
POLRMT as a Novel Susceptibility Gene for Cardiotoxicity in Epirubicin Treatment of Breast Cancer Patients

 ,
,  , , ,
, , , 

Abstract
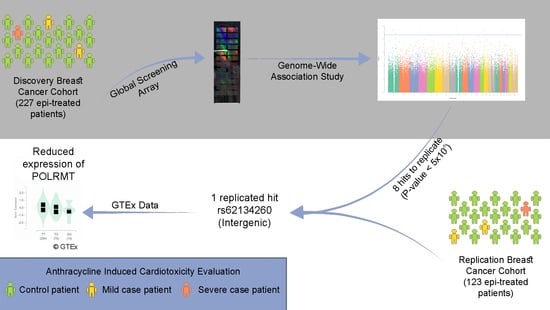
:
1. Introduction
2. Materials and Methods
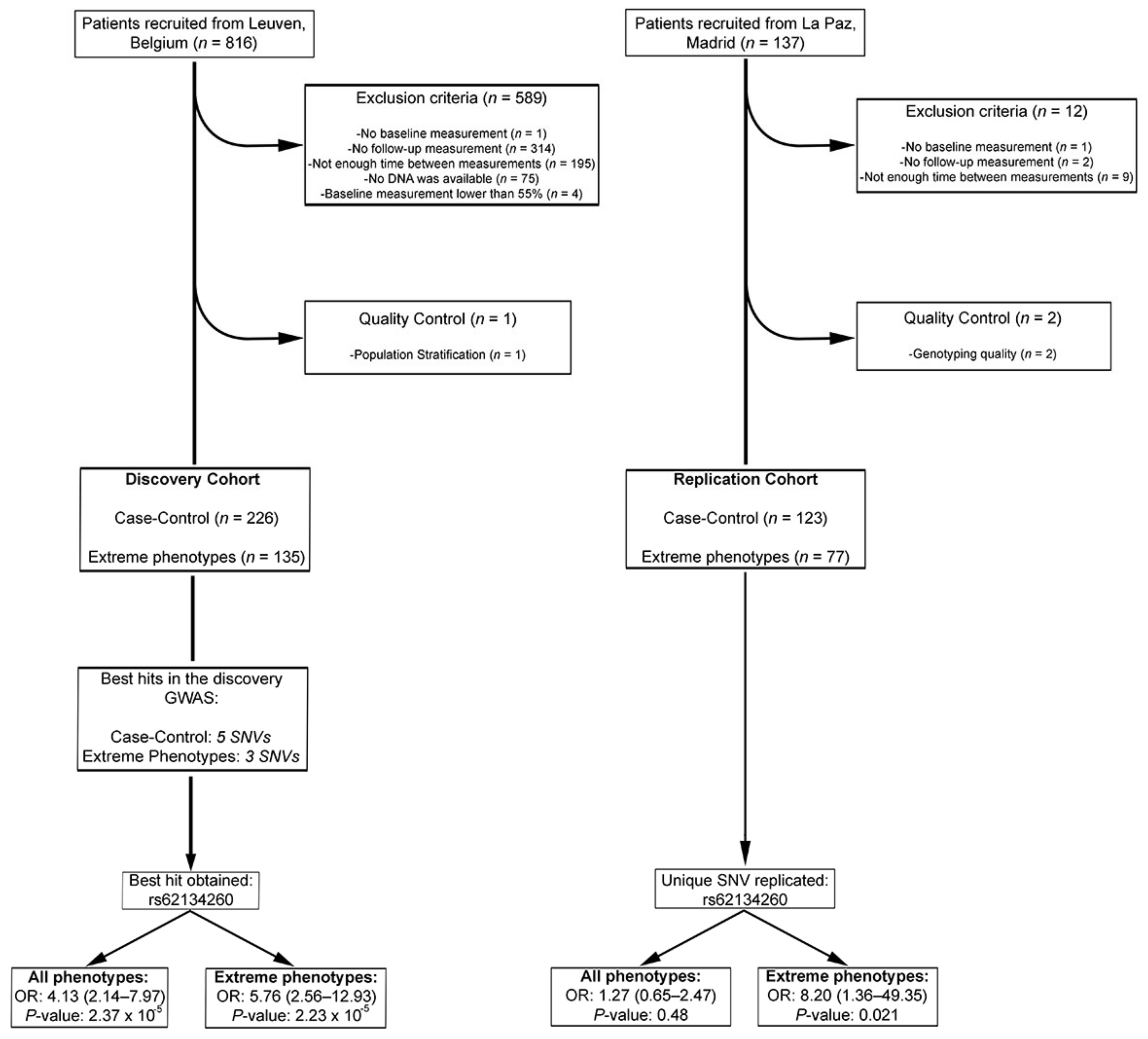
2.1. Patients
2.2. Genotyping and Quality Control
2.3. Data Imputation
2.4. Statistical Analysis
2.5. Functional annotation
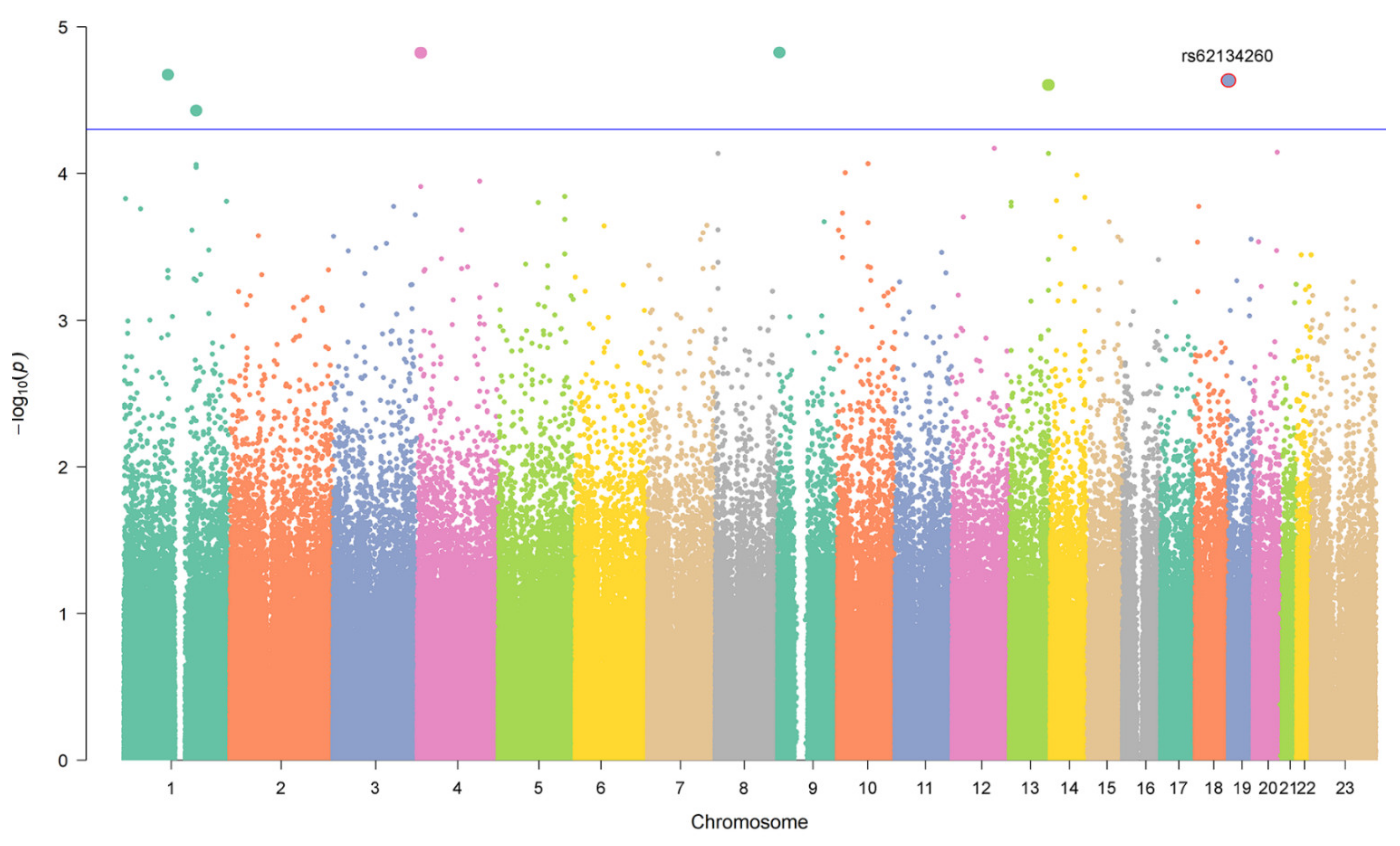
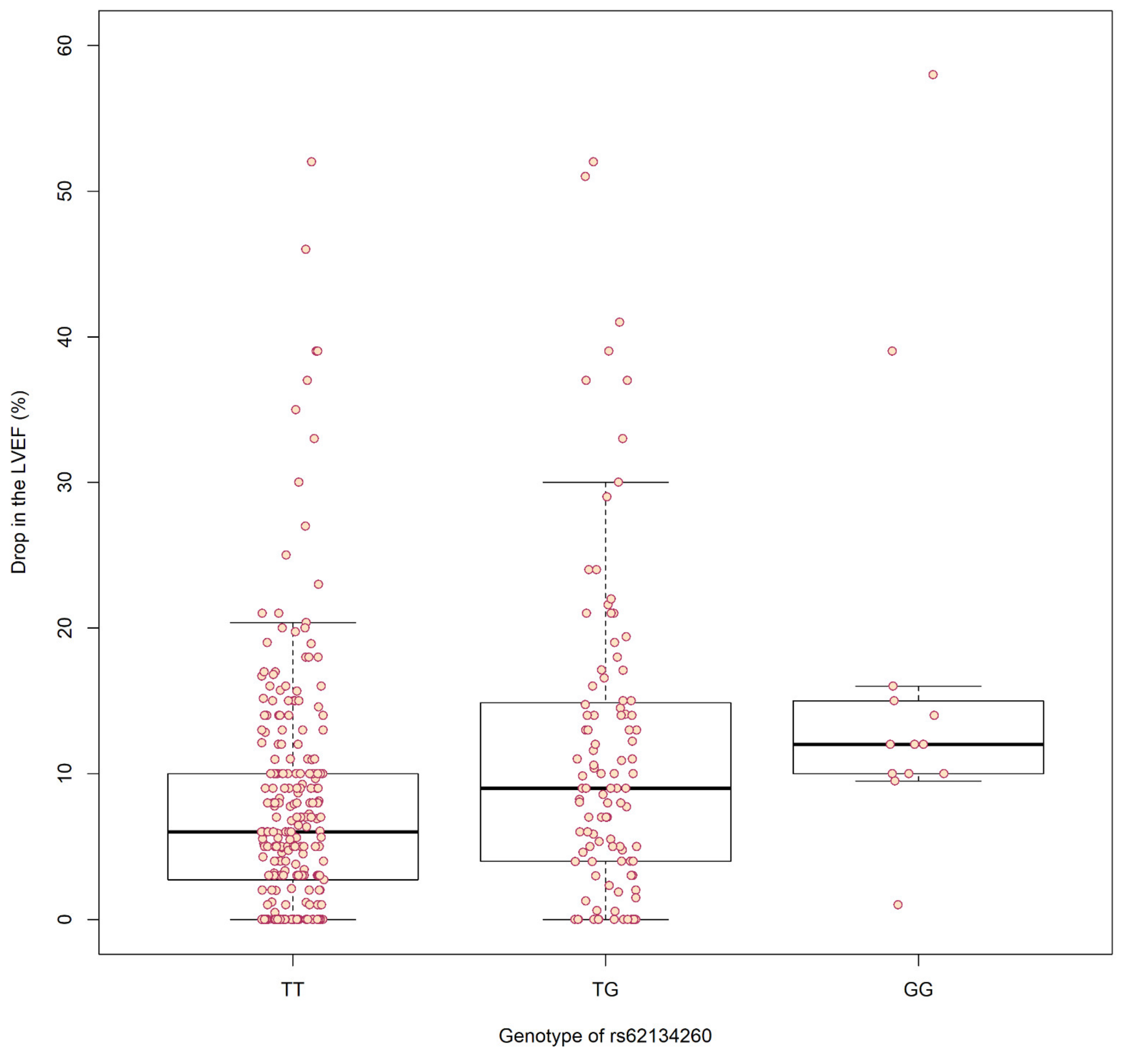
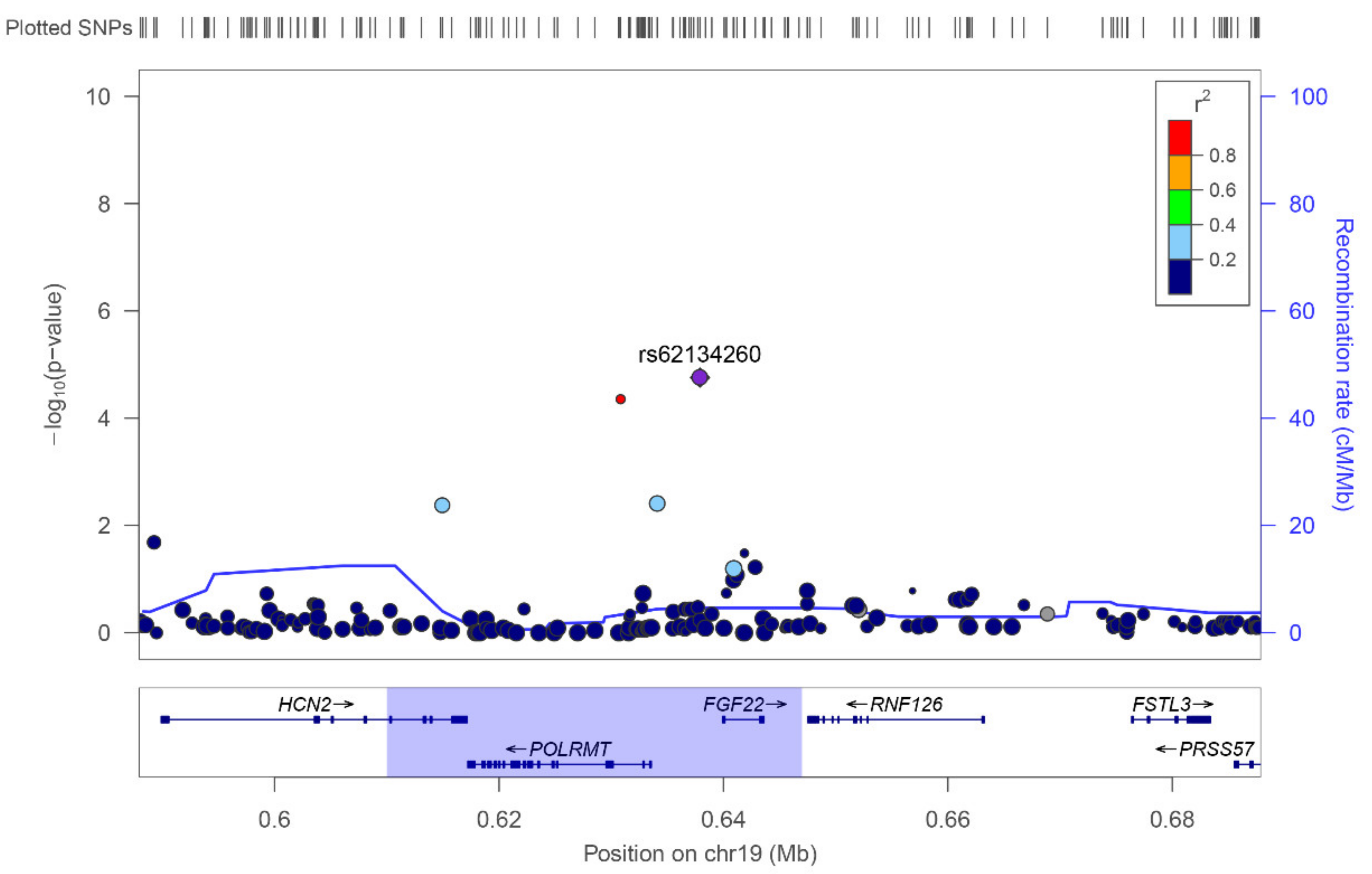
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Valcovici, M.; Andrica, F.; Serban, C.; Dragan, S. Cardiotoxicity of anthracycline therapy: Current perspectives. Arch. Med. Sci. 2016, 2, 428–435. [Google Scholar] [CrossRef] [Green Version]
- Bansal, N.; Blanco, J.G.; Sharma, U.C.; Pokharel, S.; Shisler, S.; Lipshultz, S.E. Cardiovascular diseases in survivors of childhood cancer. Cancer Metastasis Rev. 2020, 39, 55–68. [Google Scholar] [CrossRef]
- Gyöngyösi, M.; Lukovic, D.; Zlabinger, K.; Spannbauer, A.; Gugerell, A.; Pavo, N.; Traxler, D.; Pils, D.; Maurer, G.; Jakab, A.; et al. Liposomal doxorubicin attenuates cardiotoxicity via induction of interferon-related DNA damage resistance. Cardiovasc. Res. 2020, 116, 970–982. [Google Scholar] [CrossRef]
- Jasra, S.; Anampa, J. Anthracycline Use for Early Stage Breast Cancer in the Modern Era: A Review. Curr. Treat. Options Oncol. 2018, 19, 30. [Google Scholar] [CrossRef]
- Khasraw, M.; Bell, R.; Dang, C. Epirubicin: Is it like doxorubicin in breast cancer? A clinical review. The Breast 2012, 21, 142–149. [Google Scholar] [CrossRef]
- Caron, J.; Nohria, A. Cardiac Toxicity from Breast Cancer Treatment: Can We Avoid This? Curr. Oncol. Rep. 2018, 20, 61. [Google Scholar] [CrossRef]
- Aminkeng, F.; Ross, C.J.D.; Rassekh, S.R.; Hwang, S.; Rieder, M.J.; Bhavsar, A.P.; Smith, A.; Sanatani, S.; Gelmon, K.A.; Bernstein, D.; et al. Recommendations for genetic testing to reduce the incidence of anthracycline-induced cardiotoxicity. Br. J. Clin. Pharmacol. 2016, 82, 683–695. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Wang, Z.; Li, Y.; Lv, D.; Zhao, X.; Gao, J.; Teng, H. Identification of differential gene expression related to epirubicin-induced cardiomyopathy in breast cancer patients. Hum. Exp. Toxicol. 2020, 39, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.; Alvarez, J.A.; Scully, R.E.; Miller, T.L.; Lipshultz, S.E. Anthracycline-induced cardiotoxicity: Course, pathophysiology, prevention and management. Expert Opin. Pharmacother. 2007, 8, 1039–1058. [Google Scholar] [CrossRef] [PubMed]
