
- Karolewicz, B.; Gajda, M.; Pluta, J.; Górniak, A. Dissolution study and thermal analysis of fenofibrate–Pluronic F127 solid dispersions. J. Therm. Anal. Calorim. 2015, 125, 751–757. [Google Scholar] [CrossRef] [Green Version]
- Mao, F.; Kong, Q.; Ni, W.; Xu, X.; Ling, D.; Lu, Z.; Li, J. Melting Point Distribution Analysis of Globally Approved and Discontinued Drugs: A Research for Improving the Chance of Success of Drug Design and Discovery. ChemistryOpen 2016, 5, 357–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Geometry [mm] | Fenofibrate (w/w) | Tablet [a.u.] | Reference [a.u.] | Percentage Error [%] | |
|---|---|---|---|---|---|
| Immediate | Channel | ||||
| I2a | 600 | 10% | 38.79 ± 1.18 | 37.01 ± 3.48 | 4.6 |
| I2b | 1220 | 10% | 38.62 ± 0.70 | 37.01 ± 3.48 | 4.2 |
| I2c | 1830 | 10% | 39.63 ± 2.44 | 37.01 ± 3.48 | 6.6 |
| I1a | 600 | 5% | 24.01 ± 1.18 | 23.63 ± 2.82 | 1.6 |
| I2a | 600 | 10% | 38.79 ± 1.18 | 31.01 ± 3.48 | 20.1 |
| I3a | 600 | 20% | 43.01 ± 2.44 | 42.47 ± 2.93 | 1.3 |
| I4a | 600 | 30% | 70.16 ± 2.44 | 70.48 ± 0.12 | 0.5 |
| Delayed | Shell | ||||
| D2a | 1 | 10% | 36.81 ± 1.83 | 37.01 ± 3.48 | 0.5 |
| D2b | 1.6 | 10% | 37.57 ± 2.00 | 37.01 ± 3.48 | 1.5 |
| D2c | 2 | 10% | 37.85 ± 1.59 | 37.01 ± 3.48 | 2.2 |
| D1a | 1 | 5% | 23.56 ± 1.87 | 23.63 ± 2.82 | 0.3 |
| D2a | 1 | 10% | 36.81 ± 1.83 | 31.01 ± 3.48 | 15.8 |
| D3a | 1 | 20% | 43.12 ± 1.13 | 42.47 ± 2.93 | 1.5 |
| D4a | 1 | 30% | 70.47 ± 0.60 | 70.48 ± 0.12 | 0.01 |
| Pulsatile | |||||
| Core | 10% | 37.95 ± 2.04 | 37.01 ± 3.48 | 2.5 | |
| Ring | 10% | 37.50 ± 2.40 | 37.01 ± 3.48 | 1.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lion, A.; Wildman, R.D.; Alexander, M.R.; Roberts, C.J. Customisable Tablet Printing: The Development of Multimaterial Hot Melt Inkjet 3D Printing to Produce Complex and Personalised Dosage Forms. Pharmaceutics 2021, 13, 1679. https://doi.org/10.3390/pharmaceutics13101679
Lion A, Wildman RD, Alexander MR, Roberts CJ. Customisable Tablet Printing: The Development of Multimaterial Hot Melt Inkjet 3D Printing to Produce Complex and Personalised Dosage Forms. Pharmaceutics. 2021; 13(10):1679. https://doi.org/10.3390/pharmaceutics13101679
Chicago/Turabian StyleLion, Anna, Ricky D. Wildman, Morgan R. Alexander, and Clive J. Roberts. 2021. "Customisable Tablet Printing: The Development of Multimaterial Hot Melt Inkjet 3D Printing to Produce Complex and Personalised Dosage Forms" Pharmaceutics 13, no. 10: 1679. https://doi.org/10.3390/pharmaceutics13101679