
Liposomal Resiquimod for Enhanced Immunotherapy of Peritoneal Metastases of Colorectal Cancer

 , ,
, ,
Abstract
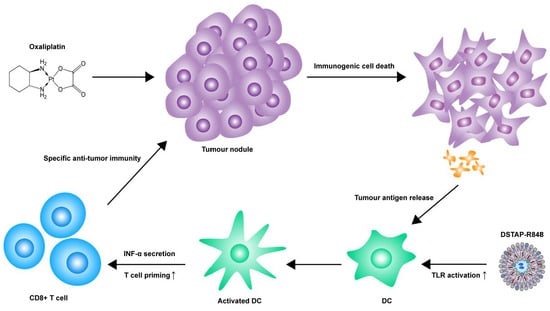
:
1. Introduction
2. Materials and Methods
2.1. Chemical and Reagents
2.2. Liposome Preparation
2.3. R848 Loading into DSTAP-Liposomes
2.4. Stewart Assay
2.5. UPLC
2.6. Cell Culture
2.7. Mice
2.8. Pharmacokinetic and Biodistribution
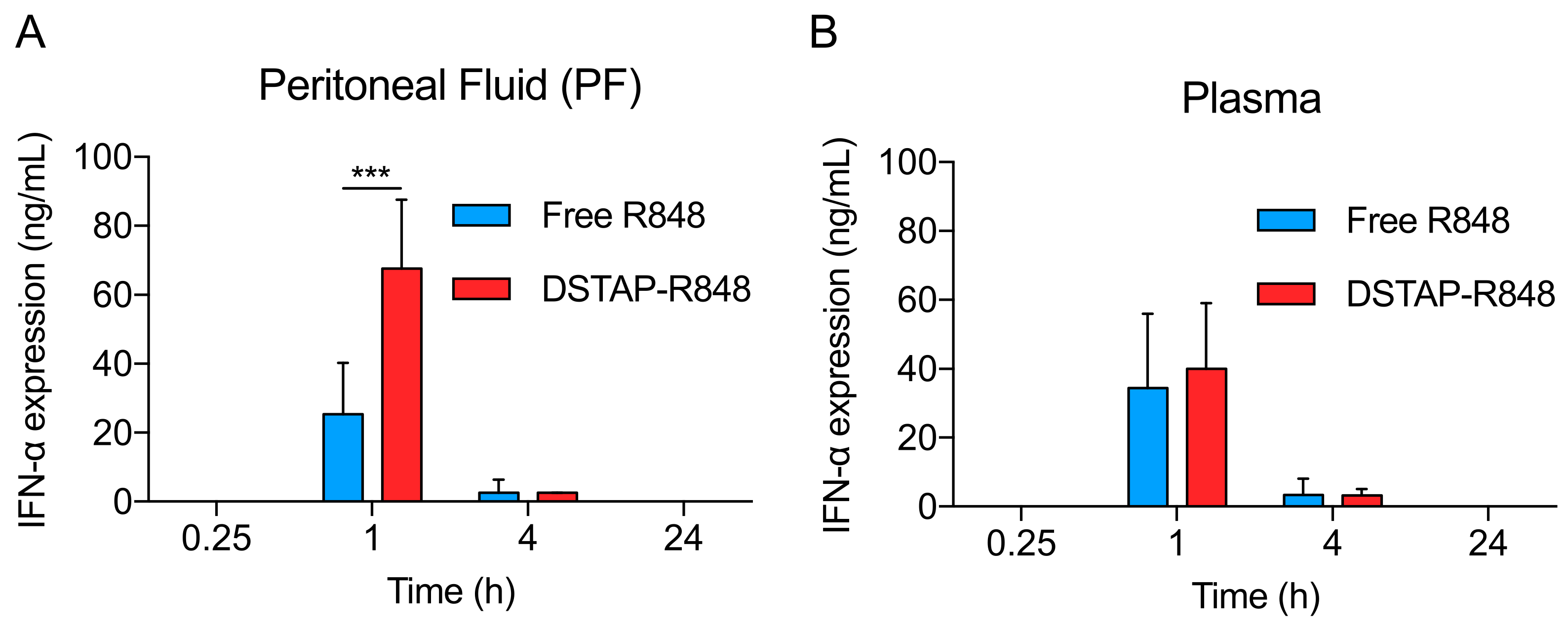
2.9. Cytokine Quantification
2.10. Cellular Uptake by IP Cells
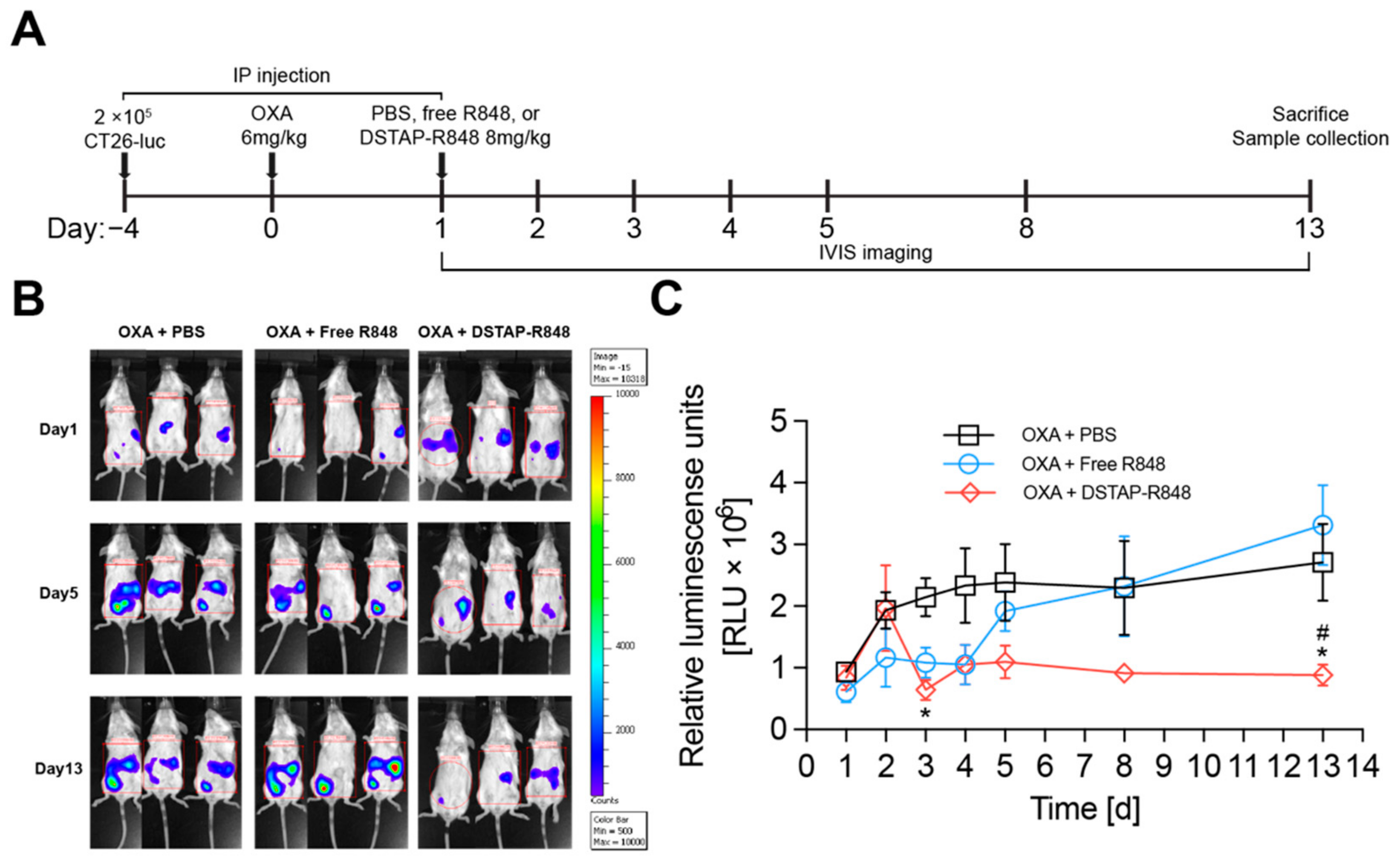
2.11. In Vivo Efficacy
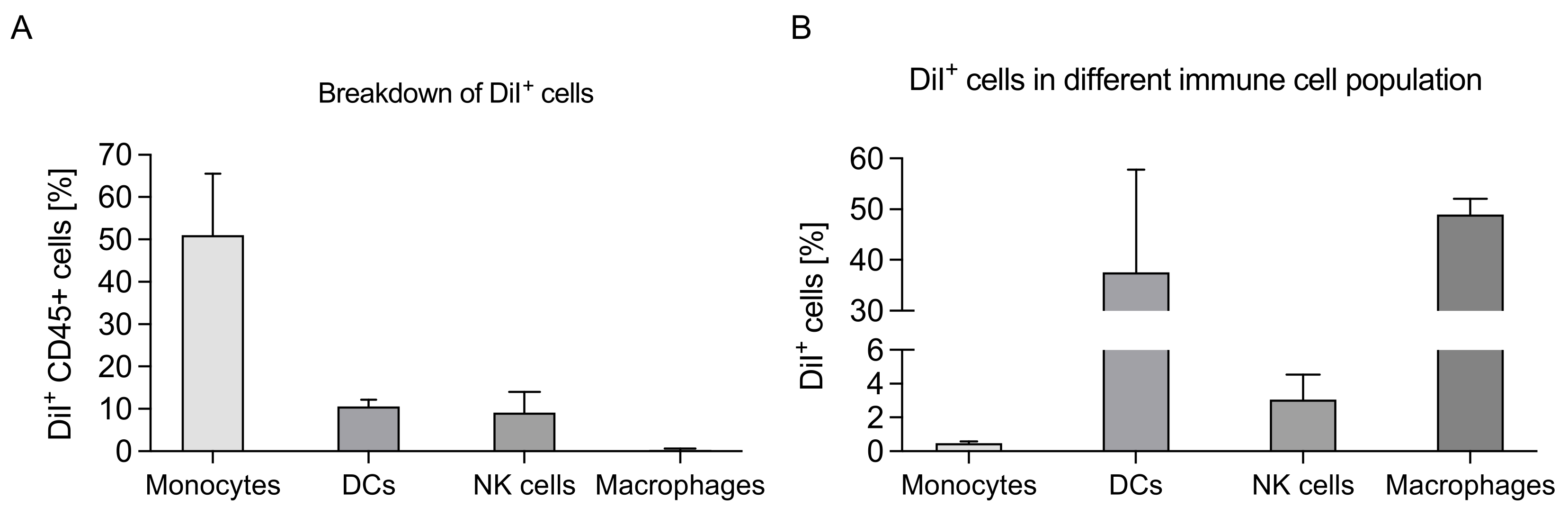
2.12. Immune Cell Uptake and Composition
2.13. Statistical Analysis
3. Results
3.1. Selection and Characterization of Liposomal Formulations
3.2. Pharmacokinetic Profile of DSTAP-R848
3.3. Delivery into Peritoneal Immune Cells
3.4. Cytokine Stimulation in the Peritoneum
3.5. Antitumor Efficacy
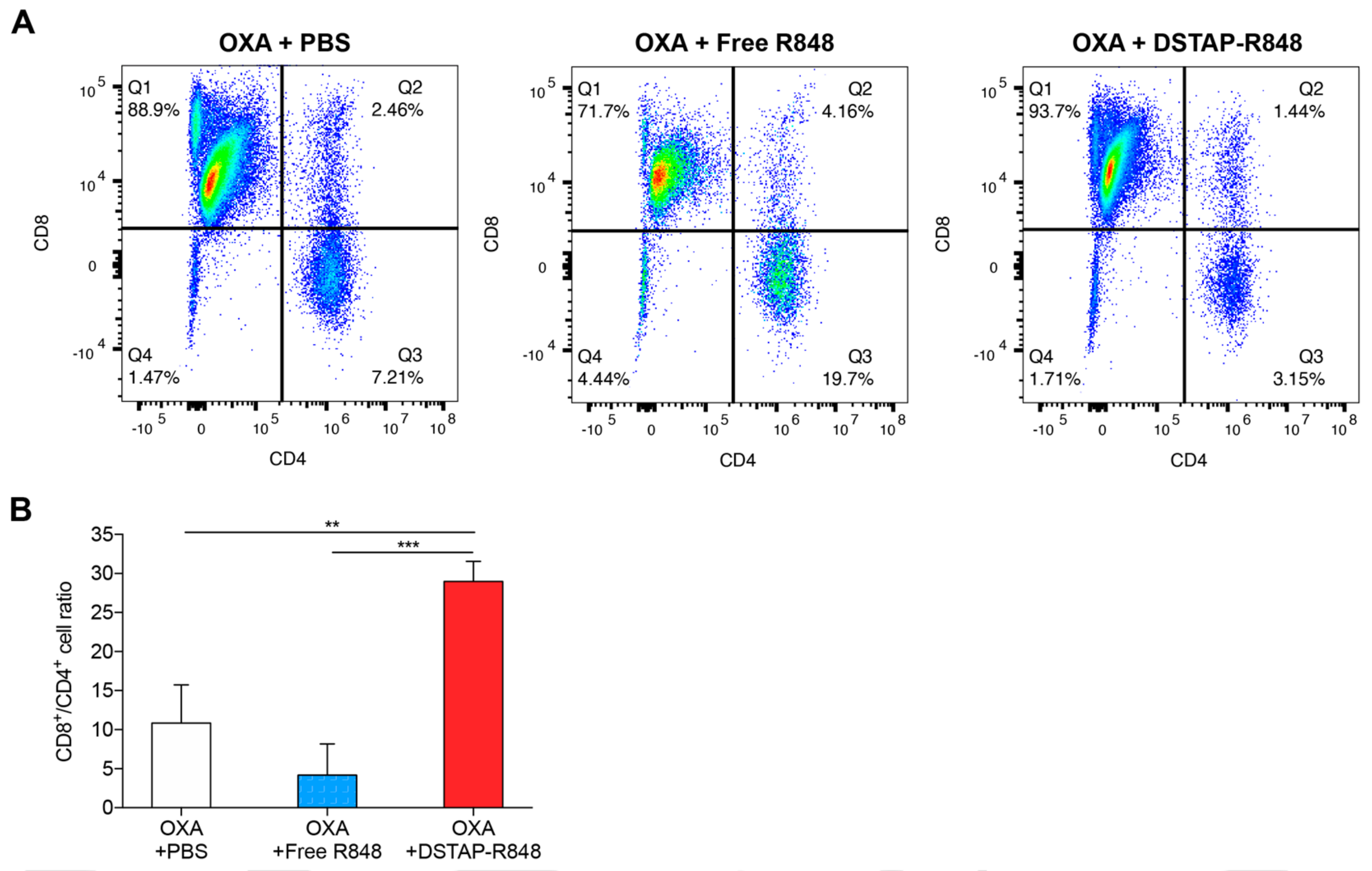
3.6. Modulation of Immune Cell Composition in PerC
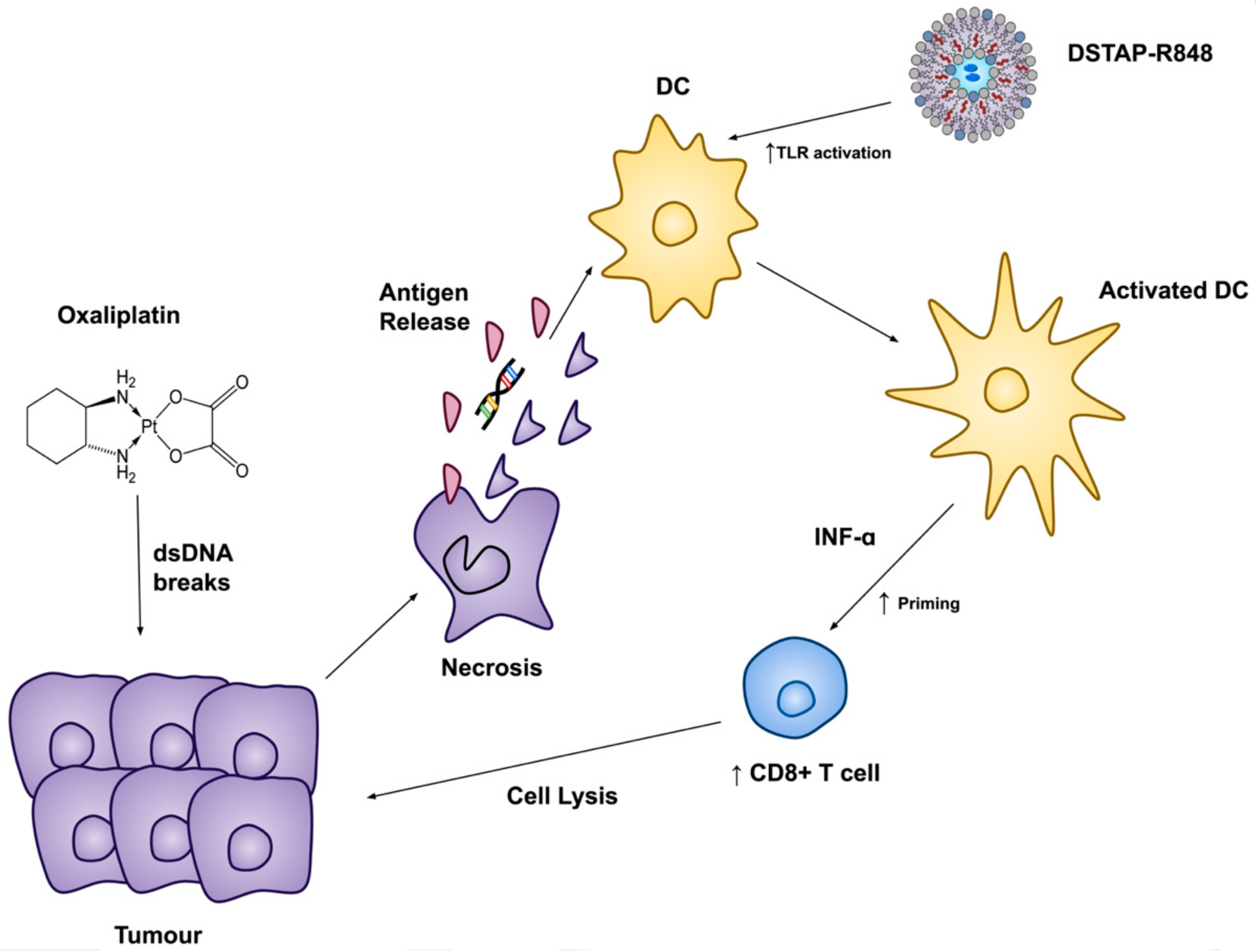
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Isaza-Restrepo, A.; Martin-Saavedra, J.S.; Velez-Leal, J.L.; Vargas-Barato, F.; Riveros-Dueñas, R. The Peritoneum: Beyond the Tissue–A Review. Front. Physiol. 2018, 9, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Wang, J.; Wientjes, M.G.; Au, J.L.-S. Intraperitoneal therapy for peritoneal cancer. Futur. Oncol. 2010, 6, 1625–1641. [Google Scholar] [CrossRef] [Green Version]
