
Activators and Inhibitors of Protein Kinase C (PKC): Their Applications in Clinical Trials

 ,
,
Abstract
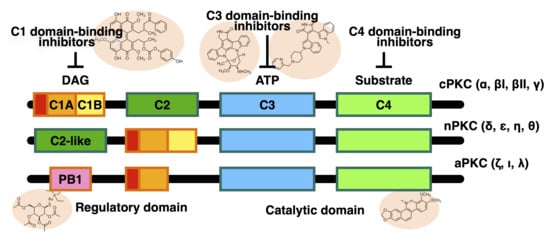
1. Introduction
2. Structure of PKC Isozymes
3. PKC Inhibitors
3.1. C1 Domain-Binding PKC Inhibitors (DAG Competitive PKC Inhibitors)
3.2. C2 Domain-Binding PKC Inhibitors (Ca2+ Competitive PKC Inhibitors)
3.3. C3 Domain (N-Lobe Domain)-Binding PKC Inhibitors (ATP Competitive PKC Inhibitors)
3.3.1. Indolocarbazole Compounds
3.3.2. Maleimide-Based Inhibitors
Bisindolylmaleimide (Bis) Compounds
Sotrastaurin
3.3.3. Other ATP Competitive PKC Inhibitors
3.4. C4 Domain (C-Lobe Domain)-Binding PKC Inhibitors (Substrate Competitive PKC Inhibitors)
3.4.1. Peptide Inhibitors
3.4.2. Other Inhibitors Binding to the C4 Domain
4. Atypical PKC Inhibitors
4.1. ZIP (PB Domain)
4.2. Auranofin and Sodium Aurothiomalate (PB1 Domain)
4.3. ICA-1 (C4, C-Terminal Lobe Domain)
4.4. ζ-Stat (C4, C-Lobe)
4.5. ACPD and DNDA (C4, C-Lobe; ACPD C3, N-Lobe for PKC-ζ)
5. Antisense Oligonucleotides
6. PKC Activators
6.1. Bryostatin-1
6.1.1. Bryostatin-1 as a PKC Inhibitor
6.1.2. Bryostatin-1 as a PKC Activator
7. Perspectives for Research and Application of PKC Inhibitors and Activators
8. Summary and Overall Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Newton, A.C. Protein kinase C: Structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem. Rev. 2001, 101, 2353–2364. [Google Scholar] [CrossRef]
- Newton, A.C. Protein kinase C: Perfectly balanced. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 208–230. [Google Scholar] [CrossRef]
- Steinberg, S.F. Structural basis of protein kinase C isoform function. Physiol. Rev. 2008, 88, 1341–1378. [Google Scholar] [CrossRef]
- Kang, J.H.; Toita, R.; Kim, C.W.; Katayama, Y. Protein kinase C (PKC) isozyme-specific substrates and their design. Biotechnol. Adv. 2012, 30, 1662–1672. [Google Scholar] [CrossRef]
- Kang, J.H. Protein kinase c (PKC) isozymes and cancer. New J. Sci. 2014, 2014, 231418. [Google Scholar] [CrossRef]
- Marrocco, V.; Bogomolovas, J.; Ehler, E.; Dos Remedios, C.G.; Yu, J.; Gao, C.; Lange, S. PKC and PKN in heart disease. J. Mol. Cell. Cardiol. 2019, 128, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Geribaldi-Doldán, N.; Gómez-Oliva, R.; Domínguez-García, S.; Nunez-Abades, P.; Castro, C. Protein kinase C: Targets to regenerate brain injuries? Front. Cell Dev. Biol. 2019, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Perander, M.; Outzen, H.; Kristiansen, K.; Øvervatn, A.; Michaelsen, E.; Bjørkøy, G.; Johansen, T. Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. J. Biol. Chem. 2003, 278, 34568–34581. [Google Scholar] [CrossRef] [PubMed]
- Hirano, Y.; Yoshinaga, S.; Ogura, K.; Yokochi, M.; Noda, Y.; Sumimoto, H.; Inagaki, F. Solution structure of atypical protein kinase C PB1 domain and its mode of interaction with ZIP/p62 and MEK5. J. Biol. Chem. 2004, 279, 31883–31890. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.; Nakano, H.; Morimoto, M.; Tamaoki, T. Calphostin C (UCN-1028C), a novel microbial compound, is a highly potent and specific inhibitor of protein kinase C. Biochem. Biophys. Res. Commun. 1989, 159, 548–553. [Google Scholar] [CrossRef]
- Shimamoto, Y.; Shimamoto, H.; Kwan, C.Y.; Daniel, E.E. Differential effects of putative protein kinase C inhibitors on contraction of rat aortic smooth muscle. Am. J. Physiol. 1993, 264, H1300–H1306. [Google Scholar] [CrossRef]
- Sullivan, J.P.; Connor, J.R.; Shearer, B.G.; Burch, R.M. NPC 15437 interacts with the C1 domain of protein kinase C. An analysis using mutant PKC constructs. FEBS Lett. 1991, 285, 120–123. [Google Scholar] [CrossRef]
- Sullivan, J.P.; Connor, J.R.; Tiffany, C.; Shearer, B.G.; Burch, R.M. 2,6-Diamino-N-([1-oxotridecyl)-2-piperidinyl]methyl)hexanamide (NPC 15437): A selective inhibitor of protein kinase C. Agents Act. 1991, 34, 142–144. [Google Scholar] [CrossRef]
- Roaten, J.B.; Kazanietz, M.G.; Caloca, M.J.; Bertics, P.J.; Lothstein, L.; Parrill, A.L.; Israel, M.; Sweatman, T.W. Interaction of the novel anthracycline antitumor agent N-benzyladriamycin-14-valerate with the C1-regulatory domain of protein kinase C: Structural requirements, isoform specificity, and correlation with drug cytotoxicity. Mol. Cancer Ther. 2002, 1, 483–492. [Google Scholar] [PubMed]
- Slater, S.J.; Seiz, J.L.; Cook, A.C.; Stagliano, B.A.; Buzas, C.J. Inhibition of protein kinase C by resveratrol. Biochim. Biophys. Acta 2003, 1637, 59–69. [Google Scholar] [CrossRef]
- Sachs, C.W.; Safa, A.R.; Harrison, S.D.; Fine, R.L. Partial inhibition of multidrug resistance by safingol is independent of modulation of P-glycoprotein substrate activities and correlated with inhibition of protein kinase C. J. Biol. Chem. 1995, 270, 26639–26648. [Google Scholar] [CrossRef]
- Schwartz, G.K.; Ward, D.; Saltz, L.; Casper, E.S.; Spiess, T.; Mullen, E.; Woodworth, J.; Venuti, R.; Zervos, P.; Storniolo, A.M.; et al. A pilot clinical/pharmacological study of the protein kinase C-specific inhibitor safingol alone and in combination with doxorubicin. Clin. Cancer Res. 1997, 3, 537–543. [Google Scholar]
- Dickson, M.A.; Carvajal, R.D.; Merrill, A.H., Jr.; Gonen, M.; Cane, L.M.; Schwartz, G.K. A phase I clinical trial of safingol in combination with cisplatin in advanced solid tumors. Clin. Cancer Res. 2011, 17, 2484–2492. [Google Scholar] [CrossRef]
- Farah, C.A.; Sossin, W.S. The role of C2 domains in PKC signaling. Adv. Exp. Med. Biol. 2012, 740, 663–683. [Google Scholar] [CrossRef]
- Ron, D.; Luo, J.; Mochly-Rosen, D. C2 region-derived peptides inhibit translocation and function of β protein kinase C in vivo. J. Biol. Chem. 1995, 270, 24180–24187. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Johnson, J.A.; Chen, L.; El-Sherif, N.; Mochly-Rosen, D.; Boutjdir, M. C2 region-derived peptides of β-protein kinase C regulate cardiac Ca2+ channels. Circ. Res. 1997, 80, 720–729. [Google Scholar] [CrossRef]
- Knight, Z.A.; Shokat, K.M. Features of selective kinase inhibitors. Chem. Biol. 2005, 12, 621–637. [Google Scholar] [CrossRef]
- Goekjian; Jirousek, M.R. Protein kinase C in the treatment of disease: Signal transduction pathways, inhibitors, and agents in development. Curr. Med. Chem. 1999, 6, 877–903. [Google Scholar] [CrossRef]
- Sánchez, C.; Méndez, C.; Salas, J.A. Indolocarbazole natural products: Occurrence, biosynthesis, and biological activity. Nat. Prod. Rep. 2006, 23, 1007–1045. [Google Scholar] [CrossRef]
- Nakano, H.; Ōmura, S. Chemical biology of natural indolocarbazole products: 30 years since the discovery of staurosporine. J. Antibiot. 2009, 62, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Park, B.S.; Abdel-Azeem, A.Z.; Al-Sanea, M.M.; Yoo, K.H.; Tae, T.S.; Lee, S. Staurosporine analogues from microbial and synthetic sources and their biological activities. Curr. Med. Chem. 2013, 20, 3872–3902. [Google Scholar] [CrossRef] [PubMed]
- Tamaoki, T.; Nomoto, H.; Takahashi, I.; Kato, Y.; Morimoto, M.; Tomita, F. Staurosporine, a potent inhibitor of phospholipid/Ca++dependent protein kinase. Biochem. Biophys. Res. Commun. 1986, 135, 397–402. [Google Scholar] [CrossRef]
- Seynaeve, C.M.; Kazanietz, M.G.; Blumberg, P.M.; Sausville, E.A.; Worland, P.J. Differential inhibition of protein kinase C isozymes by UCN-01, a staurosporine analogue. Mol. Pharmacol. 1994, 45, 1207–1214. [Google Scholar]
- Mizuno, K.; Noda, K.; Ueda, Y.; Hanaki, H.; Saido, T.C.; Ikuta, T.; Kuroki, T.; Tamaoki, T.; Hirai, S.; Osada, S.; et al. UCN-01, an anti-tumor drug, is a selective inhibitor of the conventional PKC subfamily. FEBS Lett. 1995, 359, 259–261. [Google Scholar] [CrossRef]
- Takahashi, I.; Asano, K.; Kawamoto, I.; Tamaoki, T.; Nakano, H. UCN-01 and UCN-02, new selective inhibitors of protein kinase C. I. Screening, producing organism and fermentation. J. Antibiot. 1989, 42, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, I.; Saitoh, Y.; Yoshida, M.; Sano, H.; Nakano, H.; Morimoto, M.; Tamaoki, T. UCN-01 and UCN-02, new selective inhibitors of protein kinase C. II. Purification, physico-chemical properties, structural determination and biological activities. J. Antibiot. 1989, 42, 571–576. [Google Scholar] [CrossRef]
- Takahashi, I.; Kobayashi, E.; Nakano, H.; Murakata, C.; Saitoh, H.; Suzuki, K.; Tamaoki, T. Potent selective inhibition of 7-O-methyl UCN-01 against protein kinase C. J. Pharmacol. Exp. Ther. 1990, 255, 1218–1221. [Google Scholar]
- Kurata, N.; Kuwabara, T.; Tanii, H.; Fuse, E.; Akiyama, T.; Akinaga, S.; Kobayashi, H.; Yamaguchi, K.; Kobayashi, S. Pharmacokinetics and pharmacodynamics of a novel protein kinase inhibitor, UCN-01. Cancer Chemother. Pharmacol. 1992, 44, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Seynaeve, C.M.; Stetler-Stevenson, M.; Sebers, S.; Kaur, G.; Sausville, E.A.; Worland, P.J. Cell cycle arrest and growth inhibition by the protein kinase antagonist UCN-01 in human breast carcinoma cells. Cancer Res. 1993, 53, 2081–2086. [Google Scholar]
