
Expression of Chimeric HPV-HIV Protein L1P18 in Pichia pastoris; Purification and Characterization of the Virus-like Particles



Abstract
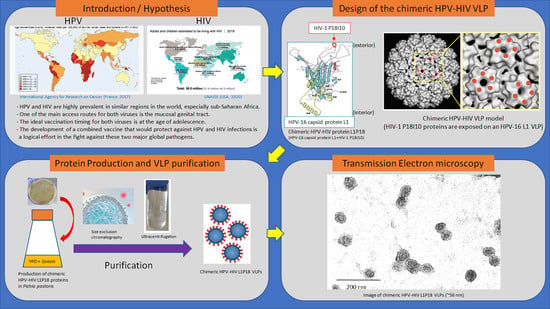
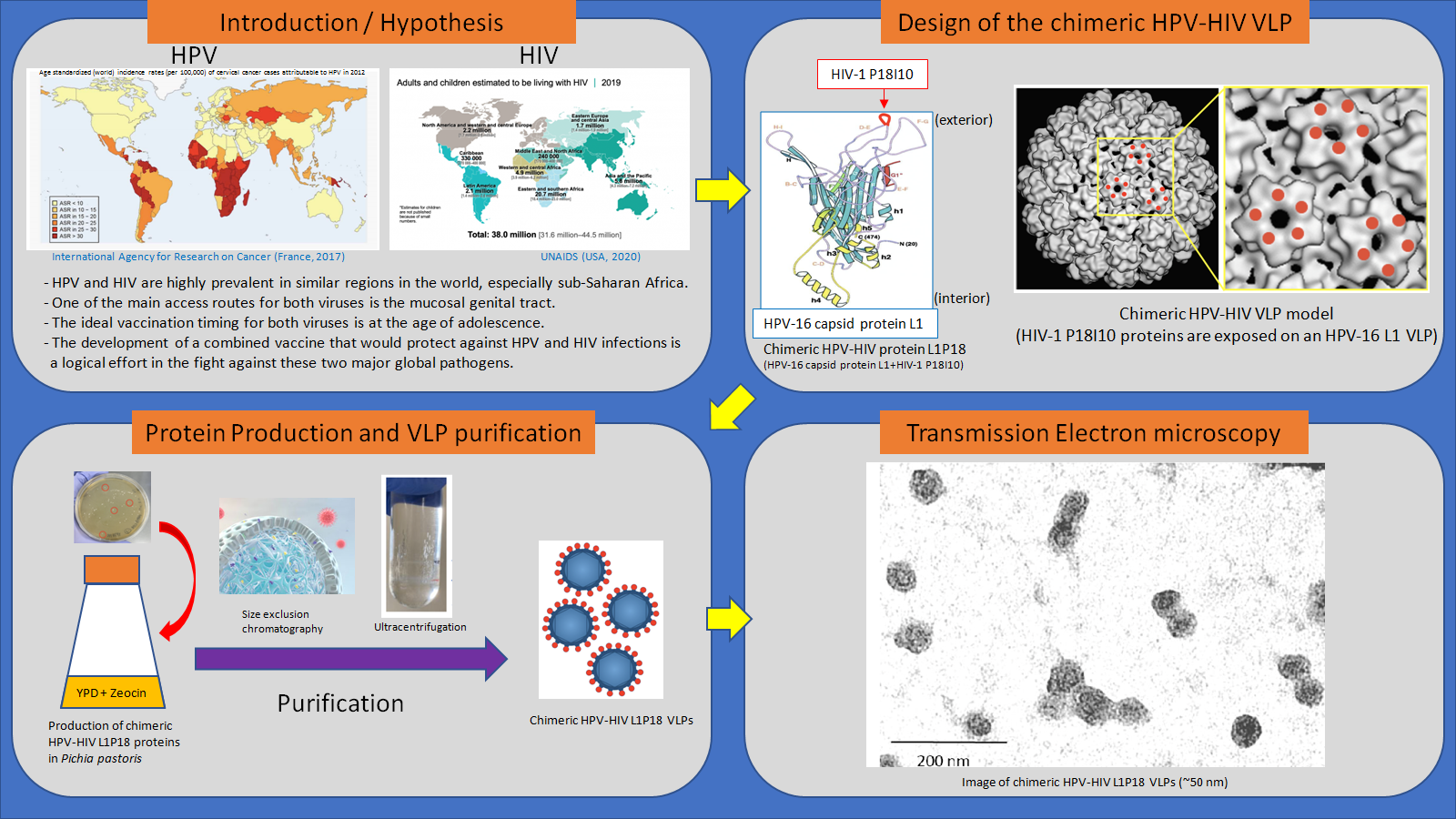
:
1. Introduction
2. Materials and Methods
2.1. Bacterial and Yeast Strains, and Cultures
