1. Introduction
Therapeutic RNA interference (RNAi) relies on the principle of silencing disease-related genes in a sequence-specific manner. After two decades of research, this promising curative modality has become a reality, since three siRNA therapeutics have been approved, i.e., ONPATTRO
® (patisiran, Sanofi Genzyme) in 2018 [
1], GIVLAARI
® (givosiran, Alnylam Pharmaceuticals) in 2019 [
2], both being commercialized, and finally OXLUMO
® (lumasiran, Alnylam Pharmaceuticals) in 2020 [
3]. In ONPATTRO
®, siRNA is formulated in lipid nanoparticles for the intravenous treatment of hereditary amyloidogenic transthyrethin amyloidosis with polyneuropathy in adults [
2]. GIVLAARI
®, an aminolevulinate synthase 1 (ALAS1)-directed siRNA used to treat acute hepatic porphyria in adults [
4], and OXLUMO
®, an siRNA targeting the mRNA for the hydroxyacid oxidase 1 gene to treat primary hyperoxaluria type 1 [
3], are both covalently linked to a ligand enabling their specific delivery to hepatocytes (Gal-NAc-siRNA conjugates), and are administered by subcutaneous injection.
The administration of nonconjugated siRNAs necessitates their formulation into vectors in order to achieve cellular delivery, since these negatively charged and water-soluble molecules, with high molecular weight and very low membrane permeability, are unable to cross cell membranes on their own. Currently, the administration route is essentially parenteral, but the oral route of delivery may be considered according to its advantages over the injectable route, i.e., ease of administration, non-invasive and painless for the patient, higher compliance, reduced health-care cost, and possibility to cure local and systemic diseases [
5]. Generally, oral delivery of drugs represents a real challenge since physiological and cellular barriers have to be overcome. The physiological barriers include (i) the pH variations along the gastrointestinal tract, ranging from harsh acidic environment in the stomach to 5–9 pH values within the intestine, (ii) the presence of bile salts and degradative enzymes such as pepsin, amylase, trypsin, lipase, (iii) fluid flow and peristaltic activity, (iv) glycocalyx and mucus layers. The cellular barriers include the intestinal mucosa (comprising three layers, i.e., epithelium, lamina propria, and muscularis mucosa), the lymphoid follicles and Peyer patches. Specifically speaking of siRNAs, under their naked form, they would theoretically not be altered at acidic pH, nor be sensitive to proteolytic enzymes such pepsin in the stomach and trypsin in the intestine, nor to glycoside hydrolases such as amylase. However, siRNAs may be degraded by endonucleases within the gastrointestinal lumen [