- Wang, Q.; Worland, P.J.; Clark, J.L.; Carlson, B.A.; Sausville, E.A. Apoptosis in 7-hydroxystaurosporine-treated T lymphoblasts correlates with activation of cyclin-dependent kinases 1 and 2. Cell Growth Differ. 1995, 6, 927–936. [Google Scholar] [PubMed]
- Akiyama, T.; Yoshida, T.; Tsujita, T.; Shimizu, M.; Mizukami, T.; Okabe, M.; Akinaga, S. G1 phase accumulation induced by UCN-01 is associated with dephosphorylation of Rb and CDK2 proteins as well as induction of CDK inhibitor p21/Cip1/WAF1/Sdi1 in p53-mutated human epidermoid carcinoma A431 cells. Cancer Res. 1997, 57, 1495–1501. [Google Scholar]
- Sugiyama, K.; Akiyama, T.; Shimizu, M.; Tamaoki, T.; Courage, C.; Gescher, A.; Akinaga, S. Decrease in susceptibility toward induction of apoptosis and alteration in G1 checkpoint function as determinants of resistance of human lung cancer cells against the antisignaling drug UCN-01 (7-Hydroxystaurosporine). Cancer Res. 1999, 59, 4406–4412. [Google Scholar] [PubMed]
- Busby, E.C.; Leistritz, D.F.; Abraham, R.T.; Karnitz, L.M.; Sarkaria, J.N. The radiosensitizing agent 7-hydroxystaurosporine (UCN-01) inhibits the DNA damage checkpoint kinase hChk1. Cancer Res. 2000, 60, 2108–2112. [Google Scholar]
- Graves, P.R.; Yu, L.; Schwarz, J.K.; Gales, J.; Sausville, E.A.; O’Connor, P.M.; Piwnica-Worms, H. The Chk1 protein kinase and the Cdc25C regulatory pathways are targets of the anticancer agent UCN-01. J. Biol. Chem. 2000, 275, 5600–5605. [Google Scholar] [CrossRef]
- Kummar, S.; Gutierrez, M.E.; Gardner, E.R.; Figg, W.D.; Melillo, G.; Dancey, J.; Sausville, E.A.; Conley, B.A.; Murgo, A.J.; Doroshow, J.H. A phase I trial of UCN-01 and prednisone in patients with refractory solid tumors and lymphomas. Cancer Chemother. Pharmacol. 2010, 65, 383–389. [Google Scholar] [CrossRef]
- Jimeno, A.; Rudek, M.A.; Purcell, T.; Laheru, D.A.; Messersmith, W.A.; Dancey, J.; Carducci, M.A.; Baker, S.D.; Hidalgo, M.; Donehower, R.C. Phase I and pharmacokinetic study of UCN-01 in combination with irinotecan in patients with solid tumors. Cancer Chemother. Pharmacol. 2008, 61, 423–433. [Google Scholar] [CrossRef]
- Gojo, I.; Perl, A.; Luger, S.; Baer, M.R.; Norsworthy, K.J.; Bauer, K.S.; Tidwell, M.; Fleckinger, S.; Carroll, M.; Sausville, E.A. Phase I study of UCN-01 and perifosine in patients with relapsed and refractory acute leukemias and high-risk myelodysplastic syndrome. Investig. New Drugs 2013, 31, 1217–1227. [Google Scholar] [CrossRef]
- Marti, G.E.; Stetler-Stevenson, M.; Grant, N.D.; White, T.; Figg, W.D.; Tohnya, T.; Jaffe, E.S.; Dunleavy, K.; Janik, J.E.; Steinberg, S.M.; et al. Phase I trial of 7-hydroxystaurosporine and fludararbine phosphate: In vivo evidence of 7-hydroxystaurosporine induced apoptosis in chronic lymphocytic leukemia. Leuk. Lymphoma 2011, 52, 2284–2292. [Google Scholar] [CrossRef] [PubMed]
- Lara, P.N., Jr.; Mackm, P.C.; Synold, T.; Frankel, P.; Longmate, J.; Gumerlock, P.H.; Doroshow, J.H.; Gandara, D.R. The cyclin-dependent kinase inhibitor UCN-01 plus cisplatin in advanced solid tumors: A California cancer consortium phase I pharmacokinetic and molecular correlative trial. Clin. Cancer Res. 2005, 11, 4444–4450. [Google Scholar] [CrossRef] [PubMed]
- Kortmansky, J.; Shah, M.A.; Kaubisch, A.; Weyerbacher, A.; Yi, S.; Tong, W.; Sowers, R.; Gonen, M.; O’reilly, E.; Kemeny, N.; et al. Phase I trial of the cyclin-dependent kinase inhibitor and protein kinase C inhibitor 7-hydroxystaurosporine in combination with fluorouracil in patients with advanced solid tumors. J. Clin. Oncol. 2005, 23, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Edelman, M.J.; Bauer, K.S., Jr.; Wu, S.; Smith, R.; Bisacia, S.; Dancey, J. Phase I and pharmacokinetic study of 7-hydroxystaurosporine and carboplatin in advanced solid tumors. Clin. Cancer Res. 2007, 13, 2667–2674. [Google Scholar] [CrossRef]
- Fracasso, P.M.; Williams, K.J.; Chen, R.C.; Picus, J.; Ma, C.X.; Ellis, M.J.; Tan, B.R.; Pluard, T.J.; Adkins, D.R.; Naughton, M.J.; et al. A phase 1 study of UCN-01 in combination with irinotecan in patients with resistant solid tumor malignancies. Cancer Chemother. Pharmacol. 2011, 67, 1225–1237. [Google Scholar] [CrossRef]
- Ma, C.X.; Ellis, M.J.; Petroni, G.R.; Guo, Z.; Cai, S.R.; Ryan, C.E.; Craig Lockhart, A.; Naughton, M.J.; Pluard, T.J.; Brenin, C.M.; et al. A phase II study of UCN-01 in combination with irinotecan in patients with metastatic triple negative breast cancer. Breast Cancer Res. Treat. 2013, 137, 483–492. [Google Scholar] [CrossRef]
- Hotte, S.J.; Oza, A.; Winquist, E.W.; Moore, M.; Chen, E.X.; Brown, S.; Pond, G.R.; Dancey, J.E.; Hirte, H.W. Phase I trial of UCN-01 in combination with topotecan in patients with advanced solid cancers: A Princess Margaret Hospital Phase II Consortium study. Ann. Oncol. 2006, 17, 334–340. [Google Scholar] [CrossRef]
- Welch, S.; Hirte, H.W.; Carey, M.S.; Hotte, S.J.; Tsao, M.S.; Brown, S.; Pond, G.R.; Dancey, J.E.; Oza, A.M. UCN-01 in combination with topotecan in patients with advanced recurrent ovarian cancer: A study of the Princess Margaret Hospital Phase II consortium. Gynecol. Oncol. 2007, 106, 305–310. [Google Scholar] [CrossRef]
- Fabbro, D.; Buchdunger, E.; Wood, J.; Mestan, J.; Hofmann, F.; Ferrari, S.; Mett, H.; O’Reilly, T.; Meyer, T. Inhibitors of protein kinases: CGP 41251, a protein kinase inhibitor with potential as an anticancer agent. Pharmacol. Ther. 1999, 82, 293–301. [Google Scholar] [CrossRef]
- Fabbro, D.; Ruetz, S.; Bodis, S.; Pruschy, M.; Csermak, K.; Man, A.; Campochiaro, P.; Wood, J.; O’Reilly, T.; Meyer, T. PKC412‒a protein kinase inhibitor with a broad therapeutic potential. Anticancer Drug Des. 2000, 15, 17–28. [Google Scholar] [PubMed]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Grundler, R.; Thiede, C.; Miething, C.; Steudel, C.; Peschel, C.; Duyster, J. Sensitivity toward tyrosine kinase inhibitors varies between different activating mutations of the FLT3 receptor. Blood 2003, 102, 646–651. [Google Scholar] [CrossRef]
- Weisberg, E.; Boulton, C.; Kelly, L.M.; Manley, P.; Fabbro, D.; Meyer, T.; Gilliland, D.G.; Griffin, J.D. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell 2002, 1, 433–443. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Metcalfe, D.D. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood 2016, 129, 1420–1427. [Google Scholar] [CrossRef]
- Gotlib, J.; Berubé, C.; Growney, J.D.; Chen, C.C.; George, T.I.; Williams, C.; Kajiguchi, T.; Ruan, J.; Lilleberg, S.L.; Durocher, J.A.; et al. Activity of the tyrosine kinase inhibitor PKC412 in a patient with mast cell leukemia with the D816V KIT mutation. Blood 2005, 106, 2865–2870. [Google Scholar] [CrossRef]
- Gleixner, K.V.; Mayerhofer, M.; Aichberger, K.J.; Derdak, S.; Sonneck, K.; Böhm, A.; Gruze, A.; Samorapoompichit, P.; Manley, P.W.; Fabbro, D.; et al. PKC412 inhibits in vitro growth of neoplastic human mast cells expressing the D816V-mutated variant of KIT: Comparison with AMN107, imatinib, and cladribine (2CdA) and evaluation of cooperative drug effects. Blood 2006, 107, 752–759. [Google Scholar] [CrossRef]
- Gleixner, K.V.; Mayerhofer, M.; Sonneck, K.; Gruze, A.; Samorapoompichit, P.; Baumgartner, C.; Lee, F.Y.; Aichberger, K.J.; Manley, P.W.; Fabbro, D.; et al. Synergistic growth-inhibitory effects of two tyrosine kinase inhibitors, dasatinib and PKC412, on neoplastic mast cells expressing the D816V-mutated oncogenic variant of KIT. Haematologica 2007, 92, 1451–1459. [Google Scholar] [CrossRef][Green Version]
- DeAngelo, D.J.; George, T.I.; Linder, A.; Langford, C.; Perkins, C.; Ma, J.; Westervelt, P.; Merker, J.D.; Berube, C.; Coutre, S.; et al. Efficacy and safety of midostaurin in patients with advanced systemic mastocytosis: 10-year median follow-up of a phase II trial. Leukemia 2018, 32, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Gotlib, J.; Kluin-Nelemans, H.C.; George, T.I.; Akin, C.; Sotlar, K.; Hermine, O.; Awan, F.T.; Hexner, E.; Mauro, M.J.; Sternberg, D.W.; et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N. Engl. J. Med. 2016, 374, 2530–2541. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, G.; Dolph, M.; Patel, S.; Brandt, P.; Forsythe, A. Cost-effectiveness analysis for midostaurin versus standard of care in acute myeloid leukemia in the United Kingdom. Cost Eff. Resour. Alloc. 2018, 16, 33. [Google Scholar] [CrossRef] [PubMed]
- Schlenk, R.F.; Weber, D.; Fiedler, W.; Salih, H.R.; Wulf, G.; Salwender, H.; Schroeder, T.; Kindler, T.; Lübbert, M.; Wolf, D.; et al. Midostaurin added to chemotherapy and continued single-agent maintenance therapy in acute myeloid leukemia with FLT3-ITD. Blood 2019, 133, 840–851. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. U.S. National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=Midostaurin&cntry=&state=&city=&dist= (accessed on 20 August 2020).