- Nadler, A.; McCart, J.A.; Govindarajan, A. Peritoneal Carcinomatosis from Colon Cancer: A Systematic Review of the Data for Cytoreduction and Intraperitoneal Chemotherapy. Clin. Colon Rectal Surg. 2015, 28, 234–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugarbaker, P.H.; Cunliffe, W.J.; Belliveau, J.; De Bruijn, E.A.; Graves, T.; Mullins, R.E.; Schlag, P. Rationale for integrating early postoperative intraperitoneal chemotherapy into the surgical treatment of gastrointestinal cancer. In Proceedings of the 3rd International Congress on Neo-Adjuvant Chemotherapy, Gabler, Paris, France, 6–9 February 1991; pp. 272–275. [Google Scholar]
- Glehen, O.; Osinsky, D.; Beaujard, A.C.; Gilly, F.N. Natural history of peritoneal carcinomatosis from nongynecologic malignancies. Surg. Oncol. Clin. North Am. 2003, 12, 729–739. [Google Scholar] [CrossRef]
- Jayne, D.G.; Fook, S.; Loi, C.; Seow-Choen, F. Peritoneal carcinomatosis from colorectal cancer. BJS 2002, 89, 1545–1550. [Google Scholar] [CrossRef]
- Minsky, B.D.; Mies, C.; Rich, T.A.; Recht, A.; Chaffey, J.T. Potentially curative surgery of colon cancer: Patterns of failure and survival. J. Clin. Oncol. 1988, 6, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.; Point, G.R.; Cunetta, M.; Thorn, M.; Guha, P.; Espat, N.J.; Boutros, C.; Hanna, N.; Junghans, R.P. Regional CAR-T cell infusions for peritoneal carcinomatosis are superior to systemic delivery. Cancer Gene Ther. 2016, 23, 142–148. [Google Scholar] [CrossRef]
- Becker, J.C.; Andersen, M.H.; Schrama, D.; Straten, P.T. Immune-suppressive properties of the tumor microenvironment. Cancer Immunol. Immunother. 2013, 62, 1137–1148. [Google Scholar] [CrossRef]