6,
7,
8]. As for lipid nanoparticles of siRNA, they could be altered by lipases, or retained by the mucus. Finally, the cellular barriers of the intestinal mucosa could represent either an obstacle to the absorption of siRNA vectors intended for a systemic effect, or a target for the local treatment of gastrointestinal diseases [
9].
Although challenging, the oral route represents an emerging route for the administration of siRNAs, and their fate in gastrointestinal conditions has been explored, either in vitro with phosphate buffers at pH 2–4.5–7 [
10], or with simulated gastric fluid SGF and simulated intestinal fluid SIF [
11,
12,
13,
14], or ex vivo with gastric and intestinal fluids harvested from rodents [
15,
16,
17], showing that siRNAs formulated into nanoparticles (NPs) could resist, to some extent, the gastrointestinal challenges. In addition, considering administered
per os in preclinical studies, some of these siRNA NPs allowed small intestine and colon epithelial cell entry [
14], or systemic translocation into liver, spleen, lung or kidney [
11,
15], and even distant biological effects in liver [
15,
16,
17,
18]. For in vivo studies, the siRNAs NPs were administered either in liquid colloidal suspensions, or loaded in microspheres (reviewed in [
5,
19]), or encapsulated in exosomes [
10]. These formulations were administered by gavage in rodents.
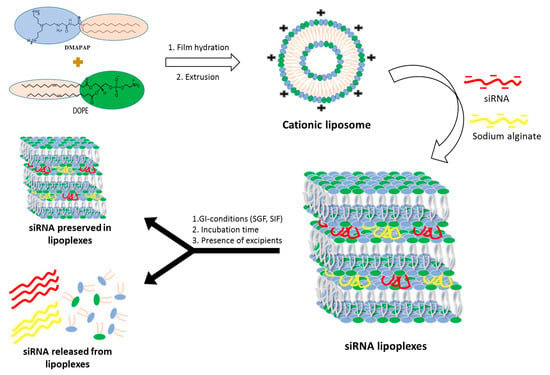
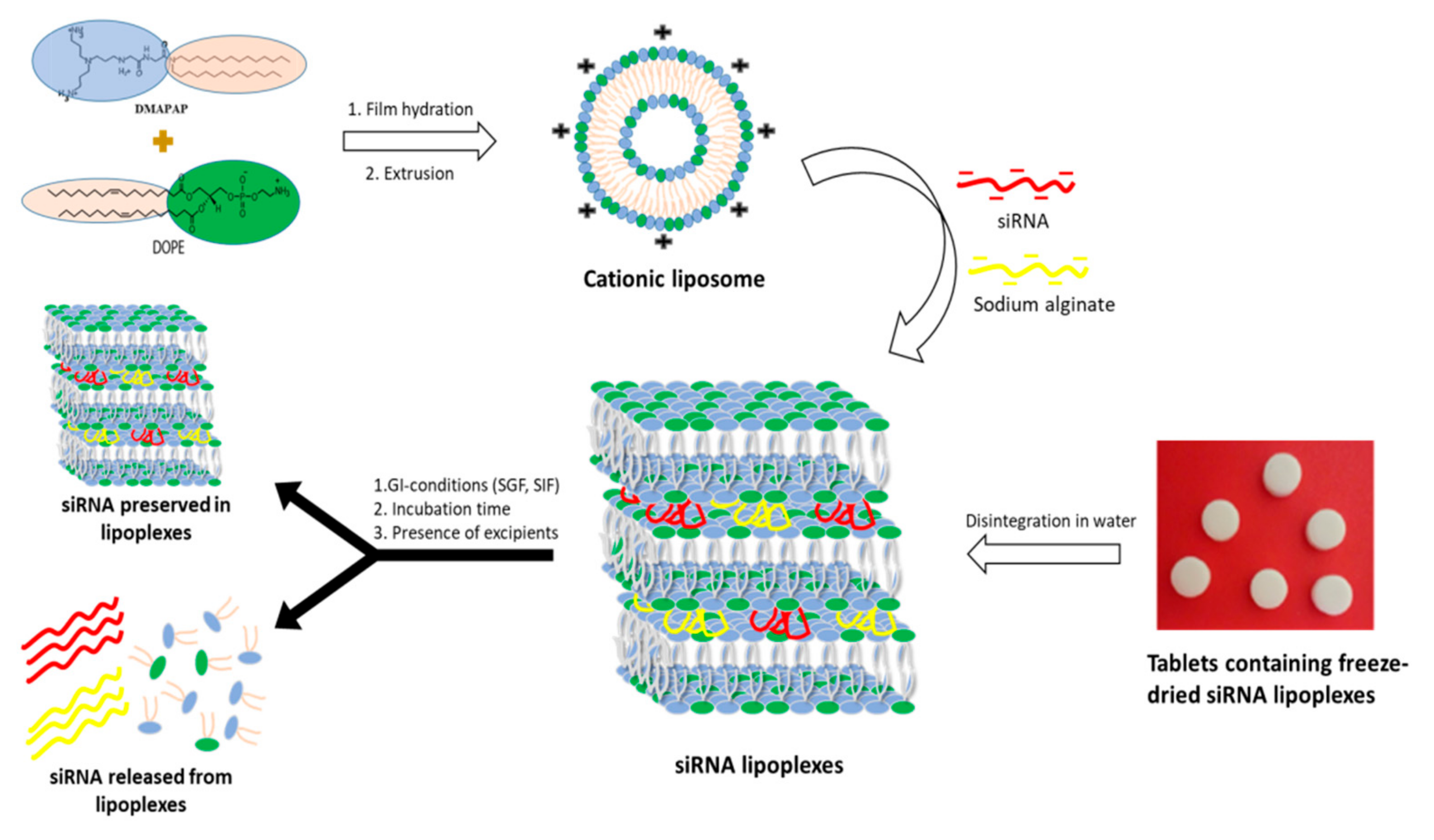
Beside these forms, our team recently patented an innovative dosage form consisting of tablets containing freeze-dried siRNA lipoplexes [
20] that retained the bilayer lipid structures of the particles as well as their gene silencing efficacy upon the freeze-drying and tableting processes [
21], thus opening promising perspectives to oral administration of siRNA-based medicines. However, there is to date no data available regarding the stability of freeze-dried and tableted siRNA lipoplexes in gastrointestinal conditions, which is a prerequisite to their efficacy in vivo.