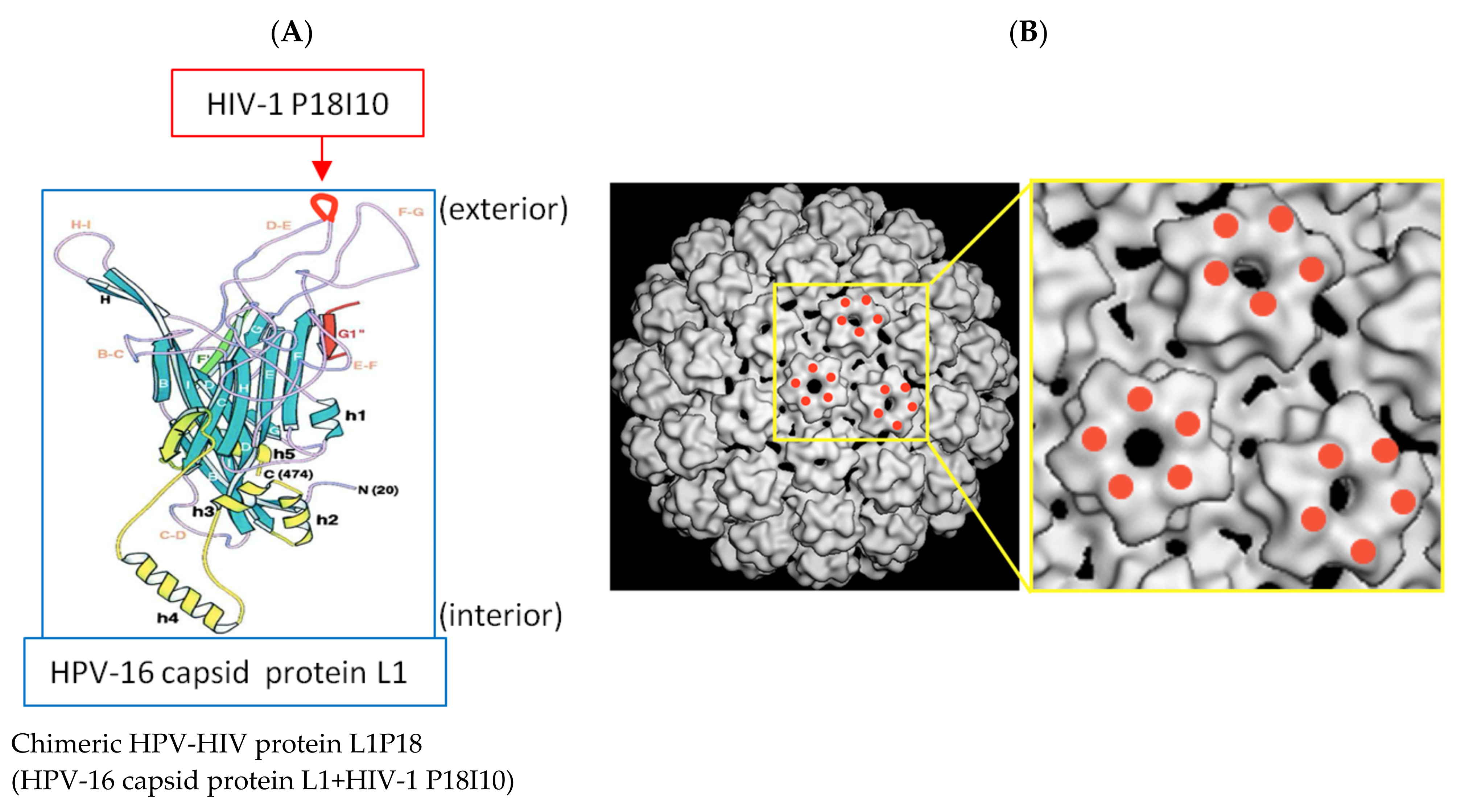
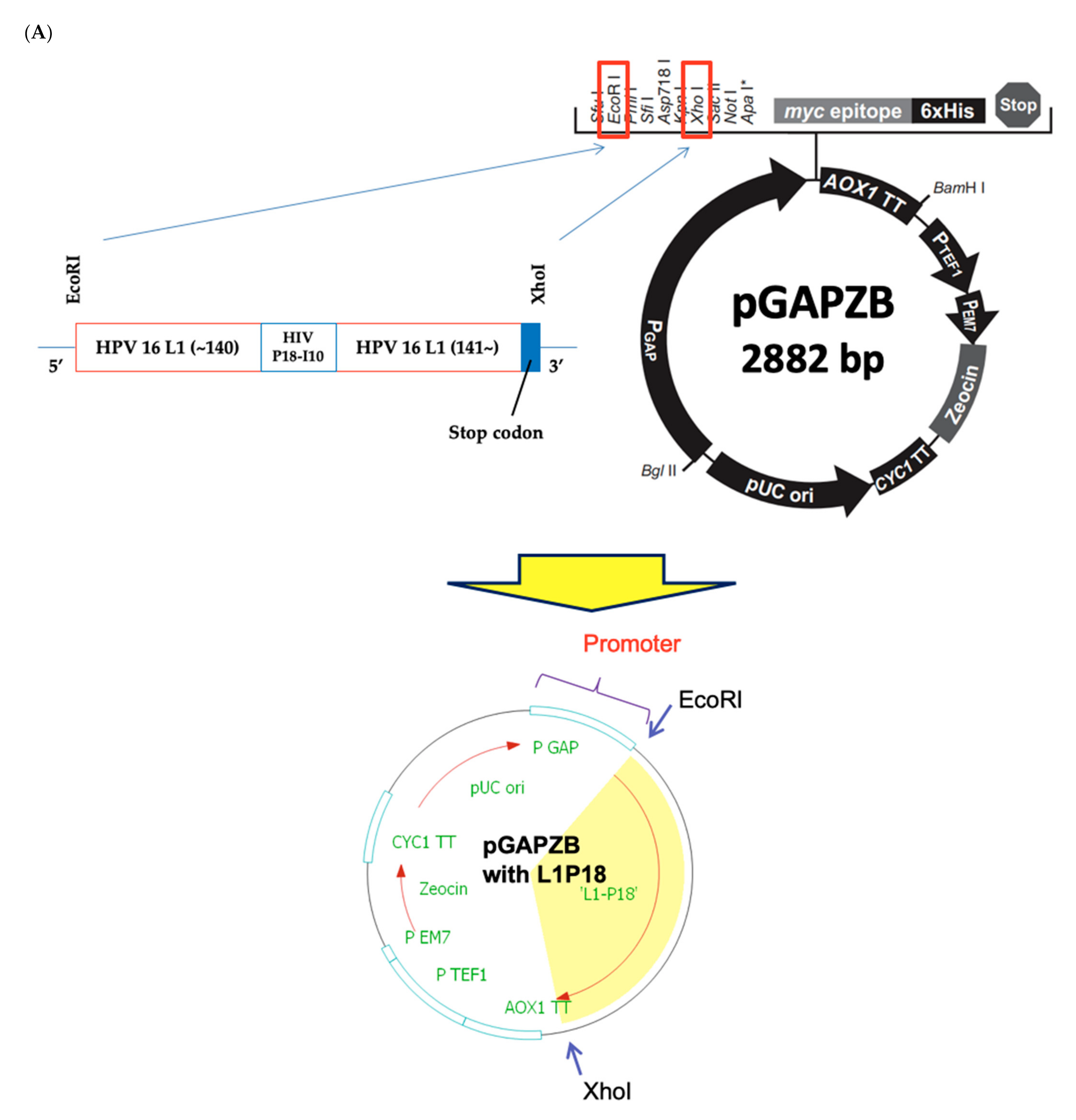
2.2. Design and Construction of Recombinant P. pastoris X-33 Strain Expressing Chimeric L1P18 Protein