- Spinetti, T.; Spagnuolo, L.; Mottas, I.; Secondini, C.; Treinies, M.; Rüegg, C.; Hotz, C.; Bourquin, C. TLR7-based cancer immunotherapy decreases intratumoral myeloid-derived suppressor cells and blocks their immunosuppressive function. OncoImmunology 2016, 5, e1230578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.-M.; Pan, W.-Y.; Wua, C.-Y.; Yeha, C.-Y.; Korupallia, C.; Luoa, P.-K.; Choua, C.-J.; Chiab, W.-T.; Sunga, H.-W. Modulation of tumor microenvironment using a TLR-7/8 agonist-loaded nanoparticle system that exerts low-temperature hyperthermia and immunotherapy for in situ cancer vaccination. Biomater. 2020, 230, 119629. [Google Scholar] [CrossRef]
- Ströhlein, M.A.; Heiss, M.M.; Jauch, K.-W. The current status of immunotherapy in peritoneal carcinomatosis. Expert Rev. Anticancer. Ther. 2016, 16, 1019–1027. [Google Scholar] [CrossRef]
- Thadi, A.; Khalili, M.; Morano, W.F.; Richard, S.D.; Katz, S.C.; Bowne, W.B. Early Investigations and Recent Advances in Intraperitoneal Immunotherapy for Peritoneal Metastasis. Vaccines 2018, 6, 54. [Google Scholar] [CrossRef] [Green Version]
- Mullins, S.R.; Vasilakos, J.P.; Deschler, K.; Grigsby, I.; Gillis, P.; John, J.; Elder, M.J.; Swales, J.; Timosenko, E.; Cooper, Z.; et al. Intratumoral immunotherapy with TLR7/8 agonist MEDI9197 modulates the tumor microenvironment leading to enhanced activity when combined with other immunotherapies. J. Immunother. Cancer 2019, 7, 244. [Google Scholar] [CrossRef]
- Engel, A.L.; Holt, G.E.; Lu, H. The pharmacokinetics of Toll-like receptor agonists and the impact on the immune system. Expert Rev. Clin. Pharmacol. 2011, 4, 275–289. [Google Scholar] [CrossRef] [Green Version]