Therefore, the objectives of the present work were to determine the stability of naked siRNA and siRNA lipoplex regarding the physiological challenges of pH and enzymes—apart from any cellular barrier—in vitro in simulated gastric and intestinal fluids. Preliminarily, the effect of the pH value, at the step of preparation of the lipoplexes, was evaluated on their size and transfection efficacy in order to set up this parameter. Then, after exposure to gastrointestinal conditions, gel electrophoresis was implemented, siRNA either released from lipoplexes or protected within the lipoplexes was quantified, and siRNA efficacy was assessed in cell transfection. This study was carried out in parallel with naked siRNA in solution, freshly prepared siRNA lipoplexes in colloidal suspension, and tableted freeze-dried siRNA lipoplexes after resuspension, with the perspective to identify whether this new tableted dosage form would require further protection for improving oral delivery of siRNA.
3. Results
Lipoplexes were prepared by mixing a DMAPAP and DOPE liposome suspension with siRNA, sodium alginate and NaCl in aqueous solution as previously reported by our research group [
21,
23,
24]. They were tested as freshly prepared siRNA lipoplexes in colloidal suspension, and tableted, freeze-dried siRNA lipoplexes after resuspension, in comparison with naked siRNA in solution (
Figure 1).
3.1. Disintegration Test
The uncoated tablets must disintegrate within 15 min to meet the standards of United States pharmacopoeia (USP) and European pharmacopoeia (EP). In this case, all six tablets that were obtained under a compression pressure of 150 MPa were disintegrated within five min, 4 min 54 s and 4 min 46 s precisely in water and SGF, respectively. Therefore, the tablets containing freeze-dried siRNA meet the standards of the USP and EP.
3.2. Effect of pH on Formation, Stability, and Gene Silencing Efficacy of Lipoplexes
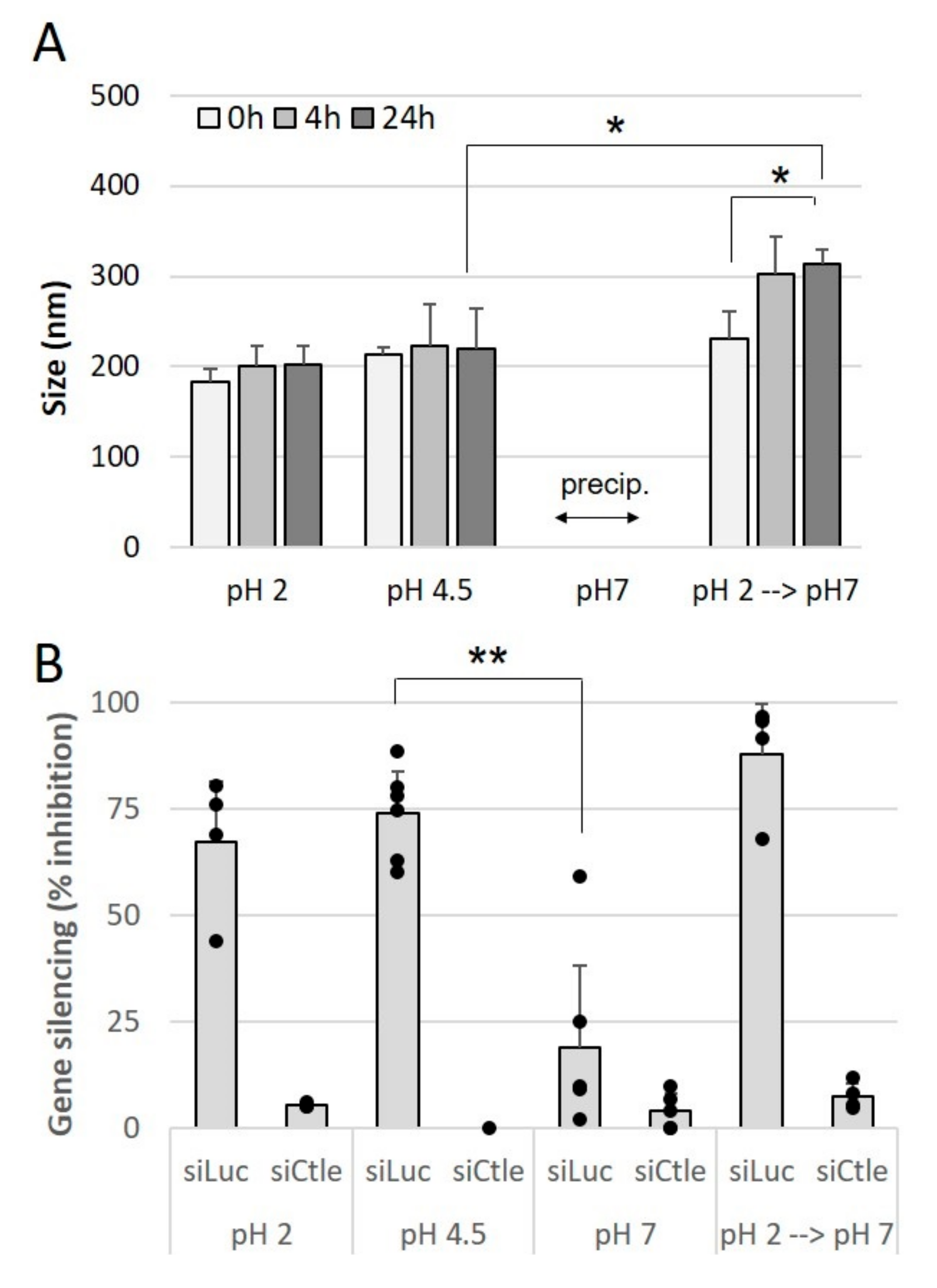
Oral administration presents a number of limitations, one of which is the acidic-to-near-neutral pH of the gastrointestinal tract. We first assessed the impact of preparing and incubating siRNA lipoplexes in acidic pH medium on the size of lipoplexes and efficacy of gene silencing.
The lipoplexes were prepared at different pH levels, namely 2, 4.5 or 7, and their average size was measured immediately after preparation and after 4 h and 24 h of storage at room temperature following their preparation as well.
Figure 3A shows that the lipoplexes prepared at acidic pH, 2 or 4.5, exhibited a size of around 200 nm which remained stable for 24 h. On the other hand, at pH 7, the lipoplexes precipitated immediately after their preparation. When they were prepared at acidic pH and brought to neutral pH, an increase in size, from 200 nm to 300 nm, was observed and these lipoplexes remained stable afterwards. Then, the impact of pH on the gene silencing efficacy of lipoplexes prepared at different pH was evaluated. As shown in