- Nysom, K.; Holm, K.; Lipsitz, S.R.; Mone, S.M.; Colan, S.D.; Orav, E.J.; Sallan, S.E.; Olsen, J.H.; Hertz, H.; Jacobsen, J.R.; et al. Relationship between cumulative anthracycline dose and late cardiotoxicity in childhood acute lymphoblastic leukemia. J. Clin. Oncol. 1998, 16, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Pinto, S.; Pita, G.; Martín, M.; Alonso-Gordoa, T.; Barnes, D.R.; Alonso, M.R.; Herraez, B.; García-Miguel, P.; Alonso, J.; Pérez-Martínez, A.; et al. Exome array analysis identifies ETFB as a novel susceptibility gene for anthracycline-induced cardiotoxicity in cancer patients. Breast Cancer Res. Treat. 2018, 167, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Maillet, A.; Tan, K.; Chai, X.; Sadananda, S.N.; Mehta, A.; Ooi, J.; Hayden, M.R.; Pouladi, M.A.; Ghosh, S.; Shim, W.; et al. Modeling Doxorubicin-Induced Cardiotoxicity in Human Pluripotent Stem Cell Derived-Cardiomyocytes. Sci. Rep. 2016, 6, 25333. [Google Scholar] [CrossRef] [Green Version]
- Raj, S.; Franco, V.I.; Lipshultz, S.E. Anthracycline-Induced Cardiotoxicity: A Review of Pathophysiology, Diagnosis, and Treatment. Curr. Treat. Options Cardiovasc. Med. 2014, 16, 315. [Google Scholar] [CrossRef] [PubMed]
- Gianni, L.; Herman, E.H.; Lipshultz, S.E.; Minotti, G.; Sarvazyan, N.; Sawyer, D.B. Anthracycline cardiotoxicity: From bench to bedside. J. Clin. Oncol. 2008, 26, 3777–3784. [Google Scholar] [CrossRef] [Green Version]
- Volkova, M.; Russell, R. Anthracycline Cardiotoxicity: Prevalence, Pathogenesis and Treatment. Curr. Cardiol. Rev. 2012, 7, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Madgy, T.; Burmeister, B.; Burridge, P. Validating the pharmacogenomics of chemotherapy-induced cardiotoxicity: What is missing? Pharmacol. Ther. 2016, 168, 113–125. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Qadir, H.; Austin, P.C.; Lee, D.S.; Amir, E.; Tu, J.V.; Thavendiranathan, P.; Fung, K.; Anderson, G.M. A population-based study of cardiovascular mortality following early-stage breast cancer. JAMA Cardiol. 2017, 2, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Edwardson, D.; Narendrula, R.; Chewchuk, S.; Mispel-Beyer, K.; Mapletoft, J.; Parissenti, A. Role of Drug Metabolism in the Cytotoxicity and Clinical Efficacy of Anthracyclines. Curr. Drug Metab. 2015, 16, 412–426. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Sun, S.; Fei, F.; Wang, J.; Wang, Y.; Zhang, R.; Wu, J.; Liu, L.; Liu, X.; Cui, Z.; et al. Screening in larval zebrafish reveals tissue-specific distribution of fifteen fluorescent compounds. Dis. Model. Mech. 2017, 10, 1155–1164. [Google Scholar] [CrossRef] [Green Version]