- Michaelis, K.A.; Norgard, M.A.; Zhu, X.; Levasseur, P.R.; Sivagnanam, S.; Liudahl, S.M.; Burfeind, K.G.; Olson, B.; Pelz, K.R.; Ramos, D.M.A.; et al. The TLR7/8 agonist R848 remodels tumor and host responses to promote survival in pancreatic cancer. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Ahonen, C.L.; Gibson, S.J.; Smith, R.M.; Pederson, L.K.; Lindh, J.M.; Tomai, M.A.; Vasilakos, J.P. Dendritic Cell Maturation and Subsequent Enhanced T-Cell Stimulation Induced with the Novel Synthetic Immune Response Modifier R-848. Cell. Immunol. 1999, 197, 62–72. [Google Scholar] [CrossRef]
- Tesniere, A.; Schlemmer, F.; Boige, V.; Kepp, O.; Martins, I.; Ghiringhelli, F.; Aymeric, L.; Michaud, M.; Apetoh, L.; Barault, L.; et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2009, 29, 482–491. [Google Scholar] [CrossRef] [Green Version]
- Pauli, G.; Tang, W.-L.; Li, S.-D. Development and Characterization of the Solvent-Assisted Active Loading Technology (SALT) for Liposomal Loading of Poorly Water-Soluble Compounds. Pharmaceutics 2019, 11, 465. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Goncalves, R.; Mosser, D.M. The Isolation and Characterization of Murine Macrophages. Curr. Protoc. Immunol. 2008, 83, 14.1.1–14.1.14. [Google Scholar] [CrossRef]
- Basha, G.; Novobrantseva, T.I.; Rosin, N.; Tam, Y.Y.C.; Hafez, I.M.; Wong, M.K.; Sugo, T.; Ruda, V.M.; Qin, J.; Klebanov, B.; et al. Influence of Cationic Lipid Composition on Gene Silencing Properties of Lipid Nanoparticle Formulations of siRNA in Antigen-Presenting Cells. Mol. Ther. 2011, 19, 2186–2200. [Google Scholar] [CrossRef]