Figure 3B, lipoplexes prepared at acidic pH, 2 or 4.5, exhibited a gene silencing efficacy of around 70%, while lipoplexes prepared at neutral pH only exhibited 20% efficacy. In addition, lipoplexes prepared at pH 2 and passed to neutral pH retained their efficacy. These results show that the most-effective lipoplexes are formed at acidic pH, and these effective lipoplexes, once prepared, retain their efficacy when they are subjected to pH variations.
3.3. Evaluation of the Stability of siRNA and Lipoplexes in Gastrointestinal (GI) Conditions
3.3.1. Effect of Simulated Gastric Conditions on the Stability of Naked siRNA and siRNA Lipoplexes
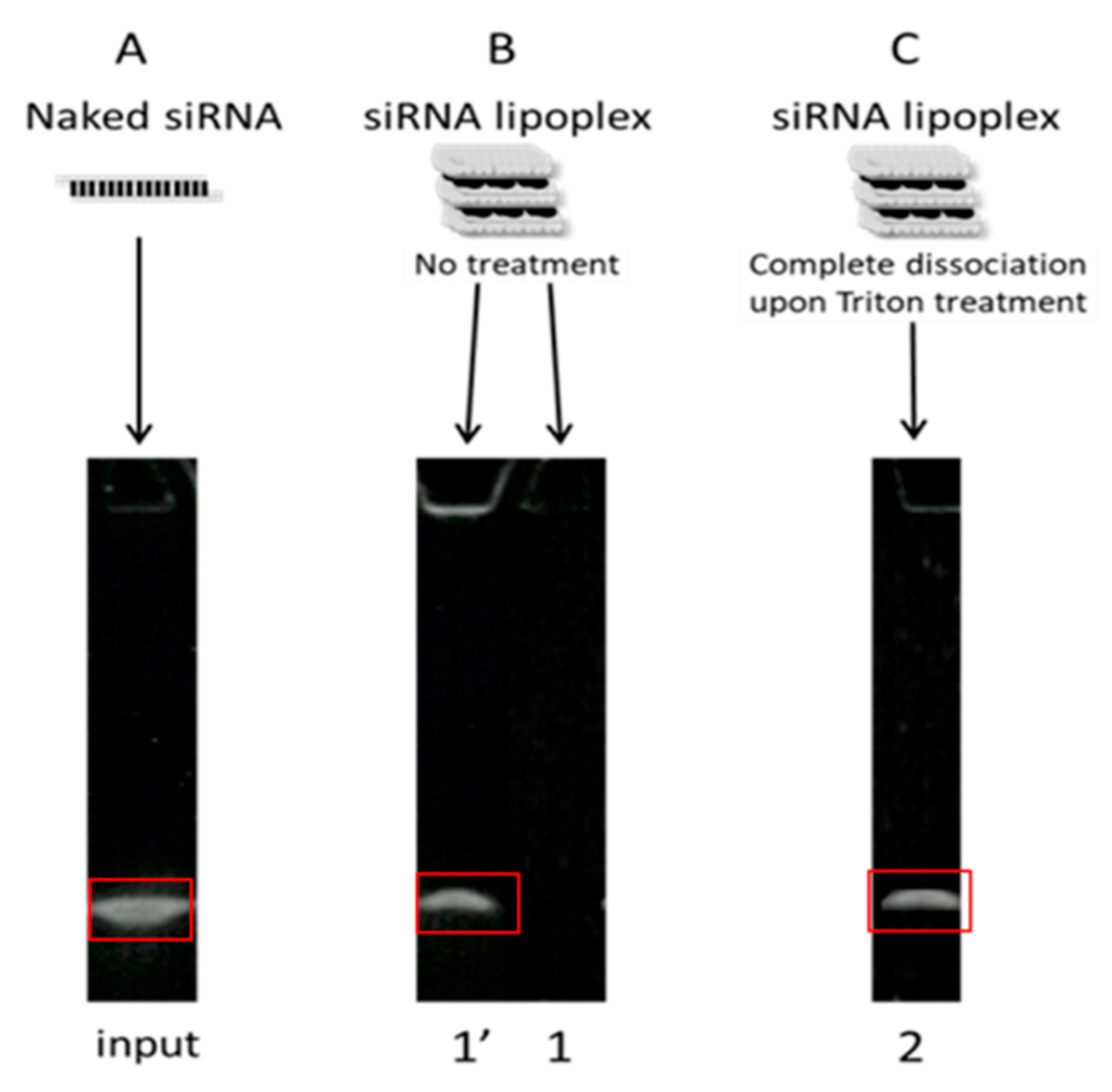
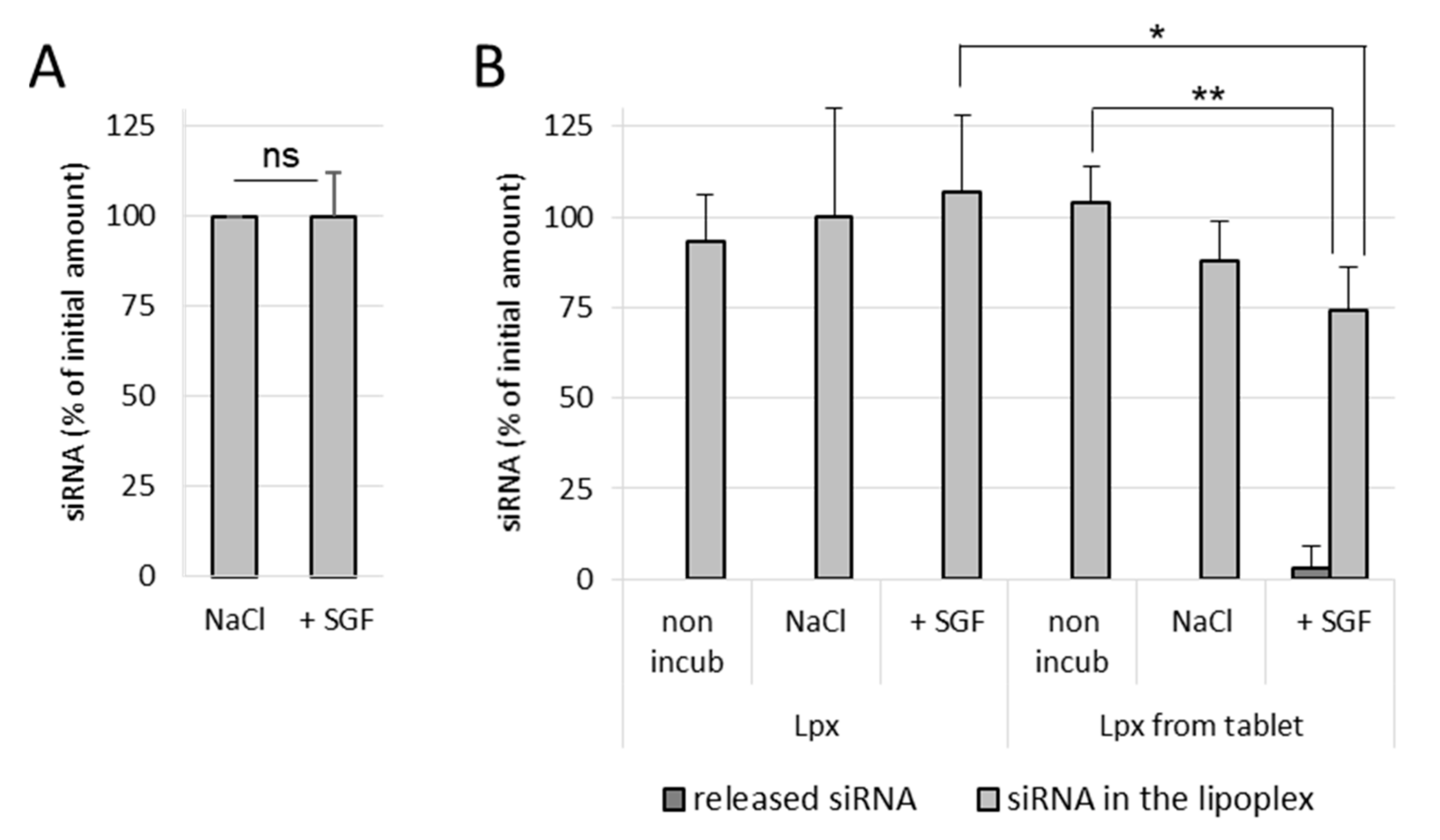
It was observed that naked siRNA remained stable in NaCl as well as in SGF, even after 1 h incubation at 37 °C, which indicates that the gastric conditions do not degrade naked siRNA (
Figure 4A). In NaCl (150 mM)-treated lipoplex samples, no release of siRNA was observed in both incubated as well as nonincubated samples, which shows that the siRNA lipoplexes (fresh lipoplexes and the ones resuspended from tablets) remain stable in NaCl (
Figure 4B). Similarly, fresh siRNA lipoplexes (in suspension) also did not show any release of siRNA after 1 h incubation in SGF with complete preservation of siRNA in lipoplexes. However, in the tableted, freeze-dried siRNA lipoplex (after disintegration of the tablet) samples, a low release of siRNA was obtained after 1 h incubation in SGF with ~74% intact siRNA preserved in lipoplexes. This shows that the siRNA is released to some extent (after 1 h incubation) from the tableted, freeze-dried lipoplexes, but the majority of siRNA content is well preserved in the tablets (
Figure 4B). Thus, it can be concluded that the siRNA and the lipoplexes (fresh as well as tableted) are rather resistant to the gastric conditions despite the pressure constraint applied on tableted siRNA lipoplexes. However, it is not possible to know whether the greater fragility of the lipoplexes resuspended from tablet, shown by the decrease in the percentage of siRNA preserved in the lipoplexes (obtained from the tableted versus the suspension sample), is due to the gastric medium or the presence of excipients (Flowlac
® 37 mg/mL, magnesium stearate 0.8 mg/mL, trehalose 90 mg/mL, mannitol 36 mg/mL).