- Cortes, J.; Perl, A.E.; Döhner, H.; Kantarjian, H.; Martinelli, G.; Kovacsovics, T.; Rousselot, P.; Steffen, B.; Dombret, H.; Estey, E.; et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: An open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol. 2018, 19, 889–903. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- Toullec, D.; Pianetti, P.; Coste, H.; Bellevergue, P.; Grand-Perret, T.; Ajakane, M.; Baudet, V.; Boissin, P.; Boursier, E.; Loriolle, F.; et al. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J. Biol. Chem. 1991, 266, 15771–15781. [Google Scholar] [CrossRef]
- Wilkinson, S.E.; Parker, P.J.; Nixon, J.S. Isoenzyme specificity of bisindolylmaleimides, selective inhibitors of protein kinase C. Biochem. J. 1993, 294, 335–357. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.R.; McNulty, A.M.; Hanna, K.R.; Konicek, B.W.; Lynch, R.L.; Bailey, S.N.; Banks, C.; Capen, A.; Goode, R.; Lewis, J.E.; et al. The protein kinase Cβ-selective inhibitor, Enzastaurin (LY317615.HCl), suppresses signaling through the AKT pathway, induces apoptosis, and suppresses growth of human colon cancer and glioblastoma xenografts. Cancer Res. 2005, 65, 7462–7469. [Google Scholar] [CrossRef]
- Jirousek, M.R.; Gillig, J.R.; Gonzalez, C.M.; Heath, W.F.; McDonald, J.H., 3rd; Neel, D.A.; Rito, C.J.; Singh, U.; Stramm, L.E.; Melikian-Badalian, A.; et al. (S)-13-[(dimethylamino)methyl]-10,11,14,15-tetrahydro-4,9:16, 21-dimetheno-1H, 13H-dibenzo[e,k]pyrrolo[3,4-h][1,4,13]oxadiazacyclohexadecene-1,3(2H)-d ione (LY333531) and related analogues: Isozyme selective inhibitors of protein kinase Cβ. J. Med Chem. 1996, 39, 2664–2671. [Google Scholar] [CrossRef]
- Davies, S.P.; Reddy, H.; Caivano, M.; Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000, 351, 95–105. [Google Scholar] [CrossRef]
- Komander, D.; Kular, G.S.; Schüttelkopf, A.W.; Deak, M.; Prakash, K.R.; Bain, J.; Elliott, M.; Garrido-Franco, M.; Kozikowski, A.P.; Alessi, D.R.; et al. Interactions of LY333531 and other bisindolyl maleimide inhibitors with PDK1. Structure 2004, 12, 215–226. [Google Scholar] [CrossRef]
- Rizvi, M.A.; Ghias, K.; Davies, K.M.; Ma, C.; Weinberg, F.; Munshi, H.G.; Krett, N.L.; Rosen, S.T. Enzastaurin (LY317615), a protein kinase Cβ inhibitor, inhibits the AKT pathway and induces apoptosis in multiple myeloma cell lines. Mol. Cancer Ther. 2006, 5, 1783–1789. [Google Scholar] [CrossRef]
- Neri, A.; Marmiroli, S.; Tassone, P.; Lombardi, L.; Nobili, L.; Verdelli, D.; Civallero, M.; Cosenza, M.; Bertacchini, J.; Federico, M.; et al. The oral protein-kinase C β inhibitor enzastaurin (LY317615) suppresses signalling through the AKT pathway, inhibits proliferation and induces apoptosis in multiple myeloma cell lines. Leuk. Lymphoma 2008, 49, 1374–1383. [Google Scholar] [CrossRef]
- Lee, S.H.; Chen, T.; Zhou, J.; Hofmann, J.; Bepler, G. Protein kinase C-β gene variants, pathway activation, and enzastaurin activity in lung cancer. Clin. Lung Cancer 2010, 11, 169–175. [Google Scholar] [CrossRef]
- Fields, A.P.; Calcagno, S.R.; Krishna, M.; Rak, S.; Leitges, M.; Murray, N.R. Protein kinase Cβ is an effective target for chemoprevention of colon cancer. Cancer Res. 2009, 69, 1643–1650. [Google Scholar] [CrossRef]
- Odia, Y.; Iwamoto, F.M.; Moustakas, A.; Fraum, T.J.; Salgado, C.A.; Li, A.; Kreisl, T.N.; Sul, J.; Butman, J.A.; Fine, H.A. A phase II trial of enzastaurin (LY317615) in combination with bevacizumab in adults with recurrent malignant gliomas. J. Neurooncol. 2016, 127, 127–135. [Google Scholar] [CrossRef]
- Wick, W.; Puduvalli, V.K.; Chamberlain, M.C.; van den Bent, M.J.; Carpentier, A.F.; Cher, L.M.; Mason, W.; Weller, M.; Hong, S.; Musib, L.; et al. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J. Clin. Oncol. 2010, 28, 1168–1174. [Google Scholar] [CrossRef]
- Crump, M.; Leppä, S.; Fayad, L.; Lee, J.J.; Di Rocco, A.; Ogura, M.; Hagberg, H.; Schnell, F.; Rifkin, R.; Mackensen, A.; et al. Randomized, double-blind, phase III trial of enzastaurin versus placebo in patients achieving remission after first-line therapy for high-risk diffuse large B-cell lymphoma. J. Clin. Oncol. 2016, 34, 2484–2492. [Google Scholar] [CrossRef]
- Jourdan, E.; Leblond, V.; Maisonneuve, H.; Benhadji, K.A.; Hossain, A.M.; Nguyen, T.S.; Wooldridge, J.E.; Moreau, P. A multicenter phase II study of single-agent enzastaurin in previously treated multiple myeloma. Leuk. Lymphoma 2014, 55, 2013–2017. [Google Scholar] [CrossRef] [PubMed]
- Grønberg, B.H.; Ciuleanu, T.; Fløtten, Ø.; Knuuttila, A.; Abel, E.; Langer, S.W.; Krejcy, K.; Liepa, A.M.; Munoz, M.; Hahka-Kemppinen, M.; et al. A placebo-controlled, randomized phase II study of maintenance enzastaurin following whole brain radiation therapy in the treatment of brain metastases from lung cancer. Lung Cancer 2012, 78, 63–69. [Google Scholar] [CrossRef]
- Usha, L.; Sill, M.W.; Darcy, K.M.; Benbrook, D.M.; Hurteau, J.A.; Michelin, D.P.; Mannel, R.S.; Hanjani, P.; De Geest, K.; Godwin, A.K. A Gynecologic Oncology Group phase II trial of the protein kinase C-β inhibitor, enzastaurin and evaluation of markers with potential predictive and prognostic value in persistent or recurrent epithelial ovarian and primary peritoneal malignancies. Gynecol. Oncol. 2011, 121, 455–461. [Google Scholar] [CrossRef]
- Morschhauser, F.; Seymour, J.F.; Kluin-Nelemans, H.C.; Grigg, A.; Wolf, M.; Pfreundschuh, M.; Tilly, H.; Raemaekers, J.; van’t Veer, M.B.; Milpied, N.; et al. A phase II study of enzastaurin, a protein kinase C β inhibitor, in patients with relapsed or refractory mantle cell lymphoma. Ann. Oncol. 2008, 19, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Mina, L.; Krop, I.; Zon, R.T.; Isakoff, S.J.; Schneider, C.J.; Yu, M.; Johnson, C.; Vaughn, L.G.; Wang, Y.; Hristova-Kazmierski, M.; et al. A phase II study of oral enzastaurin in patients with metastatic breast cancer previously treated with an anthracycline and a taxane containing regimen. Investig. New Drugs 2009, 27, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Querfeld, C.; Kuzel, T.M.; Kim, Y.H.; Porcu, P.; Duvic, M.; Musiek, A.; Rook, A.H.; Mark, L.A.; Pinter-Brown, L.; Hamid, O.; et al. Multicenter phase II trial of enzastaurin in patients with relapsed or refractory advanced cutaneous T-cell lymphoma. Leuk. Lymphoma 2011, 52, 1474–1480. [Google Scholar] [CrossRef]
- Clément-Duchêne, C.; Natale, R.B.; Jahan, T.; Krupitskaya, Y.; Osarogiagbon, R.; Sanborn, R.E.; Bernstein, E.D.; Dudek, A.Z.; Latz, J.E.; Shi, P.; et al. A phase II study of enzastaurin in combination with erlotinib in patients with previously treated advanced non-small cell lung cancer. Lung Cancer 2012, 78, 57–62. [Google Scholar] [CrossRef]
- Butowski, N.; Chang, S.M.; Lamborn, K.R.; Polley, M.Y.; Pieper, R.; Costello, J.F.; Vandenberg, S.; Parvataneni, R.; Nicole, A.; Sneed, P.K.; et al. Phase II and pharmacogenomics study of enzastaurin plus temozolomide during and following radiation therapy in patients with newly diagnosed glioblastoma multiforme and gliosarcoma. Neuro-Oncology 2011, 13, 1331–1338. [Google Scholar] [CrossRef]
- Dreicer, R.; Garcia, J.; Rini, B.; Vogelzang, N.; Srinivas, S.; Somer, B.; Shi, P.; Kania, M.; Raghavan, D. A randomized, double-blind, placebo-controlled, Phase II study with and without enzastaurin in combination with docetaxel-based chemotherapy in patients with castration-resistant metastatic prostate cancer. Investig. New Drugs. 2013, 31, 1044–1050. [Google Scholar] [CrossRef]
- Vergote, I.B.; Chekerov, R.; Amant, F.; Harter, P.; Casado, A.; Emerich, J.; Bauknecht, T.; Mansouri, K.; Myrand, S.P.; Nguyen, T.S.; et al. Randomized, phase II, placebo-controlled, double-blind study with and without enzastaurin in combination with paclitaxel and carboplatin as first-line treatment followed by maintenance treatment in advanced ovarian cancer. J. Clin. Oncol. 2013, 31, 3127–3132. [Google Scholar] [CrossRef] [PubMed]
- Wolff, R.A.; Fuchs, M.; Di Bartolomeo, M.; Hossain, A.M.; Stoffregen, C.; Nicol, S.; Heinemann, V. A double-blind, randomized, placebo-controlled, phase 2 study of maintenance enzastaurin with 5-fluorouracil/leucovorin plus bevacizumab after first-line therapy for metastatic colorectal cancer. Cancer 2012, 118, 4132–4138. [Google Scholar] [CrossRef] [PubMed]
- Chiappori, A.; Bepler, G.; Barlesi, F.; Soria, J.C.; Reck, M.; Bearz, A.; Barata, F.; Scagliotti, G.; Park, K.; Wagle, A.; et al. Phase II, double-blinded, randomized study of enzastaurin plus pemetrexed as second-line therapy in patients with advanced non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.A.; Kuefler, P.R.; Becerra, C.; Wilfong, L.S.; Gersh, R.H.; Boehm, K.A.; Zhan, F.; Asmar, L.; Myrand, S.P.; Hozak, R.R.; et al. Gemcitabine plus enzastaurin or single-agent gemcitabine in locally advanced or metastatic pancreatic cancer: Results of a phase II, randomized, noncomparative study. Investig. New Drugs 2011, 29, 144–153. [Google Scholar] [CrossRef]
- Kilburn, L.B.; Kocak, M.; Decker, R.L.; Wetmore, C.; Chintagumpala, M.; Su, J.; Goldman, S.; Banerjee, A.; Gilbertson, R.; Fouladi, M.; et al. A phase 1 and pharmacokinetic study of enzastaurin in pediatric patients with refractory primary central nervous system tumors: A pediatric brain tumor consortium study. Neuro-Oncology 2015, 17, 303–311. [Google Scholar] [CrossRef][Green Version]
- Nonaka, A.; Kiryu, J.; Tsujikawa, A.; Yamashiro, K.; Miyamoto, K.; Nishiwaki, H.; Honda, Y.; Ogura, Y. PKC-β inhibitor (LY333531) attenuates leukocyte entrapment in retinal microcirculation of diabetic rats. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2702–2706. [Google Scholar]