- Uemura, Y.; Naoi, T.; Kanai, Y.; Kobayashi, K. The efficiency of lipid nanoparticles with an original cationic lipid as a siRNA delivery system for macrophages and dendritic cells. Pharm. Dev. Technol. 2019, 24, 263–268. [Google Scholar] [CrossRef]
- Ghosn, E.E.B.; Cassado, A.A.; Govoni, G.R.; Fukuhara, T.; Yang, Y.; Monack, D.M.; Bortoluci, K.R.; Almeida, S.R.; Herzenberg, L.A. Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proc. Natl. Acad. Sci. USA 2010, 107, 2568–2573. [Google Scholar] [CrossRef] [Green Version]
- Bartneck, M.; Keul, H.A.; Zwadlo-Klarwasser, G.; Groll, J. Phagocytosis Independent Extracellular Nanoparticle Clearance by Human Immune Cells. Nano Lett. 2010, 10, 59–63. [Google Scholar] [CrossRef]
- Melichar, B.; Savary, C.A.; Patenia, R.; Templin, S.; Melicharova, K.; Freedman, R.S. Phenotype and antitumor activity of ascitic fluid monocytes in patients with ovarian carcinoma. Int. J. Gynecol. Cancer 2003, 13, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Park, C.-S.; Lee, Y.-R.; Im, S.-A.; Song, S.; Lee, C.-K. Resiquimod, a TLR7/8 agonist, promotes differentiation of myeloid-derived suppressor cells into macrophages and dendritic cells. Arch. Pharmacal Res. 2014, 37, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Rodell, C.B.; Arlauckas, S.P.; Cuccarese, M.F.; Garris, C.S.; Li, R.; Ahmed, M.S.; Kohler, R.H.; Pittet, M.J.; Weissleder, R. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2018, 2, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Smits, E.; Ponsaerts, P.; Berneman, Z.; Van Tendeloo, V. The Use of TLR7 and TLR8 Ligands for the Enhancement of Cancer Immunotherapy. Oncol. 