- Sawicki, K.T.; Sala, V.; Prever, L.; Hirsch, E.; Ardehali, H.; Ghigo, A. Preventing and Treating Anthracycline Cardiotoxicity: New Insights. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 309–332. [Google Scholar] [CrossRef]
- Lyon, A.R.; Dent, S.; Stanway, S.; Earl, H.; Brezden-Masley, C.; Cohen-Solal, A.; Tocchetti, C.G.; Moslehi, J.J.; Groarke, J.D.; Bergler-Klein, J.; et al. Baseline cardiovascular risk assessment in cancer patients scheduled to receive cardiotoxic cancer therapies: A position statement and new risk assessment tools from the Cardio-Oncology Study Group of the Heart Failure Association of the European Society. Eur. J. Heart Fail. 2020, 22, 1945–1960. [Google Scholar] [CrossRef]
- Zamorano, J.L.; Lancellotti, P.; Rodriguez Muñoz, D.; Aboyans, V.; Asteggiano, R.; Galderisi, M.; Habib, G.; Lenihan, D.J.; Lip, G.Y.H.; Lyon, A.R.; et al. 2016 European Society of Cardiology position paper on cancer treatments and cardiovascular toxicity. Eur. Heart J. 2016, 37, 2768–2801. [Google Scholar] [CrossRef] [PubMed]
- López-Sendón, J.; Álvarez-Ortega, C.; Zamora Auñon, P.; Buño Soto, A.; Lyon, A.R.; Farmakis, D.; Cardinale, D.; Canales Albendea, M.; Feliu Batlle, J.; Rodríguez Rodríguez, I.; et al. Classification, prevalence, and outcomes of anticancer therapy-induced cardiotoxicity: The CARDIOTOX registry. Eur. Heart J. 2020, 41, 1720–1729. [Google Scholar] [CrossRef] [PubMed]
- Berkman, A.M.; Hildebrandt, M.A.T.; Landstrom, A.P. The genetic underpinnings of anthracycline-induced cardiomyopathy predisposition. Clin. Genet. 2021, 100, 132–143. [Google Scholar] [CrossRef]
- Volkan-Salanci, V.; Aksoy, H.; Kiratli, P.Ö.; Tülümen, E.; Güler, N.; Öksüzoglu, B.; Tokgözoğlu, L.; Erbaş, B.; Alikaşifoğlu, M. The relationship between changes in functional cardiac parameters following anthracycline therapy and carbonyl reductase 3 and glutathione S transferase Pi polymorphisms. J. Chemother. 2012, 24, 285–291. [Google Scholar] [CrossRef]
- Hertz, D.L.; Caram, M.V.; Kidwell, K.M.; Thibert, J.N.; Gersch, C.; Seewald, N.J.; Smerage, J.; Rubenfire, M.; Henry, N.L.; Cooney, K.A.; et al. Evidence for association of SNPs in ABCB1 and CBR3, but not RAC2, NCF4, SLC28A3 or TOP2B, with chronic cardiotoxicity in a cohort of breast cancer patients treated with anthracyclines. Pharmacogenomics 2016, 17, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Lang, J.K.; Karthikeyan, B.; Quiñones-Lombraña, A.; Blair, R.H.; Early, A.P.; Levine, E.G.; Sharma, U.C.; Blanco, J.G.; O’Connor, T. CBR3 V244M is associated with LVEF reduction in breast cancer patients treated with doxorubicin. Cardio-Oncology 2021, 7, 17. [Google Scholar] [CrossRef]