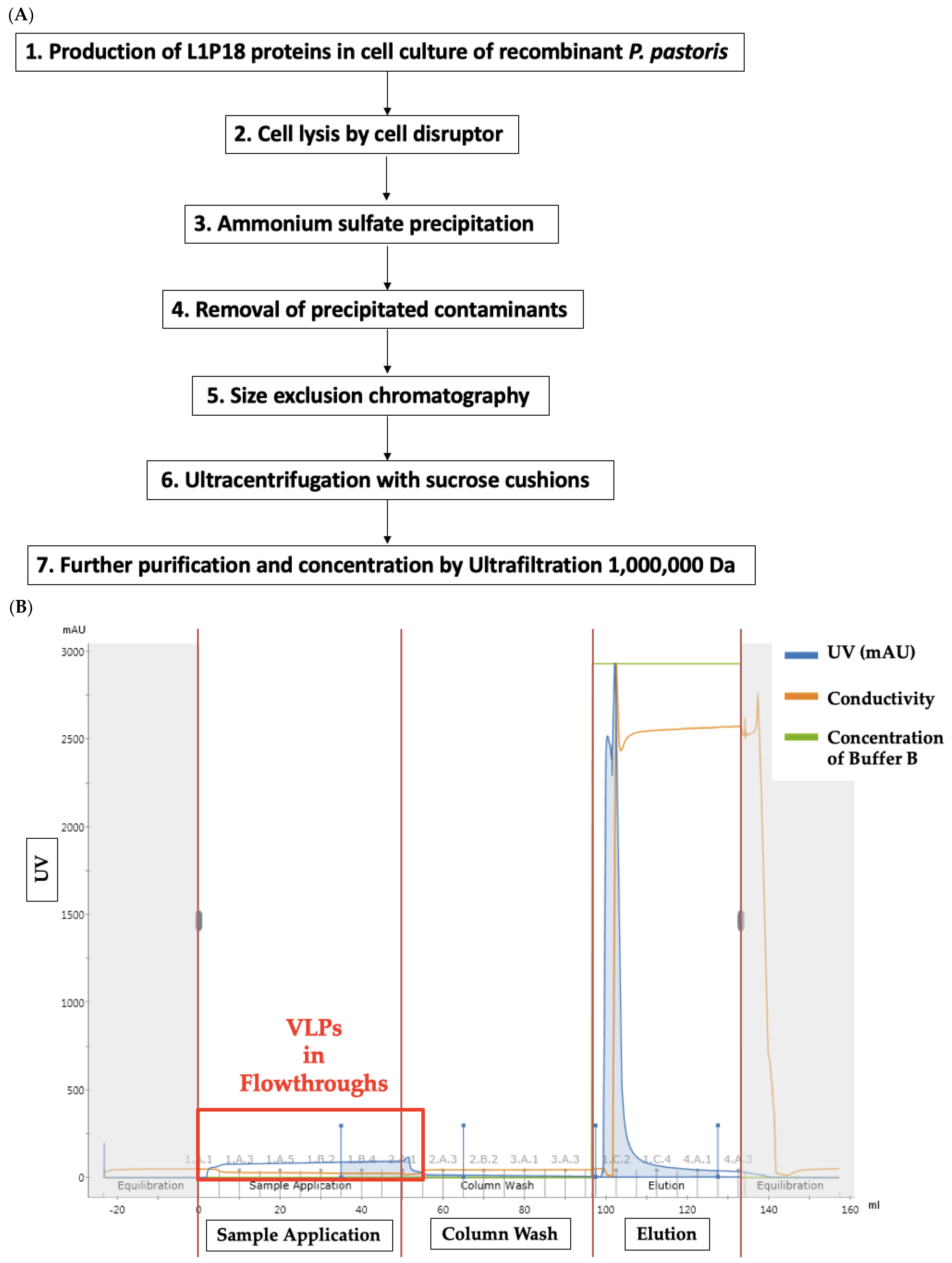
2.3. Production and Purification of L1P18 VLPs
2.4. Immunodot Analysis
2.5. Sodium Dodecyl