3.3.2. Effect of Simulated Intestinal Conditions on the Stability of Naked siRNA and siRNA Lipoplexes
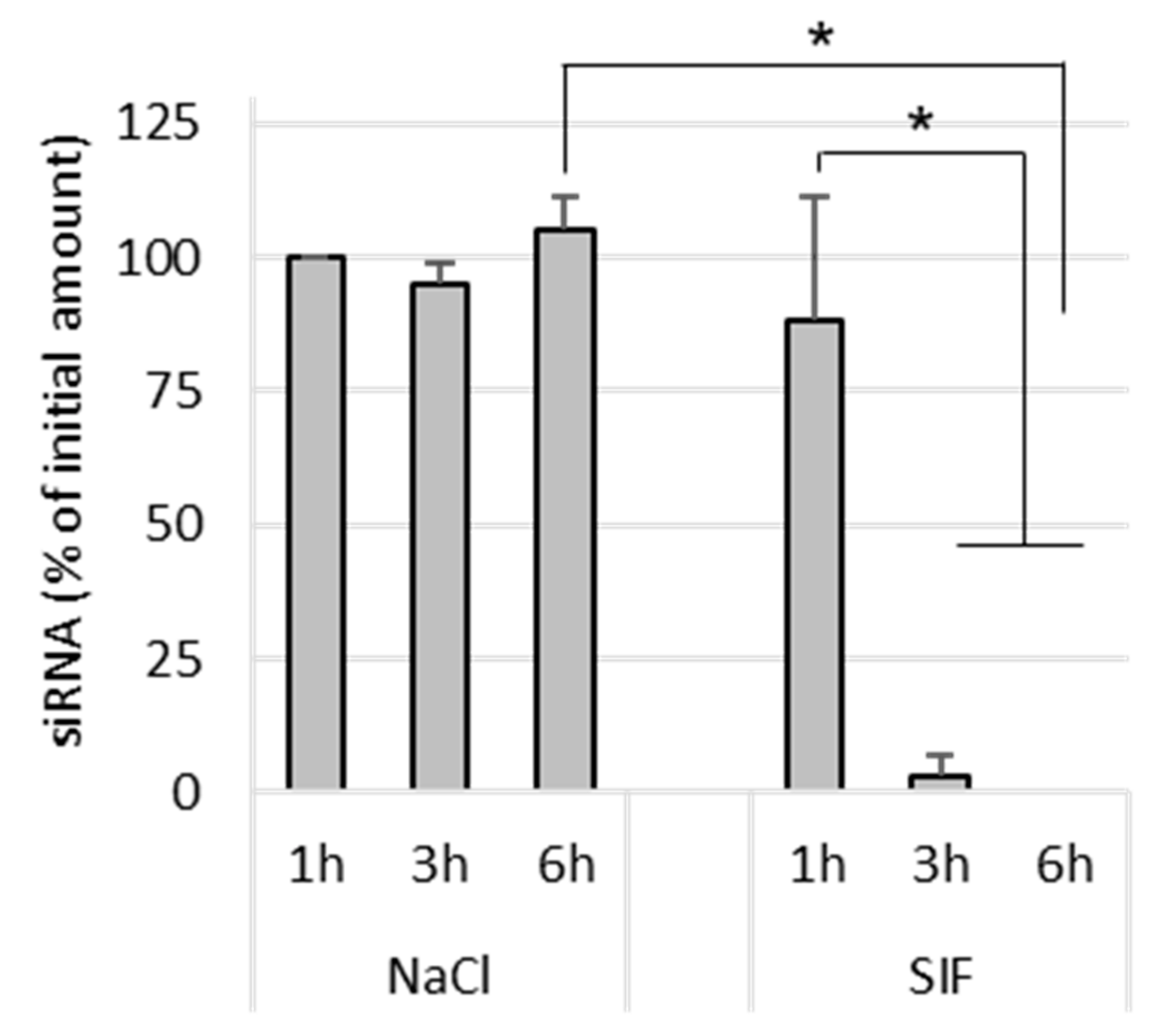
Naked siRNA remained stable in NaCl after 6 h incubation at 37 °C. In SIF-treated samples, naked siRNA was completely degraded after 3 h incubation (
Figure 5). This shows that the siRNA is degraded by its exposure to the intestinal environment, probably by enzymes.
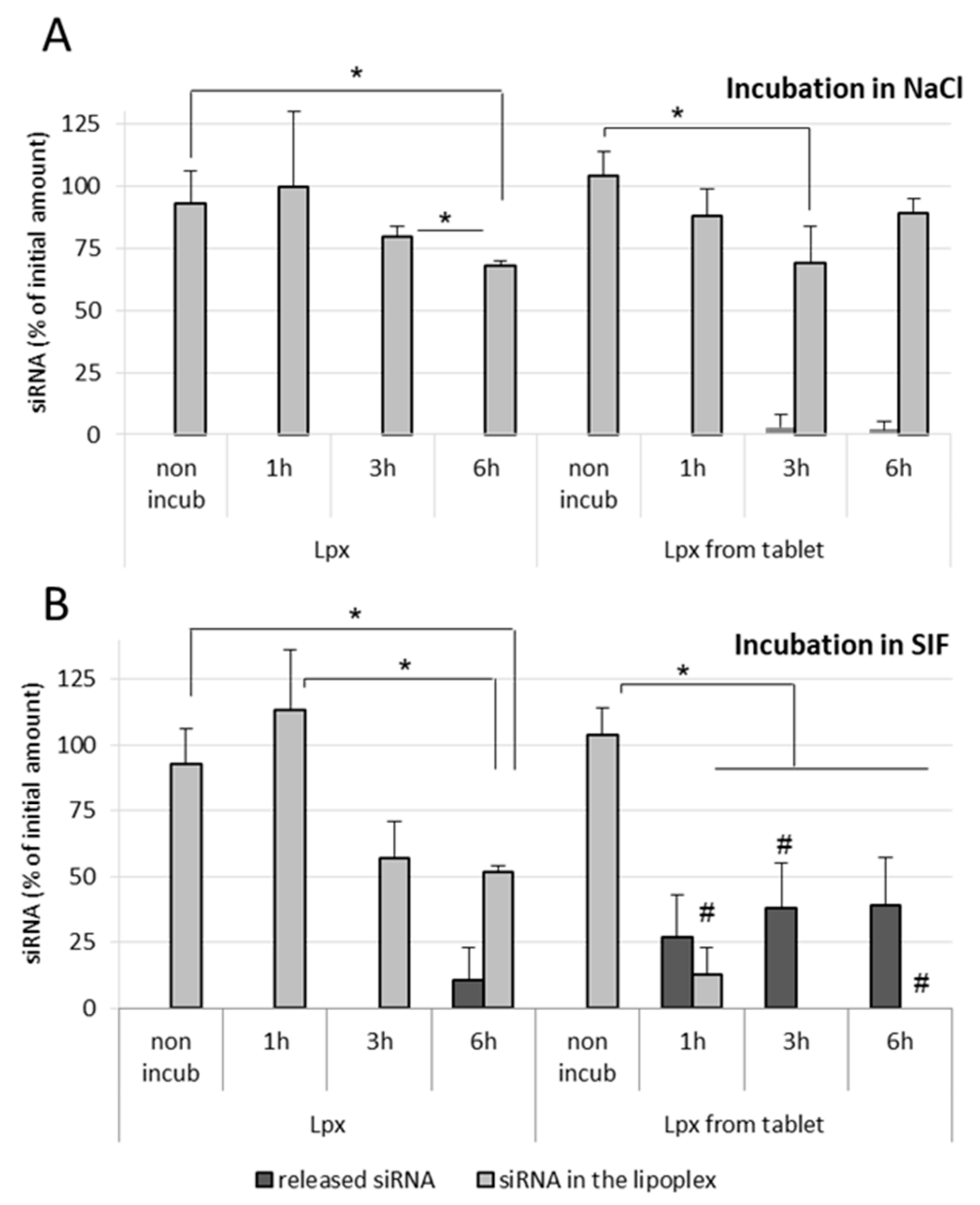
In the case of NaCl (150 mM)-treated fresh lipoplex samples, no release of siRNA was observed in both nonincubated as well as incubated samples, even after 6 h incubation at 37 °C. However, tableted, freeze-dried siRNA lipoplexes showed a very low release of siRNA after 3 h as well as after 6 h incubation (
Figure 6A). In SIF-treated fresh lipoplex samples, no release of siRNA was observed after 1 h and 3 h incubation, however an almost 50% decrease in intact siRNA within lipoplexes was obtained after 3 h incubation in SIF. After 6 h incubation in SIF, a significant amount of siRNA (>10%) was released from the fresh lipoplexes with further decrease in the intact siRNA content within lipoplexes (
Figure 6B). grotected by the lipoplexectrophoresiswas.
In the case of tableted lipoplexes, release of siRNA was observed after 1 h incubation in SIF (>25%), which kept increasing after 3 h and remained stable after 6 h incubation, respectively. The amount of intact siRNA present within the tableted lipoplexes was found around 13% after 1 h incubation in SIF, with no intact siRNA recovered from tableted lipoplexes after 3 h incubation (