- Blanco, J.G.; Leisenring, W.M.; Gonzalez-Covarrubias, V.M.; Kawashima, T.I.; Davies, S.M.; Relling, M.V.; Robison, L.L.; Sklar, C.A.; Stovall, M.; Bhatia, S. Genetic polymorphisms in the carbonyl reductase 3 geneCBR3 and the NAD(P)H:quinone oxidoreductase 1 geneNQO1 in patients who developed anthracycline-related congestive heart failure after childhood cancer. Cancer 2008, 112, 2789–2795. [Google Scholar] [CrossRef]
- Blanco, J.G.; Sun, C.-L.; Landier, W.; Chen, L.; Esparza-Duran, D.; Leisenring, W.; Mays, A.; Friedman, D.L.; Ginsberg, J.P.; Hudson, M.M.; et al. Anthracycline-Related Cardiomyopathy After Childhood Cancer: Role of Polymorphisms in Carbonyl Reductase Genes—A Report From the Children’s Oncology Group. J. Clin. Oncol. 2012, 30, 1415–1421. [Google Scholar] [CrossRef] [Green Version]
- Bains, O.S.; Karkling, M.J.; Lubieniecka, J.M.; Grigliatti, T.A.; Reid, R.E.; Riggs, K.W. Naturally Occurring Variants of Human CBR3 Alter Anthracycline In Vitro Metabolism. J. Pharmacol. Exp. Ther. 2010, 332, 755–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaupp, C.M.; White, C.C.; Merrill, G.F.; Kavanagh, T.J. Metabolism of doxorubicin to the cardiotoxic metabolite doxorubicinol is increased in a mouse model of chronic glutathione deficiency: A potential role for carbonyl reductase 3. Chem. Biol. Interact. 2015, 234, 154–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, B.P.; Shen, F.; Gardner, L.; Radovich, M.; Li, L.; Miller, K.D.; Jiang, G.; Lai, D.; O’Neill, A.; Sparano, J.A.; et al. Genome-Wide Association Study for Anthracycline-Induced Congestive Heart Failure. Clin. Cancer Res. 2017, 23, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, Q.S.; Veatch, O.J.; Fessel, J.P.; Joon, A.Y.; Levinson, R.T.; Mosley, J.D.; Held, E.P.; Lindsay, C.S.; Shaffer, C.M.; Weeke, P.E.; et al. Genome-wide association and pathway analysis of left ventricular function after anthracycline exposure in adults. Pharmacogenet. Genomics 2017, 27, 247–254. [Google Scholar] [CrossRef]
- Vulsteke, C.; Pfeil, A.M.; Maggen, C.; Schwenkglenks, M.; Pettengell, R.; Szucs, T.D.; Lambrechts, D.; Dieudonné, A.S.; Hatse, S.; Neven, P.; et al. Clinical and genetic risk factors for epirubicin-induced cardiac toxicity in early breast cancer patients. Breast Cancer Res. Treat. 2015, 152, 67–76. [Google Scholar] [CrossRef]
- López-Fernández, T.; Martín García, A.; Santaballa Beltrán, A.; Montero Luis, Á.; García Sanz, R.; Mazón Ramos, P.; Velasco del Castillo, S.; López de Sá Areses, E.; Barreiro-Pérez, M.; Hinojar Baydes, R.; et al. Cardio-Onco-Hematology in Clinical Practice. Position Paper and Recommendations. Rev. Española Cardiol. 2017, 70, 474–486. [Google Scholar] [CrossRef]