Sulphate–Polyacrylamide gel Electrophoresis and Western Blot Analysis
2.6. Transmission Electron Microscopy of L1P18 VLPs
2.7. Heterologous Protein and Total Protein Quantification
2.8. Ethics Statement
3. Results
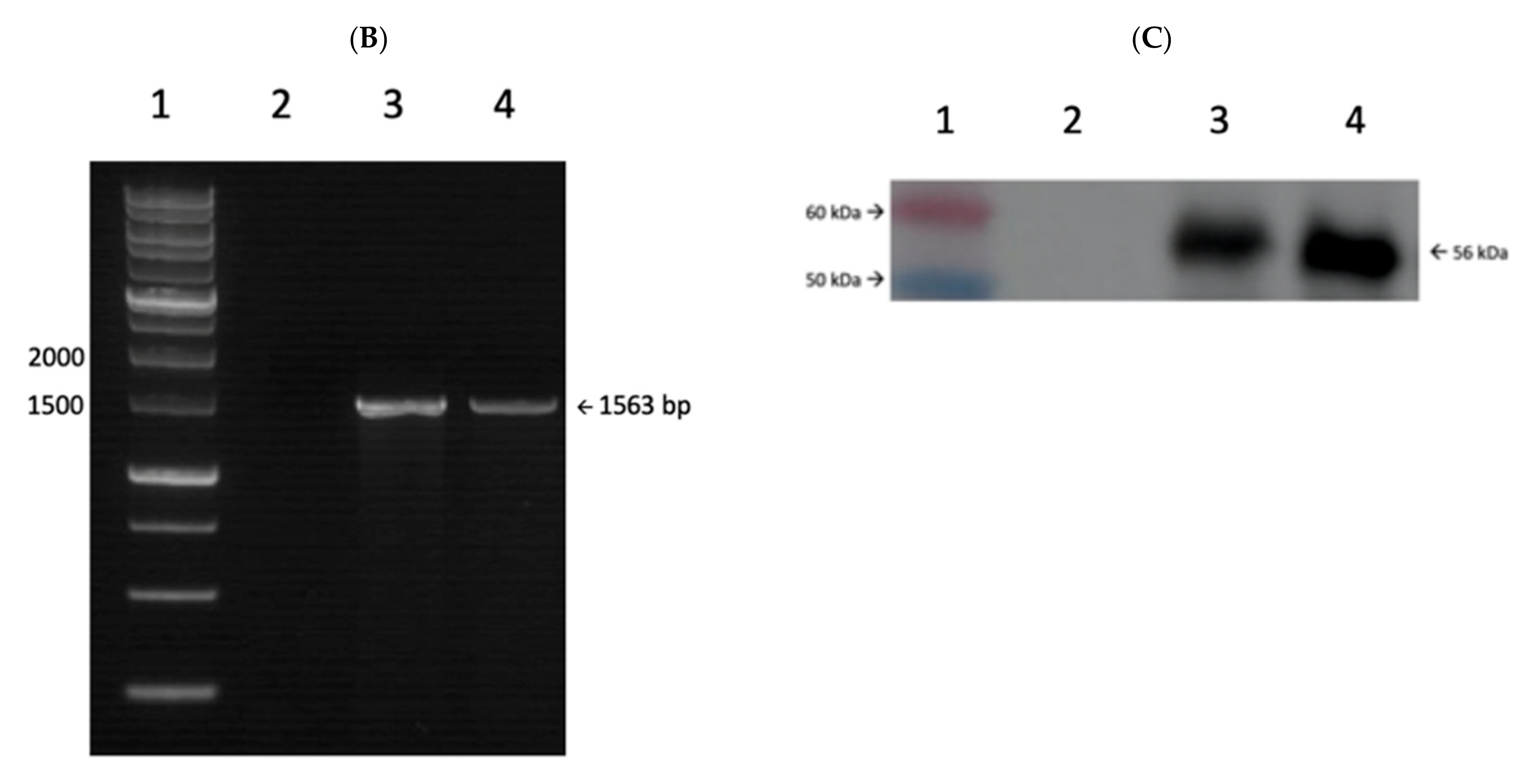
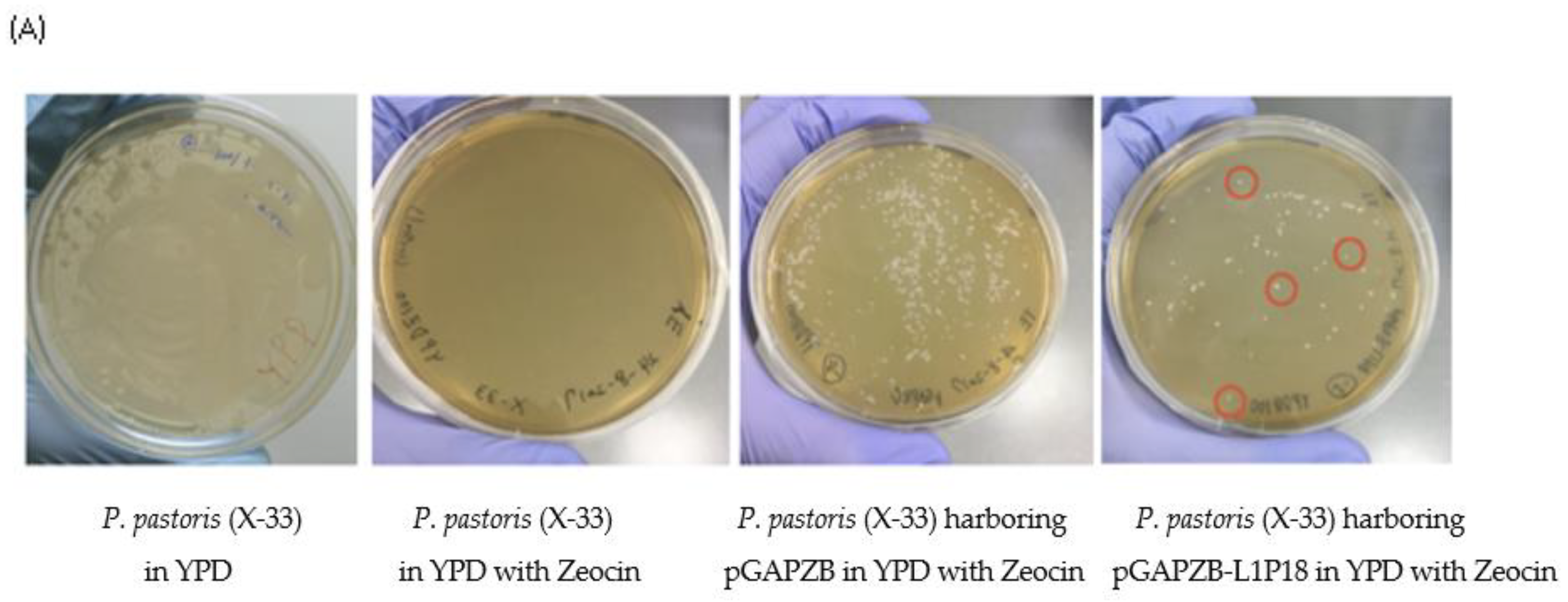
3.1. Construction of Recombinant