- Abiko, T.; Abiko, A.; Clermont, A.C.; Shoelson, B.; Horio, N.; Takahashi, J.; Adamis, A.P.; King, G.L.; Bursell, S.E. Characterization of retinal leukostasis and hemodynamics in insulin resistance and diabetes: Role of oxidants and protein kinase-C activation. Diabetes 2003, 52, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Ma, R.C.; Park, J.Y.; Isshiki, K.; Sotiropoulos, K.B.; Rauniyar, R.K.; Bornfeldt, K.E.; King, G.L. Role of protein kinase C on the expression of platelet-derived growth factor and endothelin-1 in the retina of diabetic rats and cultured retinal capillary pericytes. Diabetes 2003, 52, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Chikaraishi, Y.; Tsuruma, K.; Shimazawa, M.; Hara, H. Ruboxistaurin, a PKCβ inhibitor, inhibits retinal neovascularization via suppression of phosphorylation of ERK1/2 and Akt. Exp. Eye Res. 2010, 90, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Said, G. Diabetic neuropathy‒a review. Nat. Clin. Pract. Neurol. 2007, 3, 331–340. [Google Scholar] [CrossRef]
- Feldman, E.L.; Callaghan, B.C.; Pop-Busui, R.; Zochodne, D.W.; Wright, D.E.; Bennett, D.L.; Bril, V.; Russell, J.W.; Viswanathan, V. Diabetic neuropathy. Nat. Rev. Dis. Primers 2019, 5, 41. [Google Scholar] [CrossRef]
- Kim, H.; Sasaki, T.; Maeda, K.; Koya, D.; Kashiwagi, A.; Yasuda, H. Protein kinase Cβ selective inhibitor LY333531 attenuates diabetic hyperalgesia through ameliorating cGMP level of dorsal root ganglion neurons. Diabetes 2003, 52, 2102–2109. [Google Scholar] [CrossRef]
- Kitada, M.; Koya, D.; Sugimoto, T.; Isono, M.; Araki, S.; Kashiwagi, A.; Haneda, M. Translocation of glomerular p47phox and p67phox by protein kinase C-β activation is required for oxidative stress in diabetic nephropathy. Diabetes 2003, 52, 2603–2614. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Yuan, H.; Xu, Z.G.; Lanting, L.; Li, S.L.; Wang, M.; Hu, M.C.; Reddy, M.A.; Natarajan, R. Role of the Akt/FoxO3a pathway in TGF-β1-mediated mesangial cell dysfunction: A novel mechanism related to diabetic kidney disease. J. Am. Soc. Nephrol. 2006, 17, 3325–3335. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Peng, F.; Zhang, B.; Ingram, A.J.; Kelly, D.J.; Gilbert, R.E.; Gao, B.; Krepinsky, J.C. PKC-β1 mediates glucose-induced Akt activation and TGF-beta1 upregulation in mesangial cells. J. Am. Soc. Nephrol. 2009, 20, 554–566. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.J.; Chanty, A.; Gow, R.M.; Zhang, Y.; Gilbert, R.E. Protein kinase Cβ inhibition attenuates osteopontin expression, macrophage recruitment, and tubulointerstitial injury in advanced experimental diabetic nephropathy. J. Am. Soc. Nephrol. 2005, 16, 1654–1660. [Google Scholar] [CrossRef] [PubMed]
- Sheetz, M.J.; Aiello, L.P.; Shahri, N.; Davis, M.D.; Kles, K.A.; Danis, R.P.; Mbdv Study Group. Effect of ruboxistaurin (RBX) On visual acuity decline over a 6-year period with cessation and reinstitution of therapy: Results of an open-label extension of the Protein Kinase C Diabetic Retinopathy Study 2 (PKC-DRS2). Retina 2011, 31, 1053–1059. [Google Scholar] [CrossRef]
- Sheetz, M.J.; Aiello, L.P.; Davis, M.D.; Danis, R.; Bek, T.; Cunha-Vaz, J.; Shahri, N.; Berg, P.H.; MBDL and MBCU Study Groups. The effect of the oral PKC β inhibitor ruboxistaurin on vision loss in two phase 3 studies. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1750–1757. [Google Scholar] [CrossRef][Green Version]
- Tesfaye, S.; Tandan, R.; Bastyr, E.J., 3rd; Kles, K.A.; Skljarevski, V.; Price, K.L.; Ruboxistaurin Study Group. Factors that impact symptomatic diabetic peripheral neuropathy in placebo-administered patients from two 1-year clinical trials. Diabetes Care 2007, 30, 2626–2632. [Google Scholar] [CrossRef]
- Skvara, H.; Dawid, M.; Kleyn, E.; Wolff, B.; Meingassner, J.G.; Knight, H.; Dumortier, T.; Kopp, T.; Fallahi, N.; Stary, G.; et al. The PKC inhibitor AEB071 may be a therapeutic option for psoriasis. J. Clin. Investig. 2008, 118, 3151–3159. [Google Scholar] [CrossRef]
- Wagner, J.; von Matt, P.; Sedrani, R.; Albert, R.; Cooke, N.; Ehrhardt, C.; Geiser, M.; Rummel, G.; Stark, W.; Strauss, A.; et al. Discovery of 3-(1H-indol-3-yl)-4-[2-(4-methylpiperazin-1-yl)quinazolin-4-yl]pyrrole-2,5-dione (AEB071), a potent and selective inhibitor of protein kinase C isotypes. J. Med. Chem. 2009, 52, 6193–6196. [Google Scholar] [CrossRef]
- Evenou, J.P.; Wagner, J.; Zenke, G.; Brinkmann, V.; Wagner, K.; Kovarik, J.; Welzenbach, K.A.; Weitz-Schmidt, G.; Guntermann, C.; Towbin, H.; et al. The potent protein kinase C-selective inhibitor AEB071 (sotrastaurin) represents a new class of immunosuppressive agents affecting early T-cell activation. J. Pharmacol. Exp. Ther. 2009, 330, 792–801. [Google Scholar] [CrossRef]
- Martina, M.N.; Ramirez Bajo, M.J.; Bañon-Maneus, E.; Moya Rull, D.; Hierro-Garcia, N.; Revuelta, I.; Campistol, J.M.; Rovira, J.; Diekmann, F. Inhibition of JAK3 and PKC via immunosuppressive drugs tofacitinib and sotrastaurin inhibits proliferation of human B lymphocytes in vitro. Transpl. Proc. 2016, 48, 3046–3052. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.C.; Lo, C.M.; Fung, J.Y. Emerging drugs for prevention of T-cell mediated rejection in liver and kidney transplantation. Expert. Opin. Emerg. Drugs 2017, 22, 123–136. [Google Scholar] [CrossRef]
- Friman, S.; Arns, W.; Nashan, B.; Vincenti, F.; Banas, B.; Budde, K.; Cibrik, D.; Chan, L.; Klempnauer, J.; Mulgaonkar, S.; et al. Sotrastaurin. a novel small molecule inhibiting protein-kinase C: Randomized phase II study in renal transplant recipients. Am. J. Transpl. 2011, 11, 1444–1455. [Google Scholar] [CrossRef] [PubMed]
- Pascher, A.; De Simone, P.; Pratschke, J.; Salamé, E.; Pirenne, J.; Isoneimi, H.; Bijarnia, M.; Krishnan, I.; Klupp, J. Protein Kinase C Inhibitor sotrastaurin in de novo liver transplant recipients: A randomized phase II trial. Am. J. Transpl. 2015, 15, 1283–1292. [Google Scholar] [CrossRef]
- Russ, G.R.; Tedesco-Silva, H.; Kuypers, D.R.; Cohney, S.; Langer, R.M.; Witzke, O.; Eris, J.; Sommerer, C.; von Zur-Mühlen, B.; Woodle, E.S.; et al. Efficacy of sotrastaurin plus tacrolimus after de novo kidney transplantation: Randomized, phase II trial results. Am. J. Transpl. 2013, 13, 1746–1756. [Google Scholar] [CrossRef] [PubMed]
- Tedesco-Silva, H.; Kho, M.M.; Hartmann, A.; Vitko, S.; Russ, G.; Rostaing, L.; Budde, K.; Campistol, J.M.; Eris, J.; Krishnan, I.; et al. Sotrastaurin in calcineurin inhibitor-free regimen using everolimus in de novo kidney transplant recipients. Am. J. Transpl. 2013, 13, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Naylor, T.L.; Tang, H.; Ratsch, B.A.; Enns, A.; Loo, A.; Chen, L.; Lenz, P.; Waters, N.J.; Schuler, W.; Dörken, B.; et al. Protein kinase C inhibitor sotrastaurin selectively inhibits the growth of CD79 mutant diffuse large B-cell lymphomas. Cancer Res. 2011, 71, 2643–2653. [Google Scholar] [CrossRef]
- Chang, G.; Zheng, J.; Xiao, W.; Chang, S.; Wei, Q.; Wu, H.; Tao, Y.; Yang, G.; Xie, B.; Lan, X.; et al. PKC inhibition of sotrastaurin has antitumor activity in diffuse large B-cell lymphoma via regulating the expression of MCT-1. Acta Biochim. Biophys. Sin. 2018, 50, 399–407. [Google Scholar] [CrossRef]
- Piperno-Neumann, S.; Larkin, J.; Carvajal, R.D.; Luke, J.J.; Schwartz, G.K.; Hodi, F.S.; Sablin, M.P.; Shoushtari, A.N.; Szpakowski, S.; Chowdhury, N.R.; et al. Genomic profiling of metastatic uveal melanoma and clinical results of a phase I study of the protein kinase C inhibitor AEB071. Mol. Cancer Ther. 2020, 19, 1031–1039. [Google Scholar] [CrossRef]
- Kulanthaivel, P.; Hallock, Y.F.; Boros, C.; Hamiton, S.M.; Janzen, W.P.; Ballas, L.M.; Loomis, C.R.; Jiang, J.B.; Katz, B.; Stainer, J.R.; et al. Balanol: A novel and potent inhibitor of protein kinase C from the fungus Verticillium balanoides. J. Am. Chem. Soc. 1993, 14, 6452–6453. [Google Scholar] [CrossRef]
- Lai, Y.S.; Mendoza, J.S.; Hubbard, F.; Kalter, K. Synthesis and PKC inhibitory activities of balanol analogs with a cyclopentane substructure. Bioorg. Med. Chem. Lett. 1995, 5, 2155–2160. [Google Scholar] [CrossRef]
- Defauw, J.M.; Murphy, M.M.; Jagdmann, G.E., Jr.; Hu, H.; Lampe, J.W.; Hollinshead, S.P.; Mitchell, T.J.; Crane, H.M.; Heerding, J.M.; Mendoza, J.S.; et al. Synthesis and protein kinase C inhibitory activities of acyclic balanol analogs that are highly selective for protein kinase C over protein kinase A. J. Med. Chem. 1996, 39, 5215–5227. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.R.; Hardianto, A.; Ranganathan, S.; Liu, F. Divergent response of homologous ATP sites to stereospecific ligand fluorination for selectivity enhancement. Org. Biomol. Chem. 2017, 15, 1570–1574. [Google Scholar] [CrossRef] [PubMed]
- O’Brian, C.A.; Ward, N.E. ATP-sensitive binding of melittin to the catalytic domain of protein kinase C. Mol. Pharmacol. 1989, 36, 355–359. [Google Scholar] [PubMed]
- Katoh, N. Inhibition by melittin of phosphorylation by protein kinase C of annexin I from cow mammary gland. J. Vet. Med. Sci. 2002, 64, 779–783. [Google Scholar] [CrossRef]
- Gravitt, K.R.; Ward, N.E.; O’Brian, C.A. Inhibition of protein kinase C by melittin: Antagonism of binding interactions between melittin and the catalytic domain by active-site binding of MgATP. Biochem. Pharmacol. 1994, 47, 425–427. [Google Scholar] [CrossRef]
- Eichholtz, T.; de Bont, D.B.; de Widt, J.; Liskamp, R.M.; Ploegh, H.L. A myristoylated pseudosubstrate peptide, a novel protein kinase C inhibitor. J. Biol. Chem. 1993, 268, 1982–1986. [Google Scholar] [CrossRef]