- Ben Abdallah, I.; Ben Nasr, S.; Chourabi, C.; Boukhris, M.; Ben Abdallah, I.; Zribi, A.; Fendri, S.; Balti, M.; Fehri, W.; Chraiet, N.; et al. The Predictive Value of 2D Myocardial Strain for Epirubicin-Induced Cardiotoxicity. J. Oncol. 2020, 2020, 5706561. [Google Scholar] [CrossRef]
- Plana, J.C.; Galderisi, M.; Barac, A.; Ewer, M.S.; Ky, B.; Scherrer-Crosbie, M.; Ganame, J.; Sebag, I.A.; Agler, D.A.; Badano, L.P.; et al. Expert Consensus for Multimodality Imaging Evaluation of Adult Patients during and after Cancer Therapy: A Report from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. 2014, 27, 911–939. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 1–16. [Google Scholar] [CrossRef]
- Purcell, S.; Chang, C. PLINK Software. Available online: www.cog-genomics.org/plink/1.9 (accessed on 26 October 2018).
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Das, S.; Forer, L.; Schönherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruim, R.J.; Welch, R.P.; Sanna, S.; Teslovich, T.M.; Chines, P.S.; Gliedt, T.P.; Boehnke, M.; Abecasis, G.R.; Willer, C.J. LocusZoom: Regional visualization of genome-wide association scan results. Bioinformatics 2010, 26, 2336–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnett, I.J.; Lee, S.; Lin, X. Detecting Rare Variant Effects Using Extreme Phenotype Sampling in Sequencing Association Studies. Genet. Epidemiol. 2013, 37, 142–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staley, J.R.; Blackshaw, J.; Kamat, M.A.; Ellis, S.; Surendran, P.; Sun, B.B.; Paul, D.S.; Freitag, D.; Burgess, S.; Danesh, J.; et al. PhenoScanner: A database of human genotype–phenotype associations. Bioinformatics 2016, 32, 3207–3209. [Google Scholar] [CrossRef] [Green Version]
- Kamat, M.A.; Blackshaw, J.A.; Young, R.; Surendran, P.; Burgess, S.; Danesh, J.; Butterworth, A.S.; Staley, J.R. PhenoScanner V2: An expanded tool for searching human genotype–phenotype associations. Bioinformatics 2019, 35, 4851–4853. [Google Scholar] [CrossRef] [Green Version]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef]
- ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [CrossRef]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef] [Green Version]
- Geiman, T.M.; Robertson, K.D. Chromatin remodeling, histone modifications, and DNA methylation?how does it all fit together? J. Cell. Biochem. 2002, 87, 117–125. [Google Scholar] [CrossRef]