Figure 6B). It can be concluded that the fresh lipoplexes are more resistant towards the intestinal environment (enzymes) when compared with tableted lipoplexes.
3.3.3. Effect of Excipients on the Stability of Naked siRNA and siRNA Lipoplexes
The percentage of siRNA released from the fresh lipoplexes and the tableted lipoplexes, once submitted to intestinal environment conditions, was found to be different.
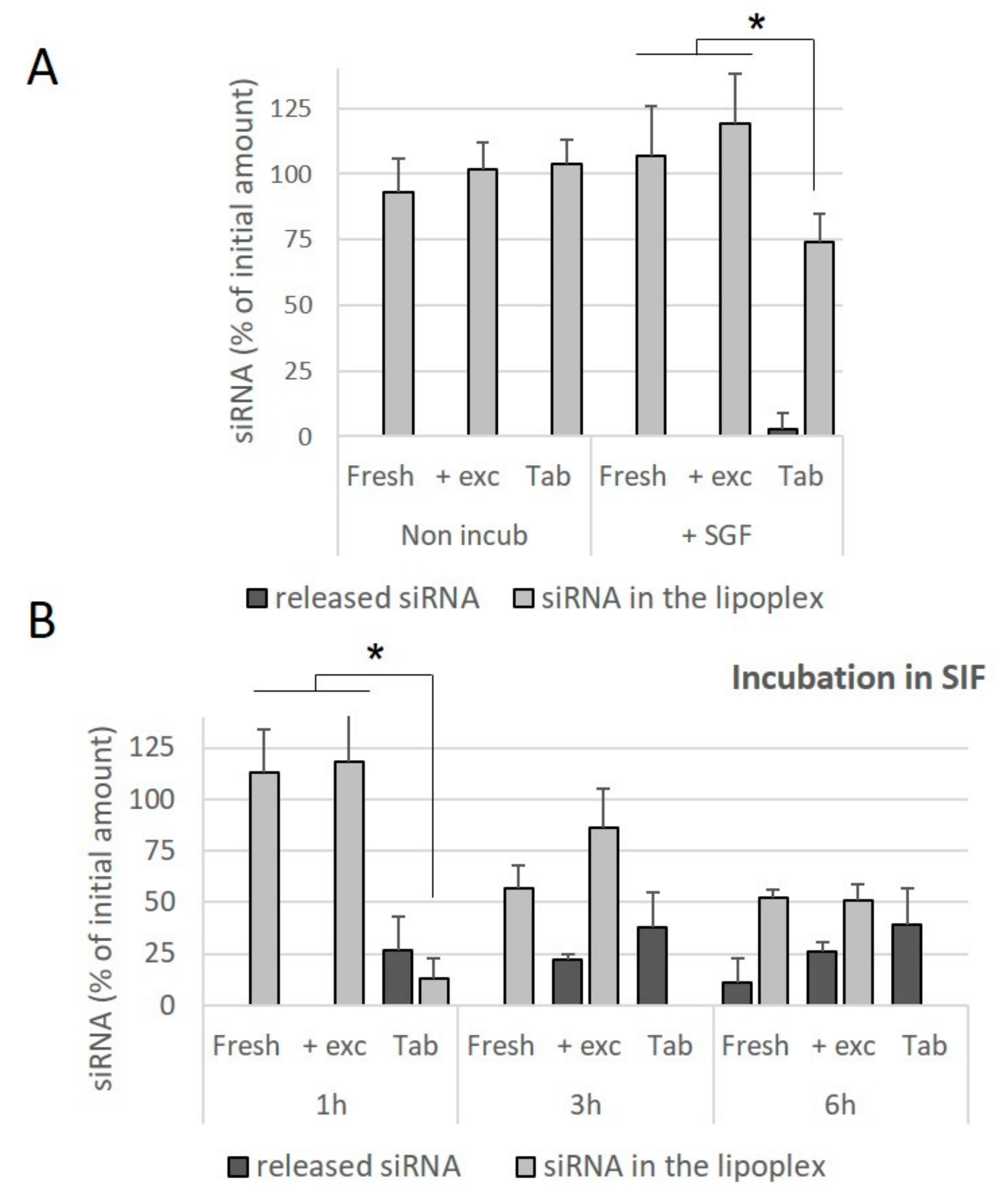
In order to assess whether this difference was due to the compressive stresses, or the presence of the excipients used for the preparation of the tablets, freshly prepared lipoplexes were incubated in an excipient solution and the effect of this incubation on the release of siRNA was evaluated. It was observed that the presence of excipients did not contribute towards the release of siRNA from the fresh lipoplexes incubated in SGF for 1 h (
Figure 7A), nor towards a decrease in the amount of intact siRNA preserved within the lipoplex. Similarly, the presence of excipients did not contribute towards a release of siRNA from lipoplexes incubated in SIF for 1 h. At 3 h and 6 h of incubation in SIF, no significant effect of the presence of excipients was observed, whereas in these conditions of incubation, no intact siRNA is preserved within lipoplexes resuspended from tablets (
Figure 7B). It can be concluded that tableted lipoplexes appeared to be weakened with respect to exposure to conditions in the intestinal environment.