- Ward, N.E.; O’Brian, C.A. Inhibition of protein kinase C by N-myristoylated peptide substrate analogs. Biochemistry 1993, 32, 11903–11909. [Google Scholar] [CrossRef]
- Hofmann, J. The potential for isoenzyme-selective modulation of protein kinase C. FASEB J. 1997, 11, 649–669. [Google Scholar] [CrossRef]
- Zaliani, A.; Pinori, M.; Ball, H.L.; DiGregorio, G.; Cremonesi, P.; Mascagni, P. The interaction of myristylated peptides with the catalytic domain of protein kinase C revealed by their sequence palindromy and the identification of a myristyl binding site. Protein Eng. 1998, 11, 803–810. [Google Scholar] [CrossRef]
- Eller, M.; Järv, J.; Toomik, R.; Ragnarsson, U.; Ekman, P.; Engström, L. Substrate specificity of protein kinase C studied with peptides containing D-amino acid residues. J. Biol. Chem. 1993, 114, 177–180. [Google Scholar] [CrossRef]
- House, C.; Kemp, B.E. Protein kinase C contains a pseudosubstrate prototope in its regulatory domain. Science 1987, 238, 1726–1728. [Google Scholar] [CrossRef]
- Baxter, G.; Oto, E.; Daniel-Issakani, S.; Strulovici, B. Constitutive presence of a catalytic fragment of protein kinase C ε in a small cell lung carcinoma cell line. J. Biol. Chem. 1992, 267, 1910–1917. [Google Scholar] [CrossRef]
- Bogard, A.S.; Tavalin, S.J. Protein kinase C (PKC)ζ pseudosubstrate inhibitor peptide promiscuously binds PKC family isoforms and disrupts conventional PKC targeting and translocation. Mol. Pharmacol. 2015, 88, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Gray, M.O.; Chen, C.H.; Mochly-Rosen, D. A protein kinase C translocation inhibitor as an isozyme-selective antagonist of cardiac function. J. Biol. Chem. 1996, 271, 24962–24966. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Thorne, S.H.; Sun, L.; Huang, B.; Mochly-Rosen, D. Sustained inhibition of PKCα reduces intravasation and lung seeding during mammary tumor metastasis in an in vivo mouse model. Oncogene 2011, 30, 323–333. [Google Scholar] [CrossRef]
- Stebbins, E.G.; Mochly-Rosen, D. Binding specificity for RACK1 resides in the V5 region of βII protein kinase C. J. Biol. Chem. 2001, 276, 29644–29650. [Google Scholar] [CrossRef]
- Chen, L.; Hahn, H.; Wu, G.; Chen, C.H.; Liron, T.; Schechtman, D.; Cavallaro, G.; Banci, L.; Guo, Y.; Bolli, R.; et al. Opposing cardioprotective actions and parallel hypertrophic effects of δPKC and ɛPKC. Proc. Natl. Acad. Sci. USA 2001, 98, 11114–11119. [Google Scholar] [CrossRef]
- Lincoff, A.M.; Roe, M.; Aylward, P.; Galla, J.; Rynkiewicz, A.; Guetta, V.; Zelizko, M.; Kleiman, N.; White, H.; McErlean, E.; et al. Inhibition of delta-protein kinase C by delcasertib as an adjunct to primary percutaneous coronary intervention for acute anterior ST-segment elevation myocardial infarction: Results of the PROTECTION AMI Randomized Controlled. Trial. Eur. Heart J. 2014, 35, 2516–2523. [Google Scholar] [CrossRef]
- Cousins, M.J.; Pickthorn, K.; Huang, S.; Critchley, L.; Bell, G. The safety and efficacy of KAI-1678- an inhibitor of epsilon protein kinase C (εPKC)-versus lidocaine and placebo for the treatment of postherpetic neuralgia: A crossover study design. Pain Med. 2013, 14, 533–540. [Google Scholar] [CrossRef][Green Version]
- Tarrant, M.K.; Cole, P.A. The chemical biology of protein phosphorylation. Annu. Rev. Biochem. 2009, 78, 797–825. [Google Scholar] [CrossRef]
- Herbert, J.M.; Augereau, J.M.; Gleye, J.; Maffrand, J.P. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem. Biophys. Res. Commun. 1990, 172, 993–999. [Google Scholar] [CrossRef]
- Chmura, S.J.; Dolan, M.E.; Cha, A.; Mauceri, H.J.; Kufe, D.W.; Weichselbaum, R.R. In vitro and in vivo activity of protein kinase C inhibitor chelerythrine chloride induces tumor cell toxicity and growth delay in vivo. Clin. Cancer Res. 2000, 6, 737–742. [Google Scholar]
- Fan, L.; Fan, Y.; Liu, L.; Tao, W.; Shan, X.; Dong, Y. Chelerythrine attenuates the inflammation of lipopolysaccharide-induced acute lung inflammation through NF-κB signaling pathway mediated by Nrf2. Front. Pharmacol. 2018, 9, 1047. [Google Scholar] [CrossRef]
- Blázquez, A.B.; Vázquez-Calvo, Á.; Martín-Acebes, M.A.; Saiz, J.C. Pharmacological inhibition of protein kinase C reduces West Nile virus replication. Viruses 2018, 10, E91. [Google Scholar] [CrossRef]
- Gong, Y.; Li, S.; Wang, W.; Li, Y.; Ma, W.; Sun, S. In vitro and in vivo activity of chelerythrine against Candida albicans and underlying mechanisms. Future Microbiol. 2019, 14, 1545–1557. [Google Scholar] [CrossRef] [PubMed]
- He, N.; Wang, P.; Wang, P.; Ma, C.; Kang, W. Antibacterial mechanism of chelerythrine isolated from root of Toddalia asiatica (Linn) Lam. BMC Complement. Altern. Med. 2018, 18, 261. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Huang, J.; Yuan, Z.; Feng, S.; Xie, Y.; Ma, W. Protein kinase C inhibitor chelerythrine selectively inhibits proliferation of triple-negative breast cancer cells. Sci. Rep. 2017, 7, 2022. [Google Scholar] [CrossRef]
- Slaninová, I.; Táborská, E.; Bochoráková, H.; Slanina, J. Interaction of benzo[c]phenanthridine and protoberberine alkaloids with animal and yeast cells. Cell Biol. Toxicol. 2001, 17, 51–63. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Yang, Z.; Zhang, L.; Song, C.; Li, Y.; Zhang, X. Additive effects of cherlerythrine chloride combination with erlotinib in human non-small cell lung cancer cells. PLoS ONE 2017, 12, e0175466. [Google Scholar] [CrossRef]
- Lee, S.K.; Qing, W.G.; Mar, W.; Luyengi, L.; Mehta, R.G.; Kawanishi, K.; Fong, H.H.; Beecher, C.W.; Kinghorn, A.D.; Pezzuto, J.M. Angoline and chelerythrine, benzophenanthridine alkaloids that do not inhibit protein kinase C. J. Biol. Chem. 1998, 273, 19829–19833. [Google Scholar] [CrossRef]
- Noh, K.M.; Hwang, J.Y.; Shin, H.C.; Koh, J.Y. A novel neuroprotective mechanism of riluzole: Direct inhibition of protein kinase C. Neurobiol. Dis. 2000, 7, 375–383. [Google Scholar] [CrossRef]
- Lamanauskas, N.; Nistri, A. Riluzole blocks persistent Na+ and Ca2+ currents and modulates release of glutamate via presynaptic NMDA receptors on neonatal rat hypoglossal motoneurons in vitro. Eur. J. Neurosci. 2008, 27, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Yoo, M.H.; Hyun, H.J.; Kob, J.Y.; Yoon, Y.H. Riluzole inhibits VEGF-induced endothelial cell proliferation in vitro and hyperoxia-induced abnormal vessel formation in vivo. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4780–4787. [Google Scholar] [CrossRef] [PubMed]
- Ekokoski, E.; Aitio, O.; Törnquist, K.; Yli-Kauhaluoma, J.; Tuominen, R.K. HIV-1 Tat-peptide inhibits protein kinase C and protein kinase A through substrate competition. Eur. J. Pharm. Sci. 2010, 40, 404–411. [Google Scholar] [CrossRef]
- Sajan, M.P.; Nimal, S.; Mastorides, S.; Acevedo-Duncan, M.; Kahn, C.R.; Fields, A.P.; Braun, U.; Leitges, M.; Farese, R.V. Correction of metabolic abnormalities in a rodent model of obesity, metabolic syndrome, and type 2 diabetes mellitus by inhibitors of hepatic protein kinase C-ι. Metabolism 2012, 61, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.M.; Hoshi, N. ATP competitive protein kinase C inhibitors demonstrate distinct state-dependent inhibition. PLoS ONE 2011, 6, e26338. [Google Scholar] [CrossRef]
- Lee, A.M.; Kanter, B.R.; Wang, D.; Lim, J.P.; Zou, M.E.; Qiu, C.; McMahon, T.; Dadgar, J.; Fischbach-Weiss, S.C.; Messing, R.O. Prkcz null mice show normal learning and memory. Nature 2013, 493, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Wu-Zhang, A.X.; Schramm, C.L.; Nabavi, S.; Malinow, R.; Newton, A.C. Cellular pharmacology of protein kinase Mζ (PKMζ) contrasts with its in vitro profile: Implications for PKMζ as a mediator of memory. J. Biol. Chem. 2012, 287, 2879–12885. [Google Scholar] [CrossRef] [PubMed]
- Tsai, L.C.; Xie, L.; Dore, K.; Xie, L.; Del Rio, J.C.; King, C.C.; Martinez-Ariza, G.; Hulme, C.; Malinow, R.; Bourne, P.E.; et al. Zeta inhibitory peptide disrupts electrostatic interactions that maintain atypical protein kinase C in its active conformation on the scaffold p62. J. Biol. Chem. 2015, 290, 21845–21856. [Google Scholar] [CrossRef] [PubMed]
- Ling, D.S.; Benardo, L.S.; Serrano, P.A.; Blace, N.; Kelly, M.T.; Crary, J.F.; Sacktor, T.C. Protein kinase Mζ is necessary and sufficient for LTP maintenance. Nat. Neurosci. 2002, 5, 295–296. [Google Scholar] [CrossRef] [PubMed]
- Volk, L.J.; Bachman, J.L.; Johnson, R.; Yu, Y.; Huganir, R.L. PKM-ζ is not required for hippocampal synaptic plasticity, learning and memory. Nature 2013, 493, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Tsokas, P.; Hsieh, C.; Yao, Y.; Lesburguères, E.; Wallace, E.J.C.; Tcherepanov, A.; Jothianandan, D.; Hartley, B.R.; Pan, L.; Rivard, B.; et al. Compensation for PKMζ in long-term potentiation and spatial long-term memory in mutant mice. Elife 2016, 5, e14846. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Shao, C.; Jothianandan, D.; Tcherepanov, A.; Shouval, H.; Sacktor, T.C. Matching biochemical and functional efficacies confirm ZIP as a potent competitive inhibitor of PKMζ in neurons. Neuropharmacology 2013, 64, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Sadeh, N.; Verbitsky, S.; Dudai, Y.; Segal, M. Zeta inhibitory peptide, a candidate inhibitor of protein kinase Mζ, is excitotoxic to cultured hippocampal neurons. J. Neurosci. 2015, 35, 12404–12411. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Chen, A.; Chen, Y.; Guo, L.; Dai, H.; Huang, Y.; Chen, Q.; Lin, C. Zeta inhibitory peptide as a novel therapy to control chronic visceral hypersensitivity in a rat model. PLoS ONE 2016, 11, e0163324. [Google Scholar] [CrossRef]