- Segal, E.; Widom, J. What controls nucleosome positions? Trends Genet. 2009, 25, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christidi, E.; Huang, H.; Shafaattalab, S.; Maillet, A.; Lin, E.; Huang, K.; Laksman, Z.; Davis, M.K.; Tibbits, G.F.; Brunham, L.R. Variation in RARG increases susceptibility to doxorubicin-induced cardiotoxicity in patient specific induced pluripotent stem cell-derived cardiomyocytes. Sci. Rep. 2020, 10, 10363. [Google Scholar] [CrossRef]
- Yang, X.; Li, G.; Yang, T.; Guan, M.; An, N.; Yang, F.; Dai, Q.; Zhong, C.; Luo, C.; Gao, Y.; et al. Possible Susceptibility Genes for Intervention against Chemotherapy-Induced Cardiotoxicity. Oxid. Med. Cell. Longev. 2020, 2020, 4894625. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, G.; Guan, M.; Bapat, A.; Dai, Q.; Zhong, C.; Yang, T.; Luo, C.; An, N.; Liu, W.; et al. Potential Gene Association Studies of Chemotherapy-Induced Cardiotoxicity: A Systematic Review and Meta-Analysis. Front. Cardiovasc. Med. 2021, 8, 458. [Google Scholar] [CrossRef]
- Bouda, E.; Stapon, A.; Garcia-Diaz, M. Mechanisms of mammalian mitochondrial transcription. Protein Sci. 2019, 28, 1594–1605. [Google Scholar] [CrossRef]
- Kühl, I.; Miranda, M.; Posse, V.; Milenkovic, D.; Mourier, A.; Siira, S.J.; Bonekamp, N.A.; Neumann, U.; Filipovska, A.; Polosa, P.L.; et al. POLRMT regulates the switch between replication primer formation and gene expression of mammalian mtDNA. Sci. Adv. 2016, 2, e1600963. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.Y. Addressing the selectivity and toxicity of antiviral nucleosides. Antivir. Chem. Chemother. 2018, 26, 204020661875852. [Google Scholar] [CrossRef]
- Varga, Z.V.; Ferdinandy, P.; Liaudet, L.; Pacher, P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Circ. Physiol. 2015, 309, H1453–H1467. [Google Scholar] [CrossRef] [Green Version]
- Burridge, P.; Li, Y.; Matsa, E.; Wu, H.; Ong, S.; Sharma, A.; Holmström, A.; Chang, A.; Coronado, M.; Ebert, A.; et al. Human Induced Pluripotent Stem Cell–Derived Cardiomyocytes Recapitulate the Predilection of Breast Cancer Patients to Doxorubicin–Induced Cardiotoxicity Paul. Nat. Med. 2016, 22, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-D.; Kim, C.H.; Rah, B.-J.; Chung, H.-I.; Shim, T.-S. Quantitative study on the relation between structural and functional properties of the hearts from three different mammals. Anat. Rec. 1994, 238, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Piquereau, J.; Caffin, F.; Novotova, M.; Lemaire, C.; Veksler, V.; Garnier, A.; Ventura-Clapier, R.; Joubert, F. Mitochondrial dynamics in the adult cardiomyocytes: Which roles for a highly specialized cell? Front. Physiol. 