2008, 13, 859–875. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, M.; Arruda-Silva, F.; Bianchetto-Aguilera, F.; Finotti, G.; Calzetti, F.; Scapini, P.; Lunardi, C.; Cassatella, M.A.; Tamassia, N. IFNα enhances the production of IL-6 by human neutrophils activated via TLR8. Sci. Rep. 2016, 6, 19674. [Google Scholar] [CrossRef] [Green Version]
- Hornung, V.; Schlender, J.; Guenthner-Biller, M.; Rothenfusser, S.; Endres, S.; Conzelmann, K.-K.; Hartmann, G. Replication-Dependent Potent IFN-α Induction in Human Plasmacytoid Dendritic Cells by a Single-Stranded RNA Virus. J. Immunol. 2004, 173, 5935–5943. [Google Scholar] [CrossRef] [Green Version]
- Wagner, T.L.; Ahonen, C.L.; Couture, A.M.; Gibson, S.J.; Miller, R.L.; Smith, R.M.; Reiter, M.J.; Vasilakos, J.P.; Tomai, M.A. Modulation of TH1 and TH2 Cytokine Production with the Immune Response Modifiers, R-848 and Imiquimod. Cell. Immunol. 1999, 191, 10–19. [Google Scholar] [CrossRef]
- Welsh, R.M.; Bahl, K.; Marshall, H.D.; Urban, S.L. Type 1 Interferons and Antiviral CD8 T-Cell Responses. PLoS Pathog. 2012, 8, e1002352. [Google Scholar] [CrossRef] [Green Version]
- Habault, J.; Poyet, J.-L. Recent Advances in Cell Penetrating Peptide-Based Anticancer Therapies. Molecules 2019, 24, 927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brockwell, N.; Parker, B.S. Tumor inherent interferons: Impact on immune reactivity and immunotherapy. Cytokine 2019, 118, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tai, Y.