3.4. Effect of Simulated Gastrointestinal Conditions on Transfection Efficacy
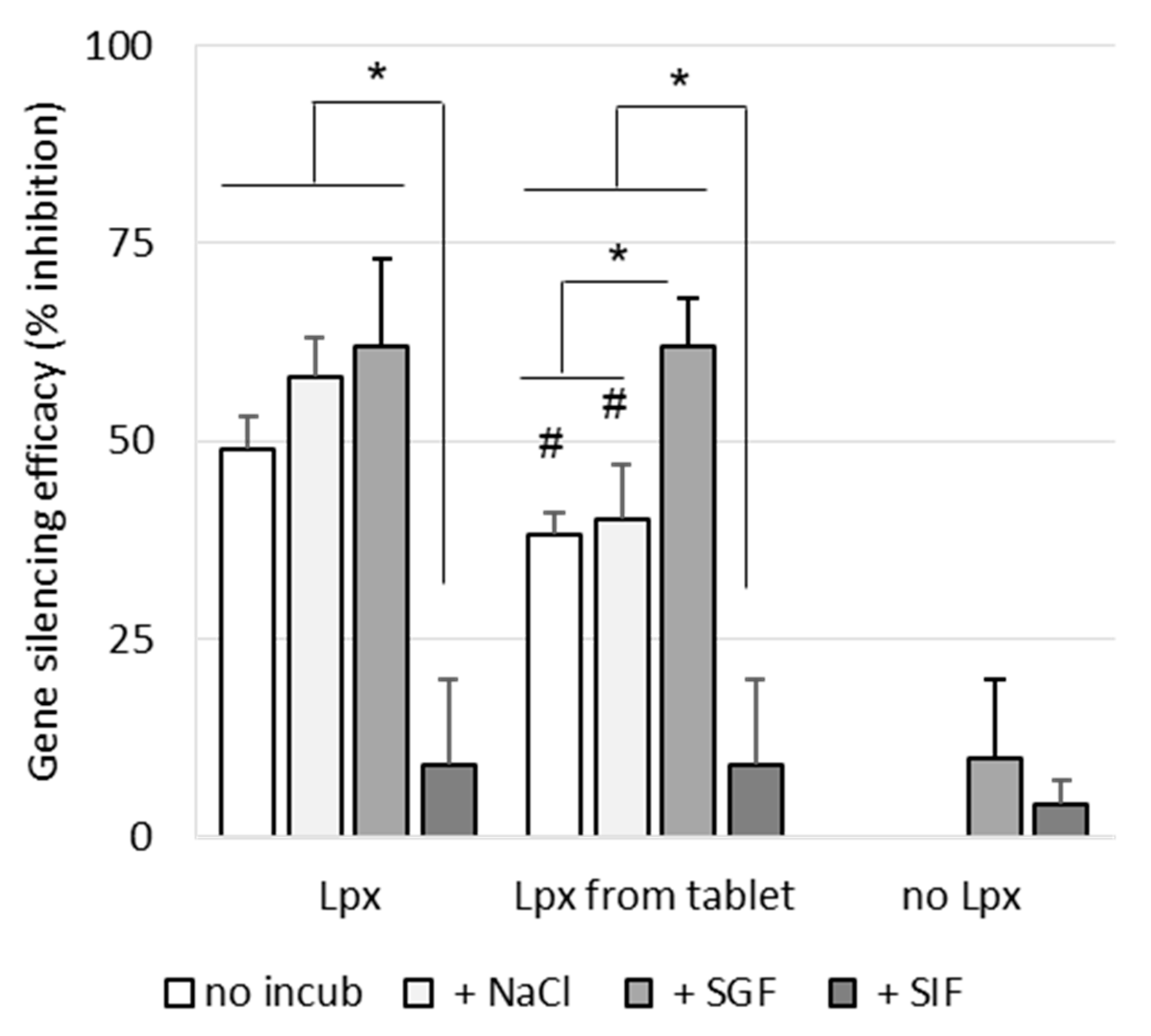
As shown in
Figure 8, incubation of siRNA lipoplexes in NaCl (150 mM) or SGF for 1 h had no effect on their gene-silencing efficacy, while incubation in SIF induced an important loss of activity. As we have already shown [
21], nonincubated lipoplexes resuspended from a tablet exhibited lower efficacy (by about 40%) than fresh lipoplexes. Incubation in saline induced no change in efficacy, while treatment with SGF even increased the observed efficacy. This increase could be due to the effect of highly acidic pH on luciferase activity, which was observed when SGF was applied to cells in the absence of lipoplexes.