2013, 4, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, K.B. Doxorubicin-Induced Cardiac Mitochondrionopathy. Pharmacol. Toxicol. 2003, 93, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, G.; Synnergren, J.; Bogestål, Y.; Améen, C.; Åkesson, K.; Holmgren, S.; Lindahl, A.; Sartipy, P. Identification of novel biomarkers for doxorubicin-induced toxicity in human cardiomyocytes derived from pluripotent stem cells. Toxicology 2015, 328, 102–111. [Google Scholar] [CrossRef]
- Weinhouse, C. Mitochondrial-epigenetic crosstalk in environmental toxicology. Toxicology 2017, 391, 5–17. [Google Scholar] [CrossRef]
- Wallace, K.B. Adriamycin-induced interference with cardiac mitochondrial calcium homeostasis. Cardiovasc. Toxicol. 2007, 7, 101–107. [Google Scholar] [CrossRef] [PubMed]



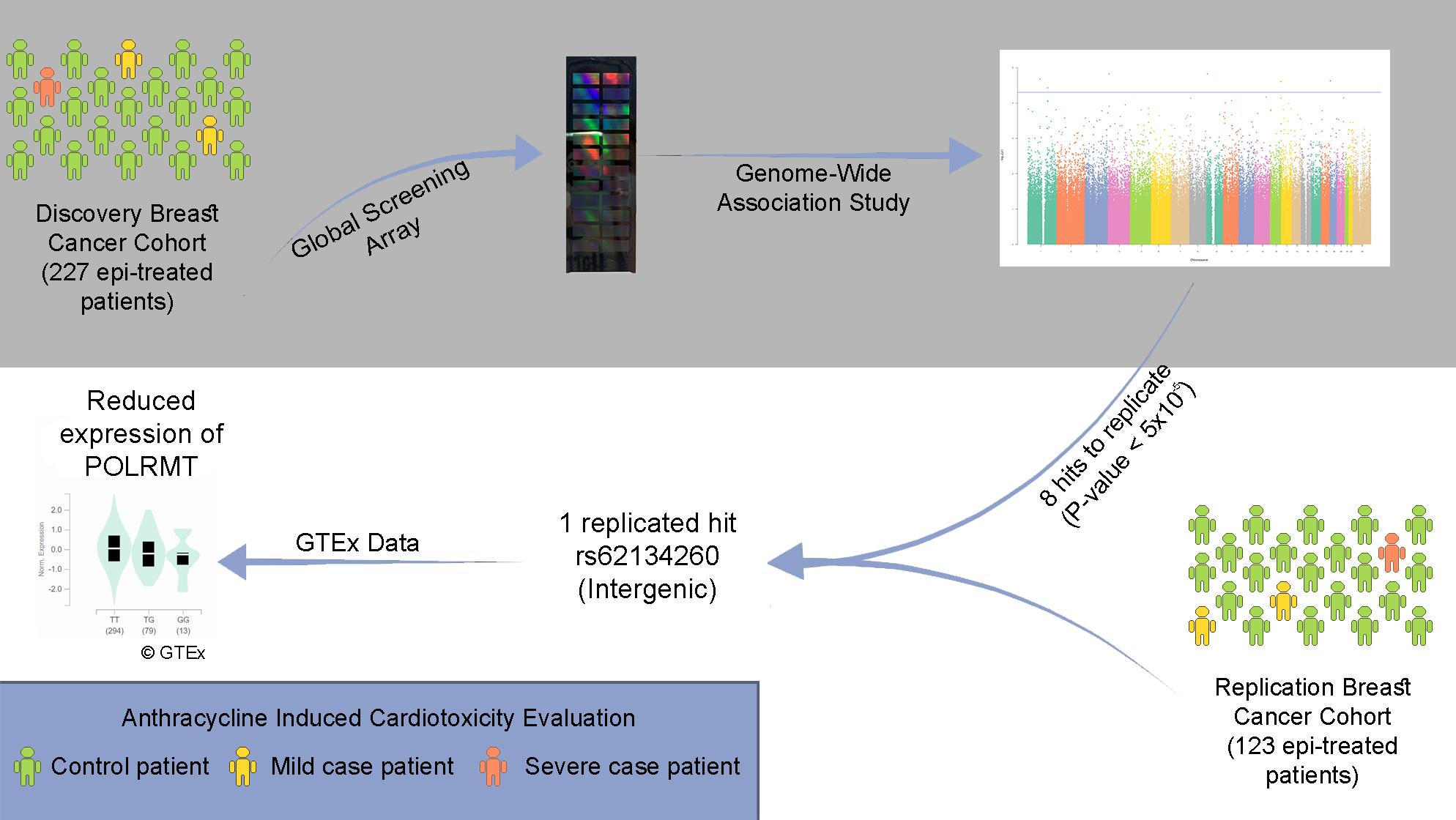{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Discovery Cohort (n = 227) | Replication Cohort (n = 123) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Controls (n = 90) | Mild Cases (n = 92) | Severe Cases (n = 45) | p-Value (Case-Control) | p-Value (Extreme Phenotypes) | Controls (n = 64) | Mild Cases (n = 49) | Severe Cases (n = 10) | p-Value (Case-Control) | p-Value (Extreme Phenotypes) | |
| Age (yr.) at diagnosis, median (IQR) | 50 (45–55.75) | 50 (44–57) | 50 (45–56) | 0.93 | 0.98 | 54 (43.75–60) | 47 (40–53) | 61.5 (47.25–67) | 0.44 | 0.16 |
| Cumulative dose (mg/m2), median (IQR) | 300 (200–600) | 600 (100–600) | 600 (100–600) | 8.7 × 10−6 | 6.5 × 10−4 | 511 (121–720) | 540 (405–765) | 486 (115–600) | 0.92 | 0.01 |
| Location of Tumor in Left Breast, No (%) | 53 (58.89) | 42 (46.15) | 23 (50.00) | 0.09 | 0.33 | 34 (53.13) | 26 (53.06) | 3 (30.00) | 0.37 | 0.06 |
| Use of Radiotherapy, No (%) | 87 (9.67) | 81 (88.04) | 46 (100.00) | 0.18 | 0.21 | 58 (90.63) | 47 (95.92) | 10 (100.00) | 0.18 | 0.32 |
| Radiotherapy treatment in Left Breast, No (%) | 49 (54.44) | 41 (44.57) | 23 (51.11) | 0.73 | 0.63 | 34 (53.13) | 26 (53.06) | 3 (30.00) | 0.61 | 0.18 |
| Bilateral Breast Cancer, No (%) | 1 (1.11) | 0 (0) | 0 (0) | - | - | 3 (4.69) | 3 (6.12) | 0 (0) | - | - |
| CHR | SNV | Gene | Allele | AF | Analysis 1 | Discovery p-Value | Cohort OR [95% CI] | Replication p-Value | Cohort OR [95% CI] |
|---|---|---|---|---|---|---|---|---|---|
| 1 | rs11185202 | 74 kb from AMY1C, 136 kb from AMY1B | T | 0.41 | Case-Control | 2.13 × 10−5 | 0.36 [0.23–0.58] | 0.88 | 0.96 [0.57–1.61] |
| 1 | rs66539320 | lncRNA | G | 0.18 | Case-Control | 3.72 × 10−5 | 0.33 [0.19–0.56] | 0.56 | 1.24 [0.60–2.54] |