-T.; Ho, M.; Qiu, L.; Anderson, K.C. Interferon-alpha-based immunotherapies in the treatment of B cell-derived hematologic neoplasms in today’s treat-to-target era. Exp. Hematol. Oncol. 2017, 6, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hervas-Stubbs, S.; Perez-Gracia, J.L.; Rouzaut, A.; Sanmamed, M.F.; Le Bon, A.; Melero, I. Direct Effects of Type I Interferons on Cells of the Immune System. Clin. Cancer Res. 2011, 17, 2619–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickow, J.; Francois, S.; Kaiserling, R.-L.; Malyshkina, A.; Drexler, I.; Westendorf, A.M.; Lang, K.S.; Santiago, M.L.; Dittmer, U.; Sutter, K. Diverse Immunomodulatory Effects of Individual IFNα Subtypes on Virus-Specific CD8+ T Cell Responses. Front. Immunol. 2019, 10, 2255. [Google Scholar] [CrossRef] [PubMed]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef]
- Auer, K.; Bachmayr-Heyda, A.; Sukhbaatar, N.; Aust, S.; Schmetterer, K.G.; Meier, S.M.; Gerner, C.; Grimm, C.; Horvat, R.; Pils, D. Role of the immune system in the peritoneal tumor spread of high grade serous ovarian cancer. Oncotarget 2016, 7, 61336–61354. [Google Scholar] [CrossRef] [Green Version]
- Valzasina, B.; Piconese, S.; Guiducci, C.; Colombo, M.P. Tumor-Induced Expansion of Regulatory T Cells by Conversion of CD4+CD25− Lymphocytes Is Thymus and Proliferation Independent. Cancer Res. 2006, 66, 4488–4495. [Google Scholar] [CrossRef] [Green Version]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the role of CD4+ T cells in cancer immunotherapy—new insights into old paradigms. Cancer Gene Ther. 2021, 28, 5–17. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R848 Conc. (mg/mL) | Total Lipid Conc. (mg/mL) | Size (nm) | Polydispersity Index (PDI) | Zeta Potential (mV) |