- Yao, J.; Zhang, L.; Lei, W.; Huang, Y.; Zhang, H.; Lu, B.; Yao, Y.; Zhao, L.; Sun, J. Intra-amygdala infusion of zeta inhibitory peptide attenuates neuropathic pain but not inflammatory pain in adult rats. Ann. Palliat. Med. 2019, 8, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zheng, R.; Ma, X.; Gong, Z.; Xia, D.; Zhou, Q. Elevated level of PKMζ underlies the excessive anxiety in an autism model. Front. Mol. Neurosci. 2019, 12, 291. [Google Scholar] [CrossRef]
- Opendak, M.; Zanca, R.M.; Anane, E.; Serrano, P.A.; Sullivan, R.M. Developmental transitions in amygdala PKC isoforms and AMPA receptor expression associated with threat memory in infant rats. Sci. Rep. 2018, 8, 14679. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ouyang, Z.; Lipina, T.V.; Wang, H.; Zhou, Q. Conditioned stimulus presentations alter anxiety level in fear-conditioned mice. Mol. Brain 2019, 12, 28. [Google Scholar] [CrossRef]
- Brückle, W.; Dexel, T.; Grasedyck, K.; Schattenkirchner, M. Treatment of psoriatic arthritis with auranofin and gold sodium thiomalate. Clin. Rheumatol. 1994, 13, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Jessop, J.D.; O’Sullivan, M.M.; Lewis, P.A.; Williams, L.A.; Camilleri, J.P.; Plant, M.J.; Coles, E.C. A long-term five-year randomized controlled trial of hydroxychloroquine, sodium aurothiomalate, auranofin and penicillamine in the treatment of patients with rheumatoid arthritis. Br. J. Rheumatol. 1998, 37, 992–1002. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Erdogan, E.; Lamark, T.; Stallings-Mann, M.; Lee, J.; Pellecchia, M.; Thompson, E.A.; Johansen, T.; Fields, A.P. Aurothiomalate inhibits transformed growth by targeting the PB1 domain of protein kinase Cι. J. Biol. Chem. 2006, 281, 28450–28459. [Google Scholar] [CrossRef]
- Butler, A.M.; Scotti Buzhardt, M.L.; Erdogan, E.; Li, S.; Inman, K.S.; Fields, A.P.; Murray, N.R. A small molecule inhibitor of atypical protein kinase C signaling inhibits pancreatic cancer cell transformed growth and invasion. Oncotarget 2015, 6, 15297–15310. [Google Scholar] [CrossRef] [PubMed]
- Stallings-Mann, M.; Jamieson, L.; Regala, R.P.; Weems, C.; Murray, N.R.; Fields, A.P. A novel small-molecule inhibitor of protein kinase Cι blocks transformed growth of non-small-cell lung cancer cells. Cancer Res. 2006, 66, 1767–1774. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Hu., J.; Wu, S.; Wang, L.; Cao, X.; Zhang, X.; Dai, B.; Cao, M.; Shao, R.; Zhang, R.; et al. Auranofin-mediated inhibition of PI3K/AKT/mTOR axis and anticancer activity in non-small cell lung cancer cells. Oncotarget 2016, 7, 3548. [Google Scholar] [CrossRef] [PubMed]
- Mirabelli, C.K.; Johnson, R.K.; Hill, D.T.; Faucette, L.F.; Girard, G.R.; Kuo, G.Y.; Sung, C.M.; Crooke, S.T. Correlation of the in vitro cytotoxic and in vivo antitumor activities of gold(I) coordination complexes. J. Med. Chem. 1986, 29, 218–223. [Google Scholar] [CrossRef]
- Regala, R.P.; Thompson, E.A.; Fields, A.P. Atypical protein kinase Cι expression and aurothiomalate sensitivity in human lung cancer cells. Cancer Res. 2008, 68, 5888–5895. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, A.S.; Fields, A.P.; Jatoi, A.; Qi, Y.; Adjei, A.A.; Erlichman, C.; Molina, J.R. Phase I dose escalation study of the PKCι inhibitor aurothiomalate for advanced non-small-cell lung cancer, ovarian cancer, and pancreatic cancer. Anticancer Drugs 2013, 24, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Jatoi, A.; Radecki Breitkopf, C.; Foster, N.R.; Block, M.S.; Grudem, M.; Wahner Hendrickson, A.; Carlson, R.E.; Barrette, B.; Karlin, N.; Fields, A.P. A mixed-methods feasibility trial of protein kinase Cι inhibition with auranofin in asymptomatic ovarian cancer patients. Oncology 2015, 88, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Kambhampati, S. Phase I and II Study of Auranofin in Chronic Lymphocytic Leukemia. 2016. Available online: https://clinicaltrials.gov/ct2/show/NCT01419691?term=auranofin&draw=2&rank=4 (accessed on 24 September 2020).
- Pillai, P.; Desai, S.; Patel, R.; Sajan, M.; Farese, R.; Ostrov, D.; Acevedo-Duncan, M. A novel PKC-ι inhibitor abrogates cell proliferation and induces apoptosis in neuroblastoma. Int. J. Biochem. Cell Biol. 2011, 43, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Ratnayake, W.S.; Apostolatos, C.A.; Apostolatos, A.H.; Schutte, R.J.; Huynh, M.A.; Ostrov, D.A.; Acevedo-Duncan, M. Oncogenic PKC-ι activates Vimentin during epithelial-mesenchymal transition in melanoma; a study based on PKC-ι and PKC-ζ specific inhibitors. Cell Adhes. Migr. 2018, 12, 47–463. [Google Scholar] [CrossRef]
- Apostolatos, A.H.; Ratnayake, W.S.; Win-Piazza, H.; Apostolatos, C.A.; Smalley, T.; Kang, L.; Salup, R.; Hill, R.; Acevedo-Duncan, M. Inhibition of atypical protein kinase C-ι effectively reduces the malignancy of prostate cancer cells by downregulating the NF-κB signaling cascade. Int. J. Oncol. 2018, 53, 1836–1846. [Google Scholar] [CrossRef]
- Acevedo-Duncan, M.E. In vivo effects of ICA-1 on breast cancer and glioma xenografts. Cancer Res. 2021, 72 (Suppl. 8), 2827. [Google Scholar]
- Islam, S.M.A.; Patel, R.; Acevedo-Duncan, M. Protein Kinase C-ζ stimulates colorectal cancer cell carcinogenesis via PKC-ζ/Rac1/Pak1/β-Catenin signaling cascade. Biochim. Biophys. Acta Mol. Cell. Res. 2018, 1865, 650–664. [Google Scholar] [CrossRef] [PubMed]
- Smalley, T.; Metcalf, R.; Patel, R.; Islam, S.M.S.; Bommareddy, R.R.; Acevedo-Duncan, M. The atypical protein kinase C small molecule inhibitor ζ-Stat, and its effects on invasion through decreases in PKC-ζ protein expression. Front. Oncol. 2020, 10, 209. [Google Scholar] [CrossRef] [PubMed]
- Ratnayake, W.S.; Apostolatos, A.H.; Ostrov, D.A.; Acevedo-Duncan, M. Two novel atypical PKC inhibitors; ACPD and DNDA effectively mitigate cell proliferation and epithelial to mesenchymal transition of metastatic melanoma while inducing apoptosis. Int. J. Oncol. 2017, 51, 1370–1382. [Google Scholar] [CrossRef]
- Chen, T.C.; Lee, R.A.; Tsai, S.L.; Kanamaluru, D.; Gray, N.E.; Yiv, N.; Cheang, R.T.; Tan, J.H.; Lee, J.Y.; Fitch, M.D.; et al. An ANGPTL4-ceramide-protein kinase Cζ axis mediates chronic glucocorticoid exposure-induced hepatic steatosis and hypertriglyceridemia in mice. J. Biol. Chem. 2019, 294, 9213–9224. [Google Scholar] [CrossRef]
- Sajan, M.P.; Ivey, R.A.; Lee, M.C.; Farese, R.V. Hepatic insulin resistance in ob/ob mice involves increases in ceramide, aPKC activity, and selective impairment of Akt-dependent FoxO1 phosphorylation. J. Lipid Res. 2015, 56, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Inokuchi, J.; Kawano, T.; Murata, M. Protein kinase Cα as a therapeutic target in cancer. In Protein Kinase C: Emerging Roles and Therapeutic Potential; Pierce, D.N., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2018; pp. 25–47. [Google Scholar]
- Grossman, S.A.; Alavi, J.B.; Supko, J.G.; Carson, K.A.; Priet, R.; Dorr, F.A.; Grundy, J.S.; Holmlund, J.T. Efficacy and toxicity of the antisense oligonucleotide aprinocarsen directed against protein kinase C-α delivered as a 21-day continuous intravenous infusion in patients with recurrent high-grade astrocytomas. Neuro-Oncology 2005, 7, 32–40. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Douillard, J.Y.; Koralewski, P.; Manegold, C.; Smit, E.F.; Reyes, J.M.; Chang, G.C.; John, W.J.; Peterson, P.M.; Obasaju, C.K.; et al. Phase III study of gemcitabine and cisplatin with or without aprinocarsen, a protein kinase C-alpha antisense oligonucleotide, in patients with advanced-stage non-small-cell lung cancer. J. Clin. Oncol. 2006, 24, 1428–1434. [Google Scholar] [CrossRef] [PubMed]
- Ritch, P.; Rudin, C.M.; Bitran, J.D.; Edelman, M.J.; Makalinao, A.; Irwin, D.; Lilenbaum, R.; Peterson, P.; John, W.J. Phase II study of PKC-α antisense oligonucleotide aprinocarsen in combination with gemcitabine and carboplatin in patients with advanced non-small cell lung cancer. Lung Cancer 2006, 52, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Advani, R.; Peethambaram, P.; Lum, B.L.; Fisher, G.A.; Hartmann, L.; Long, H.J.; Halsey, J.; Holmlund, J.T.; Dorr, A.; Sikic, B.I. A Phase II trial of aprinocarsen, an antisense oligonucleotide inhibitor of protein kinase C α, administered as a 21-day infusion to patients with advanced ovarian carcinoma. Cancer 2004, 100, 321–326. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Reyno, L.; Venner, P.M.; Ernst, S.D.; Moore, M.; Geary, R.S.; Chi, K.; Hall, S.; Walsh, W.; Dorr, A.; et al. A randomized phase II and pharmacokinetic study of the antisense oligonucleotides ISIS 3521 and ISIS 5132 in patients with hormone-refractory prostate cancer. Clin. Cancer Res. 2002, 8, 2530–2535. [Google Scholar]
- Marshall, J.L.; Eisenberg, S.G.; Johnson, M.D.; Hanfelt, J.; Dorr, F.A.; El-Ashry, D.; Oberst, M.; Fuxman, Y.; Holmlund, J.; Malik, S. A phase II trial of ISIS 3521 in patients with metastatic colorectal cancer. Clin. Colorectal Cancer 2004, 4, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Watkins, D.; Cunningham, D.; Dunlop, D.; Johnson, P.; Selby, P.; Hancock, B.W.; Fegan, C.; Culligan, D.; Schey, S.; et al. Phase II study of ISIS 3521, an antisense oligodeoxynucleotide to protein kinase C alpha, in patients with previously treated low-grade non-Hodgkin’s lymphoma. Ann. Oncol. 2004, 15, 1413–1418. [Google Scholar] [CrossRef] [PubMed]
- Strair, R.K.; Schaar, D.; Goodell, L.; Aisner, J.; Chin, K.V.; Eid, J.; Senzon, R.; Cui, X.X.; Han, Z.T.; Knox, B.; et al. Administration of a phorbol ester to patients with hematological malignancies: Preliminary results from a phase I clinical trial of 12-O-tetradecanoylphorbol-13-acetate. Clin. Cancer Res. 2002, 8, 2512–2518. [Google Scholar]