| 1 | rs382092 | lncRNA | T | 0.36 | Extreme Phenotypes | 2.32 × 10−5 | 4.03 [2.11–7.68] | 0.31 | 0.57 [0.19–1.68] |
| 2 | rs17687727 | lncRNA | A | 0.16 | Extreme Phenotypes | 4.73 × 10−5 | 4.92 [2.29–10.66] | 0.80 | 1.17 [0.34–4.07] |
| 4 | rs2270271 | GPR78 | T | 0.46 | Case-Control | 1.51 × 10−5 | 0.38 [0.25–0.59] | 0.84 | 0.95 [0.58–.’54] |
| 9 | rs377186 | RCL1 | A | 0.44 | Case-Control | 1.50 × 10−5 | 0.35 [0.22–0.57] | 0.38 | 0.81 [0.50–1.30] |
| 13 | rs8000668 | 98 kb from ARHGEF7, and 101 kb from ANKRD10 | T | 0.52 | Case-Control | 2.50 × 10−5 | 0.40 [0.26–0.62] | 0.41 | 0.79 [0.46–1.38] |
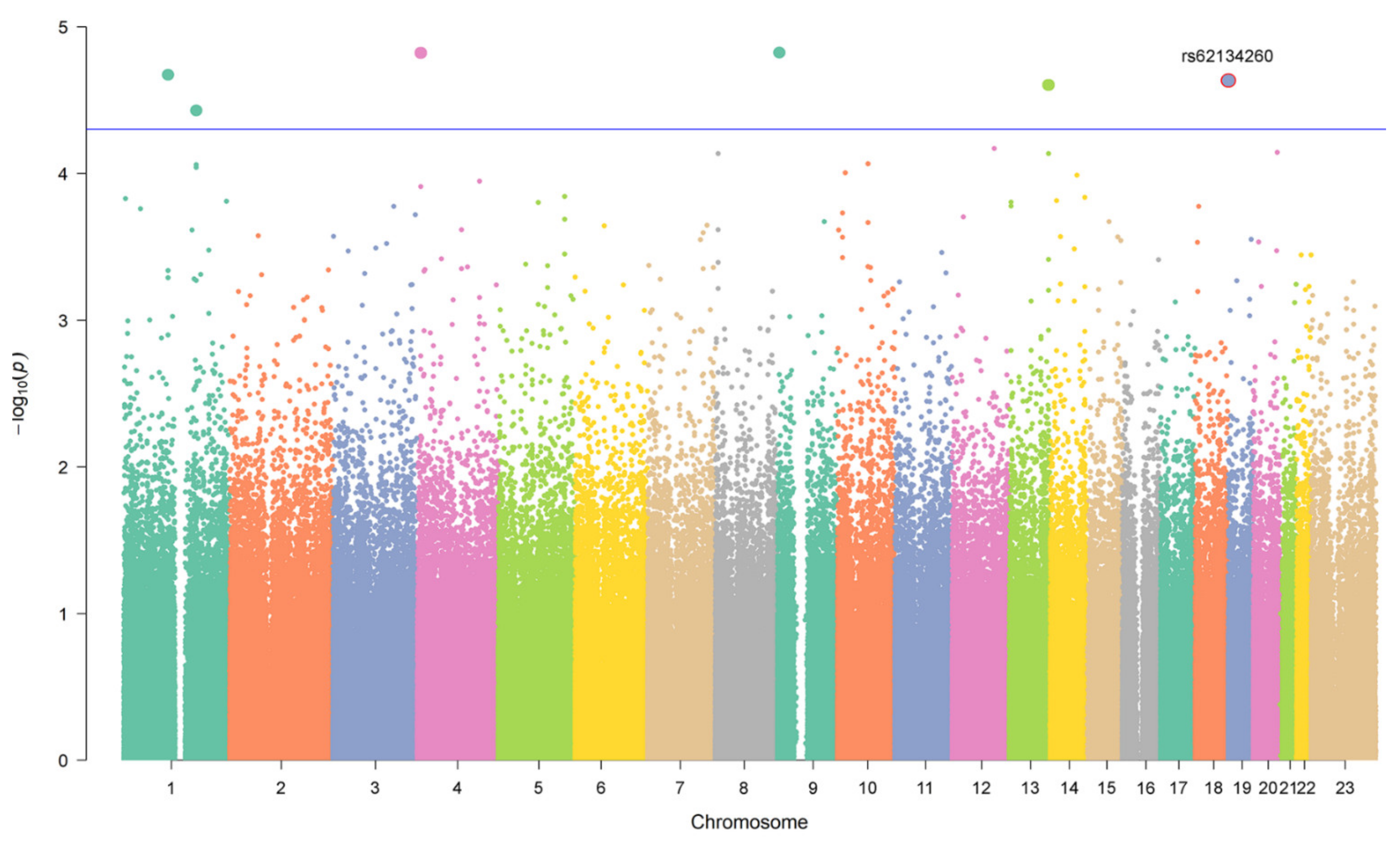
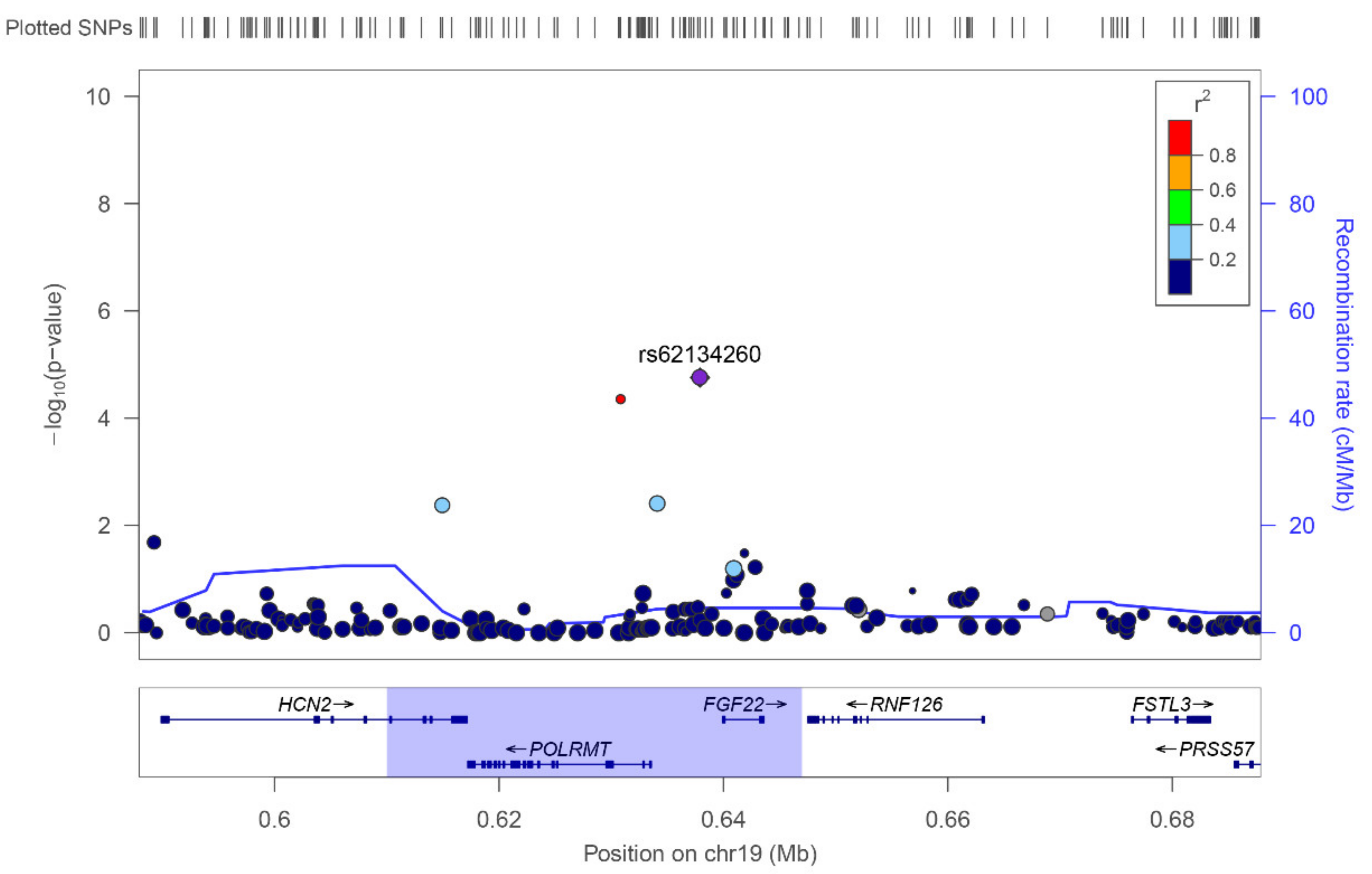
| 19 | rs62134260 | 4 kb from POLRMT, and 2 kb from FGF22 | G | 0.16 | Case-Control | 2.37 × 10−5 | 4.13 [2.14–7.97] | 0.48 | 1.27 [0.65–2.47] |
| Extreme Phenotypes | 2.23 × 10−5 | 5.76 [2.56–12.93] | 0.021 | 8.2 [1.36–49.35] | |||||
| 20 | rs6099854 | 225 kb from PMEPA1, and 214 kb from C20orf85 | A | 0.14 | Extreme Phenotypes | 3.77 × 10−5 | 6.57 [2.69–16.09] | 0.42 | 0.42 [0.05–3.47] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velasco-Ruiz, A.; Nuñez-Torres, R.; Pita, G.; Wildiers, H.; Lambrechts, D.; Hatse, S.; Delombaerde, D.; Van Brussel, T.; Alonso, M.R.; Alvarez, N.; et al. POLRMT as a Novel Susceptibility Gene for Cardiotoxicity in Epirubicin Treatment of Breast Cancer Patients. Pharmaceutics 2021, 13, 1942. https://doi.org/10.3390/pharmaceutics13111942
Velasco-Ruiz A, Nuñez-Torres R, Pita G, Wildiers H, Lambrechts D, Hatse S, Delombaerde D, Van Brussel T, Alonso MR, Alvarez N, et al. POLRMT as a Novel Susceptibility Gene for Cardiotoxicity in Epirubicin Treatment of Breast Cancer Patients. Pharmaceutics. 2021; 13(11):1942. https://doi.org/10.3390/pharmaceutics13111942
Chicago/Turabian StyleVelasco-Ruiz, Alejandro, Rocio Nuñez-Torres, Guillermo Pita, Hans Wildiers, Diether Lambrechts, Sigrid Hatse, Danielle Delombaerde, Thomas Van Brussel, M. Rosario Alonso, Nuria Alvarez, and et al. 2021. "POLRMT as a Novel Susceptibility Gene for Cardiotoxicity in Epirubicin Treatment of Breast Cancer Patients" Pharmaceutics 13, no. 11: 1942. https://doi.org/10.3390/pharmaceutics13111942
APA StyleVelasco-Ruiz, A., Nuñez-Torres, R., Pita, G., Wildiers, H., Lambrechts, D., Hatse, S., Delombaerde, D., Van Brussel, T., Alonso, M. R., Alvarez, N., Herraez, B., Vulsteke, C., Zamora, P., Lopez-Fernandez, T., & Gonzalez-Neira, A. (2021). POLRMT as a Novel Susceptibility Gene for Cardiotoxicity in Epirubicin Treatment of Breast Cancer Patients. Pharmaceutics, 13(11), 1942. https://doi.org/10.3390/pharmaceutics13111942