As with fresh lipoplexes, incubation in SIF completely collapsed their efficacy. In summary, whether the lipoplexes were fresh or resuspended from a tablet, their efficacy was maintained after SGF challenge but a significant loss of activity was observed after SIF challenge.
4. Discussion
Oral delivery of siRNAs has been considered ever since the Nobel Prize was awarded to RNA interference in 2006 [
6]. Hence, the first study reporting successful systemic delivery of siRNA after oral gavage of glucan-encapsulated siRNA microparticles was published in 2009 [
29]. Since then, to our best knowledge, relatively few authors [
10,
11,
12,
13,
14,
15,
16,
17,
18] have explored the capacity of siRNAs and their carriers to stand the challenging transit in the gastrointestinal tract and reach their intracellular target site intact and active, which is the prerequisite for efficient oral siRNA therapies. The stability of siRNAs in gastric and intestinal media can be considered at three levels: the naked siRNA molecule in solution, the siRNA loaded in a nanoparticulate vector in suspension, and the siRNA nanocarrier incorporated in a solid pharmaceutical oral dosage form.
In order to assess siRNA stability, we used an original method previously published by our research group that allowed quantification of both siRNA retained inside and released from its nanocarrier by assaying siRNA lipoplexes through gel electrophoresis without or with NaCl-Triton treatment, measuring integrated optical density for each siRNA band, and further calculating the percentages of intact siRNA either released in the medium or remaining in the lipoplex, relative to naked siRNA total load [
28]. This method, completed with gene-silencing assays in cultured cells after siRNA (naked and formulated) exposure to simulated gastric and intestinal fluids, provided an informative view of siRNA’s fate in gastrointestinal conditions when simply loaded in lipoplexes or further incorporated in a tablet.
In our study, the naked siRNA in solution could resist gastric challenges for one hour at physiological temperature, but it was progressively altered in intestinal conditions, with a total degradation at 3 h. When documented in the literature, the stability of naked siRNA in gastric media varied according to the experimental conditions of the assays. Hence, siRNA tested in simulated gastric fluid at pH 1.2 without pepsin was stable for one hour at room temperature [
11], while siRNA tested in gastric fluids from rodents was totally degraded within 20 min [
15], or one hour at 37 °C [
16,
17,
18]. If the temperature could explain partially these differences, the presence of nucleases in the fluids harvested from animals could also be involved in the degradation of naked siRNA at acidic pH. Indeed, Guo et al. have shown that naked siRNA, stable in simulated gastric fluid, was considerably degraded when extra nucleases were added into the medium [
30]. The alteration of siRNA observed in our study in simulated intestinal fluid at pH 6.8 is in agreement with previously published data reporting a major degradation in simulated intestinal fluid within 30 min at 37 °C [
30], and for a total degradation in intestinal fluids from rodents within 2 h at 37 °C [
15,
16,
17,
18]. This could be attributed to the presence of nucleases in the pancreatin extract added in the simulated intestinal fluid in our work and Guo’s study, and in the intestinal fluids collected in animals in the other studies. One single publication carried out at ambient temperature in simulated intestinal fluid without pancreatin showed indeed the stability of naked siRNA [
11]. In summary, pH in itself, between 1 and 7, does not alter naked siRNA, but nucleases are deleterious at gastric and intestinal levels.
Since naked siRNA in itself cannot enter cells to exert RNA interference, its incorporation into nanovectors is mandatory to allow for its intracellular delivery. Several nanosystems of siRNA have been shown to protect, to some extent, siRNA from gastro-intestinal challenge, mainly including polymeric nanoparticles [
11,
15,
16,
17,
18], but also cell-penetrating peptides–siRNA noncovalent nanocomplexes [
12], amphiphilic polyallylamine-based polymeric micelles [
30], lipidoid nanoparticles [
14], and milk exosomes [
10]. To our best knowledge, we report here for the first time the fate of polymeric–lipidic nanoparticles, namely lipoplexes, in gastrointestinal conditions. Interestingly, these nanoparticles were able to keep their siRNA load intact and unreleased for one hour in simulated gastric fluid at 37 °C; in addition, their transfection efficiency was totally preserved. However, in simulated intestinal fluid, despite the fact that no release of siRNA from lipoplexes could be detected before six hours of incubation, silencing efficacy was dramatically decreased after one hour exposure, showing that this medium was deleterious to siRNA lipoplex stability. This could be due to presence of bile salts in simulated intestinal fluid that could progressively destabilize the lipoplex structure and expose siRNA to degradative nucleases. In the same way, lipidoid nanoparticles could resist gastric challenge but they partially lost silencing efficacy when exposed to “fed” concentrations of pepsin and bile salts [
14]. As a consequence, such nanocarriers should be protected from the intestinal environment in order to exert full therapeutic efficacy. This objective might be achieved through their inclusion into dosage forms devoted to oral administration, such as tablets and capsules.
Currently, the incorporation of siRNA nanovectors in solid oral dosage forms and further determination of their fate in gastrointestinal conditions are scarce. Indeed, to our best knowledge, one single article reported the formulation of cell-penetrating peptides–siRNA nanocomplexes into solid dispersions intended for oral delivery that are stable in simulated gastric conditions [
12]. No compacted solid dosage forms such as tablets could be found in the literature as siRNA oral delivery forms. In our study, tablets obtained from freeze-dried lipoplexes were shown to efficiently protect siRNA in simulated gastric fluid—although a minimal release could be observed at one hour—allowing preservation of the majority of the gene-silencing efficacy. Exposure to simulated intestinal fluid for one hour also permitted preservation of about 13% of intact siRNA within the tableted lipoplexes. However, lipoplexes from tablets could not resist longer simulated intestinal fluid challenges, which resulted in a marked siRNA release and decreased gene-silencing efficacy, thus showing that the freeze-drying and tableting processes further weakened siRNA lipoplexes. Importantly, the tablets tested here were obtained under a compression pressure in the medium range of pressures previously tested between 50 and 250 MPa [
21]. Amongst several advantages, tableted dosage forms can easily modulate the release kinetics of active ingredients through an adequate formulation. Two options might be considered in order to improve this promising oral dosage form and to bring tableted siRNAs to clinical applications. On the one hand, the lyophilization and compression parameters, for example, sample temperature during sublimation and lower compaction pressure, could be further optimized in order to reduce their impact on lipoplex stability. On the other hand, tablet formulation could be optimized by using excipients able to resist intestinal conditions, either under a matrix form, or as external film coatings.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