|---|---|---|---|---|
| 1.76 ± 0.64 | 9.59 ± 2.06 | 104.6 ± 7.2 | 0.084 ± 0.001 | 43.0 ± 5.3 |
| Parameter | Free R848 | DSTAP-R848 | Significance |
|---|---|---|---|
| AUC0.25−4h (h • µg/mL) in peritoneal fluid | 5.29 ± 4.10 | 71.06 ± 17.70 | p < 0.005 |
| AUC0.25−4h (h • µg/mL) in plasma | 0.12 ± 0.05 | 0.13 ± 0.05 | p > 0.05 |
| AUC0.25−4h (h • µg) in IP cells | 0 | 0.46 ± 0.16 | p < 0.005 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pauli, G.; Chao, P.-H.; Qin, Z.; Böttger, R.; Lee, S.E.; Li, S.-D. Liposomal Resiquimod for Enhanced Immunotherapy of Peritoneal Metastases of Colorectal Cancer. Pharmaceutics 2021, 13, 1696. https://doi.org/10.3390/pharmaceutics13101696
Pauli G, Chao P-H, Qin Z, Böttger R, Lee SE, Li S-D. Liposomal Resiquimod for Enhanced Immunotherapy of Peritoneal Metastases of Colorectal Cancer. Pharmaceutics. 2021; 13(10):1696. https://doi.org/10.3390/pharmaceutics13101696
Chicago/Turabian StylePauli, Griffin, Po-Han Chao, Zhu Qin, Roland Böttger, Suen Ern Lee, and Shyh-Dar Li. 2021. "Liposomal Resiquimod for Enhanced Immunotherapy of Peritoneal Metastases of Colorectal Cancer" Pharmaceutics 13, no. 10: 1696. https://doi.org/10.3390/pharmaceutics13101696
APA StylePauli, G., Chao, P.-H., Qin, Z., Böttger, R., Lee, S. E., & Li, S.-D. (2021). Liposomal Resiquimod for Enhanced Immunotherapy of Peritoneal Metastases of Colorectal Cancer. Pharmaceutics, 13(10), 1696. https://doi.org/10.3390/pharmaceutics13101696