- Schaar, D.; Goodell, L.; Aisner, J.; Cui, X.X.; Han, Z.T.; Chang, R.; Martin, J.; Grospe, S.; Dudek, L.; Riley, J.; et al. A phase I clinical trial of 12-O-tetradecanoylphorbol-13-acetate for patients with relapsed/refractory malignancies. Cancer Chemother. Pharmacol. 2006, 57, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Kortmansky, J.; Schwartz, G.K. Bryostatin-1: A novel PKC inhibitor in clinical development. Cancer Investig. 2003, 21, 924–936. [Google Scholar] [CrossRef]
- Propper, D.J.; Macaulay, V.; O’Byrne, K.J.; Braybrooke, J.P.; Wilner, S.M.; Ganesan, T.S.; Talbot, D.C.; Harris, A.L. A phase II study of bryostatin 1 in metastatic malignant melanoma. Br. J. Cancer 1998, 78, 1337–1341. [Google Scholar] [CrossRef]
- Bedikian, A.Y.; Plager, C.; Stewart, J.R.; O’Brian, C.A.; Herdman, S.K.; Ross, M.; Papadopoulos, N.; Eton, O.; Ellerhorst, J.; Smith, T. Phase II evaluation of bryostatin-1 in metastatic melanoma Phase II evaluation of bryostatin-1 in metastatic melanoma. Melanoma Res. 2001, 11, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Tozer, R.G.; Burdette-Radoux, S.; Berlanger, K.; Davis, M.L.; Lohmann, R.C.; Rusthoven, J.R.; Wainman, N.; Zee, B.; Seymour, L.; National Cancer Institute of Canada Clinical Trials Group. A randomized phase II study of two schedules of bryostatin-1 (NSC339555) in patients with advanced malignant melanoma: A National Cancer Institute of Canada Clinical Trials Group Study. Investig. New Drugs 2002, 20, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Pagliaro, L.; Daliani, D.; Amato, R.; Tu, S.M.; Jones, D.; Smith, T.; Logothetis, C.; Millikan, R. A phase II trial of bryostatin-1 for patients with metastatic renal cell carcinoma. Cancer 2000, 89, 615–618. [Google Scholar] [CrossRef]
- Madhusudan, S.; Protheroe, A.; Propper, D.; Han, C.; Corrie, P.; Earl, H.; Hancock, B.; Vasey, P.; Turner, A.; Balkwill, F.; et al. A multicentre phase II trial of bryostatin-1 in patients with advanced renal cancer. Br. J. Cancer 2003, 89, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Zonder, J.A.; Shields, A.F.; Zalupski, M.; Chaplen, R.; Heilbrun, L.K.; Arlauskas, P.; Philip, P.A. A phase II trial of bryostatin 1 in the treatment of metastatic colorectal cancer. Clin. Cancer Res. 2001, 7, 38–42. [Google Scholar] [PubMed]
- Blackhall, F.H.; Ranson, M.; Radford, J.A.; Hancock, B.W.; Soukop, M.; McGown, A.T.; Robbins, A.; Halbert, G.; Jayson, G.C.; Cancer Research Campaign Phase I/II Committee. A phase II trial of bryostatin 1 in patients with non-Hodgkin’s lymphoma. Br. J. Cancer 2001, 84, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Varterasian, M.L.; Pemberton, P.A.; Hulburd, K.; Rodriguez, D.H.; Murgo, A.; Al-Katib, A.M. Phase II study of bryostatin 1 in patients with relapsed multiple myeloma. Investig. New Drugs 2001, 19, 245–247. [Google Scholar] [CrossRef]
- Brockstein, B.; Samuels, B.; Humerickhouse, R.; Arietta, R.; Fishkin, P.; Wade, J.; Sosman, J.; Vokes, E.E. Phase II studies of bryostatin-1 in patients with advanced sarcoma and advanced head and neck cancer. Investig. New Drugs 2001, 19, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Pfister, D.G.; McCaffrey, J.; Zahalsky, A.J.; Schwartz, G.K.; Lis, E.; Gerald, W.; Huvos, A.; Shah, J.; Kraus, D.; Shaha, A.; et al. A phase II trial of bryostatin-1 in patients with metastatic or recurrent squamous cell carcinoma of the head and neck. Investig. New Drugs 2002, 20, 123–127. [Google Scholar] [CrossRef]
- Armstrong, D.K.; Blessing, J.A.; Rader, J.; Sorosky, J.L.; Gynecologic Oncology Group Study. A randomized phase II evaluation of bryostatin-1 (NSC #339555) in persistent or recurrent squamous cell carcinoma of the cervix: A Gynecologic Oncology Group Study. Investig. New Drugs 2003, 21, 453–457. [Google Scholar] [CrossRef]
- Clamp, A.R.; Blackhall, F.H.; Vasey, P.; Soukop, M.; Coleman, R.; Halbert, G.; Robson, L.; Jayson, G.C.; Cancer Research UK Phase I/II Committee. A phase II trial of bryostatin-1 administered by weekly 24-hour infusion in recurrent epithelial ovarian carcinoma. Br. J. Cancer 2003, 89, 1152–1154. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.P.; Sparano, J.A.; Vinciguerra, V.; Ocean, A.J.; Christos, P.; Hochster, H.; Camacho, F.; Goel, S.; Mani, S.; Kaubisch, A. Phase II study of paclitaxel plus the protein kinase C inhibitor bryostatin-1 in advanced pancreatic carcinoma. Am. J. Clin. Oncol. 2010, 33, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Winegarden, J.D.; Mauer, A.M.; Gajewski, T.F.; Hoffman, P.C.; Krauss, S.; Rudin, C.M.; Vokes, E.E. A phase II study of bryostatin-1 and paclitaxel in patients with advanced non-small cell lung cancer. Lung Cancer 2003, 39, 191–196. [Google Scholar] [CrossRef]
- Nezhat, F.; Wadler, S.; Muggia, F.; Mandeli, J.; Goldberg, G.; Rahaman, J.; Runowicz, C.; Murgo, A.J.; Gardner, G.J. Phase II trial of the combination of bryostatin-1 and cisplatin in advanced or recurrent carcinoma of the cervix: A New York Gynecologic Oncology Group study. Gynecol. Oncol. 2004, 93, 144–148. [Google Scholar] [CrossRef]
- Peterson, A.C.; Harlin, H.; Karrison, T.; Vogelzang, N.J.; Knost, J.A.; Kugler, J.W.; Lester, E.; Vokes, E.; Gajewski, T.F.; Stadler, W.M.; et al. A randomized phase II trial of interleukin-2 in combination with four different doses of bryostatin-1 in patients with renal cell carcinoma. Investig. New Drugs 2006, 24, 141–149. [Google Scholar] [CrossRef]
- Ajani, J.A.; Jiang, Y.; Faust, J.; Chang, B.B.; Ho, L.; Yao, J.C.; Rousey, S.; Dakhil, S.; Cherny, R.C.; Craig, C.; et al. A multi-center phase II study of sequential paclitaxel and bryostatin-1 (NSC 339555) in patients with untreated, advanced gastric or gastroesophageal junction adenocarcinoma. Investig. New Drugs 2006, 24, 353–357. [Google Scholar] [CrossRef]
- Ku, G.Y.; Ilson, D.H.; Schwartz, L.H.; Capanu, M.; O’Reilly, E.; Shah, M.A.; Kelsen, D.P.; Schwartz, G.K. Phase II trial of sequential paclitaxel and 1 h infusion of bryostatin-1 in patients with advanced esophageal cancer. Cancer Chemother. Pharmacol. 2008, 62, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.J.; Leong, L.; Chow, W.; Gandara, D.; Frankel, P.; Garcia, A.; Lenz, H.J.; Doroshow, J.H. Phase II trial of bryostatin-1 in combination with cisplatin in patients with recurrent or persistent epithelial ovarian cancer: A California cancer consortium study. Investig. New Drugs 2012, 30, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Barr, P.M.; Lazarus, H.M.; Cooper, B.W.; Schluchter, M.D.; Panneerselvam, A.; Jacobberger, J.W.; Hsu, J.W.; Janakiraman, N.; Simic, A.; Dowlati, A.; et al. Phase II study of bryostatin 1 and vincristine for aggressive non-Hodgkin lymphoma relapsing after an autologous stem cell transplant. Am. J. Hematol. 2009, 84, 484–487. [Google Scholar] [CrossRef]
- Hongpaisan, J.; Sun, M.K.; Alkon, D.L. PKC ε activation prevents synaptic loss, Aβ elevation, and cognitive deficits in Alzheimer’s disease transgenic mice. J. Neurosci. 2011, 31, 630–643. [Google Scholar] [CrossRef]
- Sen, A.; Nelson, T.J.; Alkon, D.L.; Hongpaisan, J. Loss in PKCɛ causes downregulation of MnSOD and BDNF expression in neurons of Alzheimer’s disease hippocampus. J. Alzheimers Dis. 2018, 63, 1173–1189. [Google Scholar] [CrossRef]
- Zhu, G.; Wang, D.; Lin, Y.H.; McMahon, T.; Koo, E.H.; Messing, R.O. Protein kinase C ϵ suppresses Aβ production and promotes activation of α-secretase. Biochem. Biophys. Res. Commun. 2001, 285, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.J.; Sun, M.K.; Lim, C.; Sen, A.; Khan, T.; Chirila, F.V.; Alkon, D.L. Bryostatin effects on cognitive function and PKCɛ in Alzheimer’s disease phase IIa and expanded access trials. J. Alzheimers Dis. 2017, 58, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Farlow, M.R.; Thompson, R.E.; Wei, L.J.; Tuchman, A.J.; Grenier, E.; Crockford, D.; Wilke, S.; Benison, J.; Alkon, D.L. A randomized, double-blind, placebo-controlled, phase II study assessing safety, tolerability, and efficacy of bryostatin in the treatment of moderately severe to severe Alzheimer’s disease. J. Alzheimers Dis. 2019, 67, 555–570. [Google Scholar] [CrossRef] [PubMed]
- Mehla, R.; Bivalkar-Mehla, S.; Zhang, R.; Handy, I.; Albrecht, H.; Giri, S.; Nagarkatti, P.; Nagarkatti, M.; Chauhan, A. Bryostatin modulates latent HIV-1 infection via PKC and AMPK signaling but inhibits acute infection in a receptor independent manner. PLoS ONE 2010, 5, e11160. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.; de Vinuesa, A.G.; Sanchez-Duffhues, G.; Marquez, N.; Bellido, M.L.; Muñoz-Fernandez, M.A.; Moreno, S.; Castor, T.P.; Calzado, M.A.; Muñoz, E. Bryostatin-1 synergizes with histone deacetylase inhibitors to reactivate HIV-1 from latency. Curr. HIV Res. 2010, 8, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, C.; Serrano-Villar, S.; Madrid-Elena, N.; Pérez-Elías, M.J.; Martín, M.E.; Barbas, C.; Ruipérez, J.; Muñoz, E.; Muñoz-Fernández, M.A.; Castor, T.; et al. Bryostatin-1 for latent virus reactivation in HIV-infected patients on antiretroviral therapy. AIDS 2016, 30, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.K.; Sen, A.; Hongpaisan, J.; Lim, C.S.; Nelson, T.J.; Alkon, D.L. PKCε deficits in Alzheimer’s disease brains and skin fibroblasts. J. Alzheimers Dis. 2015, 43, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Talman, V.; Pascale, A.; Jäntti, M.; Amadio, M.; Tuominen, R.K. Protein kinase C activation as a potential therapeutic strategy in Alzheimer’s disease: Is there a role for embryonic lethal abnormal vision-like proteins? Basic Clin. Pharmacol. Toxicol. 2016, 119, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Zhang, H. Par3 and aPKC regulate BACE1 endosome-to-TGN trafficking through PACS1. Neurobiol. Aging 2017, 60, 129–140. [Google Scholar] [CrossRef]
- Sajan, M.P.; Hansen, B.C.; Higgs, M.G.; Kahn, C.R.; Braun, U.; Leitges, M.; Park, C.R.; Diamond, D.M.; Farese, R.V. Atypical PKC, PKCλ/ι, activates β-secretase and increases Aβ1–40/42 and phospho-tau in mouse brain and isolated neuronal cells, and may link hyperinsulinemia and other aPKC activators to development of pathological and memory abnormalities in Alzheimer’s disease. Neurobiol. Aging 2018, 61, 225–237. [Google Scholar] [CrossRef]
- Du, Y.; Zhao, Y.; Li, C.; Zheng, Q.; Tian, J.; Li, Z.; Huang, T.Y.; Zhang, W.; Xu, H. Inhibition of PKCδ reduces amyloid-β levels and reverses Alzheimer disease phenotypes. J. Exp. Med. 2018, 215, 1665–1677. [Google Scholar] [CrossRef]
- Boucau, J.; Das, J.; Joshi, N.; Le Gall, S. Latency reversal agents modulate HIV antigen processing and presentation to CD8 T cells. PLoS Pathog. 2020, 16, e1008442. [Google Scholar] [CrossRef] [PubMed]
- Sloane, J.L.; Benner, N.L.; Keenan, K.N.; Zang, X.; Soliman, M.S.A.; Wu, X.; Dimapasoc, M.; Chun, T.W.; Marsden, M.D.; Zack, J.A.; et al. Prodrugs of PKC modulators show enhanced HIV latency reversal and an expanded therapeutic window. Proc. Natl. Acad. Sci. USA 2020, 117, 10688–10698. [Google Scholar] [CrossRef]
- Zaikos, T.D.; Painter, M.M.; Sebastian Kettinger, N.T.; Terry, V.H.; Collins, K.L. Class 1-Selective Histone Deacetylase (HDAC) Inhibitors enhance HIV latency reversal while preserving the activity of HDAC isoforms necessary for maximal HIV gene expression. J. Virol. 2018, 92, e02110-17. [Google Scholar] [CrossRef] [PubMed]
- De la Torre-Tarazona, H.E.; Jiménez, R.; Bueno, P.; Camarero, S.; Román, L.; Fernández-García, J.L.; Beltrán, M.; Nothias, L.F.; Cachet, X.; Paolini, J.; et al. 4-Deoxyphorbol inhibits HIV-1 infection in synergism with antiretroviral drugs and reactivates viral reservoirs through PKC/MEK activation synergizing with vorinostat. Biochem. Pharmacol. 2020, 177, 113937. [Google Scholar] [CrossRef]
- Palaniyandi, S.S.; Sun, L.; Ferreira, J.C.; Mochly-Rosen, D. Protein kinase C in heart failure: A therapeutic target? Cardiovasc. Res. 2009, 82, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.M.; Cummings, E.; Pantos, C.; Singh, J. Protein kinase C and cardiac dysfunction: A review. Heart Fail. Rev. 2017, 22, 843–859. [Google Scholar] [CrossRef]
- Bates, E.; Bode, C.; Costa, M.; Gibson, C.M.; Granger, C.; Green, C.; Grimes, K.; Harrington, R.; Huber, K.; Kleiman, N.; et al. Intracoronary KAI-9803 as an adjunct to primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction. Circulation 2008, 117, 886–896. [Google Scholar] [CrossRef]
- Garg, R.; Benedetti, L.G.; Abera, M.B.; Wang, H.; Abba, M.; Kazanietz, M.G. Protein kinase C and cancer: What we know and what we do not. Oncogene 2014, 33, 5225–5237. [Google Scholar] [CrossRef]
- Cooke, M.; Magimaidas, A.; Casado-Medrano, V.; Kazanietz, M.G. Protein kinase C in cancer: The top five unanswered questions. Mol. Carcinog. 2017, 56, 1531–1542. [Google Scholar] [CrossRef] [PubMed]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Sapon-Cousineau, V.; Sapon-Cousineau, S.; Assouline, S. PI3K inhibitors and their role as novel agents for targeted therapy in lymphoma. Curr. Treat Options Oncol. 2020, 21, 51. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, D.A.; Sagrillo, F.S.; Fraga, C.A.M. Duvelisib: A 2018 novel FDA-approved small molecule inhibiting phosphoinositide 3-kinases. Pharmaceuticals 2019, 12, 69. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diseases | PKC Activators or Inhibitors/Other Agents | Phases | Clinical Benefits | References |
|---|---|---|---|---|
| Cancer | ||||
| Metastatic triple negative breast cancer | UCN-01/irinotecan | II | No significant clinical benefits | [48] |
| Advanced ovarian cancer | UCN-01/topotecan | II | No significant clinical benefits | [51] |
| Mutant FLT3-positive acute myeloid leukemia | Midostaurin/standard chemotherapy | III | Significantly prolonged overall and event-free survival | [56] |
| Advanced systemic mastocytosis | Midostaurin | II | Significant clinical benefits and no unexpected toxicity | [61] |
| High-risk diffuse large B-cell lymphoma | Enzastaurin (LY317615) | III | No significant clinical benefits | [80] |
| Multiple myeloma, lung cancer with brain metastases, epithelial ovarian or primary peritoneal carcinoma, metastatic breast cancer, and relapsed or refractory mantle cell lymphoma and advanced cutaneous T-cell lymphoma | Enzastaurin | II | No significant clinical benefits | [81,82,83,84,85,86] |
| Advanced non-small-cell lung cancer | Enzastaurin/erlotinib | II | No significant clinical benefits | [87] |
| Glioblastoma multiforme and gliosarcoma | Enzastaurin/temozolomide + radiation therapy | II | No significant clinical benefits | [88] |
| Castration-resistant metastatic prostate cancer | Enzastaurin/docetaxel + prednisone | II | No significant clinical benefits | [89] |
| Advanced ovarian cancer | Enzastaurin/paclitaxel + carboplatin | II | No significant clinical benefits | [90] |
| Metastatic colorectal cancer | Enzastaurin/5-fluorouracil + leucovorin + bevacizumab | II | No significant clinical benefits | [91] |
| Advanced non-small cell lung cancer | Enzastaurin/pemetrexed | II | No significant clinical benefits | [92] |
| Advanced or metastatic pancreatic cancer | Enzastaurin/gemcitabine | II | No significant clinical benefits | [93] |
| Metastatic malignant melanoma, renal cell carcinoma, and colorectal cancer, non-Hodgkin’s lymphoma, relapsed multiple myeloma, advanced sarcoma and advanced head and neck cancer, metastatic or recurrent squamous cell carcinoma of the head and neck, squamous cell carcinoma of the cervix, and recurrent epithelial ovarian carcinoma | Bryostatin-1 | II | No significant clinical benefits | [205,206,207,208,209,210,211,212,213,214] |
| Advanced pancreatic carcinoma, non-small cell lung cancer, and esophageal cancer, advanced or recurrent carcinoma of the cervix, and advanced gastric or gastroesophageal junction adenocarcinoma | Bryostatin-1/paclitaxel | II | No significant clinical benefits | [215,216,217,219,220] |
| Renal cell carcinoma | Bryostatin-1/interleukin-2 | II | No significant clinical benefits | [218] |
| Recurrent platinum-sensitive or resistant ovarian cancer | Bryostatin-1/cisplatin | II | Modest response rate but high toxicity in platinum-pretreated patients | [221] |
| Aggressive B-cell non-Hodgkin lymphoma relapsing after autologous stem cell transplantation | Bryostatin-1/vincristine | II | Overall response rate of 31% (efficacy in select patients) | [222] |
| Diabetic retinopathy and neuropathy | ||||
| Moderate to severe non-proliferative diabetic retinopathy | Ruboxistaurin (LY 333531) | III | Reduced occurrence of sustained moderate vision loss but not significant | [106] |
| Diabetic retinopathy (retinopathy level 20 to 47D or 35B to 53E) | Ruboxistaurin | III | Approximately 50% reduction of sustained moderate vision loss but not significant | [107] |
| Diabetes and symptomatic diabetic peripheral neuropathy | Ruboxistaurin | III | No significant and progressive improvement in symptoms | [108] |
| Neurological diseases | ||||
| Alzheimer’s disease | Bryostatin-1 | IIa | Cognitive improvement in the first 24 weeks | [226] |
| Bryostatin-1 | II | Improved the full analysis set and the Severe Impairment Battery scores at 15 weeks | [227] | |
| Postherpetic neuralgia | KAI-1678 | II | No significant reduction in pain intensity | [141] |
| Transplant rejection | ||||
| De novo kidney transplantation | Sotrastaurin (AEB071) | II | Low efficacy and high adverse events | [114] |
| De novo kidney transplantation | Sotrastaurin/tacrolimus | II | Limited benefits over standard immunosuppressive therapy and high adverse events | [116] |
| De novo kidney transplantation | Sotrastaurin/everolimus | II | High efficacy failure rates and adverse events | [117] |
| De novo liver transplantation | Sotrastaurin/tacrolimus | II | High efficacy failure rates and adverse events | [115] |
| Cardiovascular diseases | ||||
| Myocardial infarction | Delcasertib (KAI-9803) | II | No significant clinical benefits | [140] |
| Infections | ||||
| Human immunodeficiency virus (HIV) infection | Bryostatin-1 | I | No effect on the transcription of latent HIV-1 | [230] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawano, T.; Inokuchi, J.; Eto, M.; Murata, M.; Kang, J.-H. Activators and Inhibitors of Protein Kinase C (PKC): Their Applications in Clinical Trials. Pharmaceutics 2021, 13, 1748. https://doi.org/10.3390/pharmaceutics13111748
Kawano T, Inokuchi J, Eto M, Murata M, Kang J-H. Activators and Inhibitors of Protein Kinase C (PKC): Their Applications in Clinical Trials. Pharmaceutics. 2021; 13(11):1748. https://doi.org/10.3390/pharmaceutics13111748
Chicago/Turabian StyleKawano, Takahito, Junichi Inokuchi, Masatoshi Eto, Masaharu Murata, and Jeong-Hun Kang. 2021. "Activators and Inhibitors of Protein Kinase C (PKC): Their Applications in Clinical Trials" Pharmaceutics 13, no. 11: 1748. https://doi.org/10.3390/pharmaceutics13111748
APA StyleKawano, T., Inokuchi, J., Eto, M., Murata, M., & Kang, J.-H. (2021). Activators and Inhibitors of Protein Kinase C (PKC): Their Applications in Clinical Trials. Pharmaceutics, 13(11), 1748. https://doi.org/10.3390/pharmaceutics13111748