P. pastoris X-33-L1P18I10 Strain
3.2. Genetic and Phenotypic Characterization of Recombinant X-33-L1P18I10 Yeast Strain
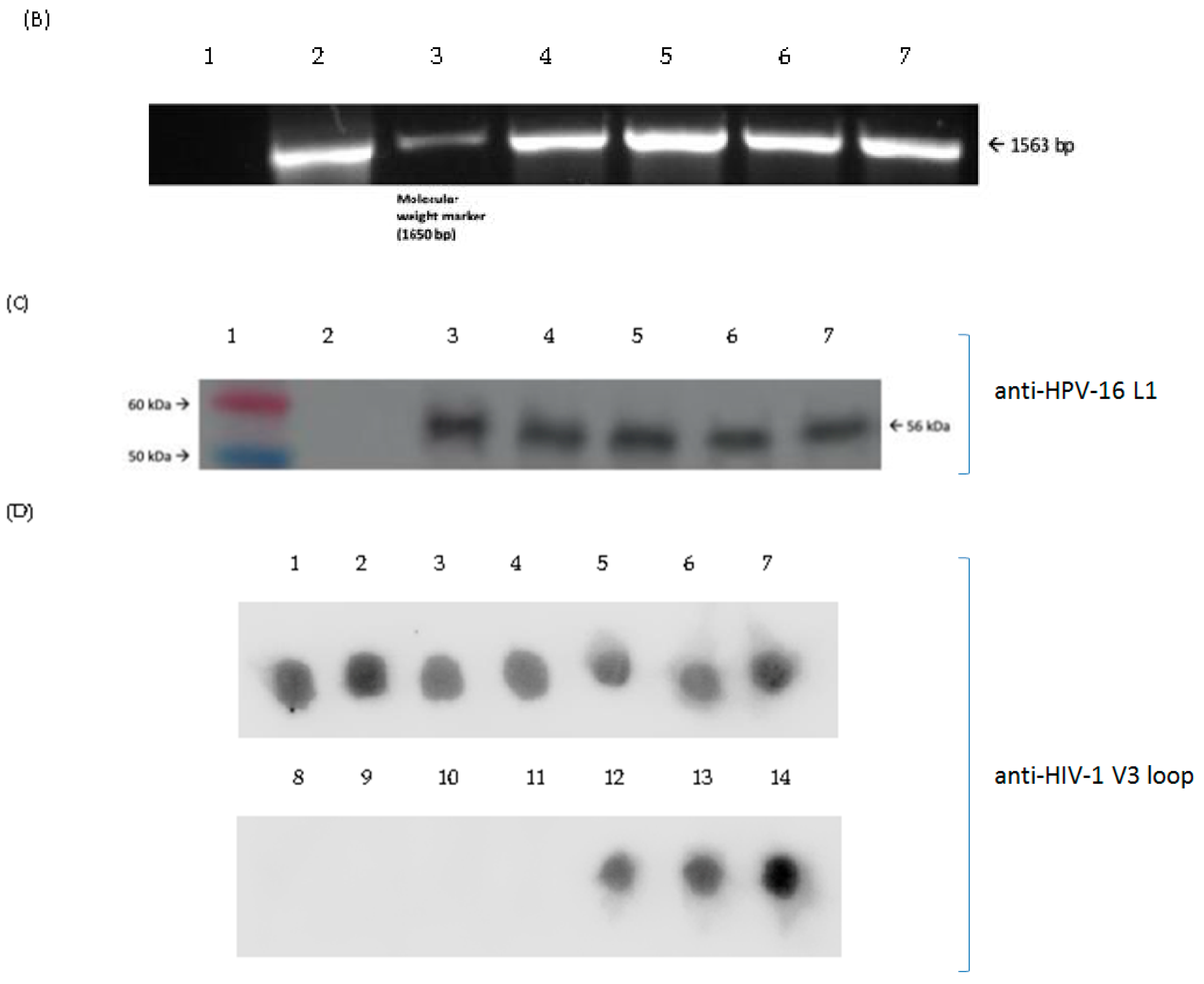
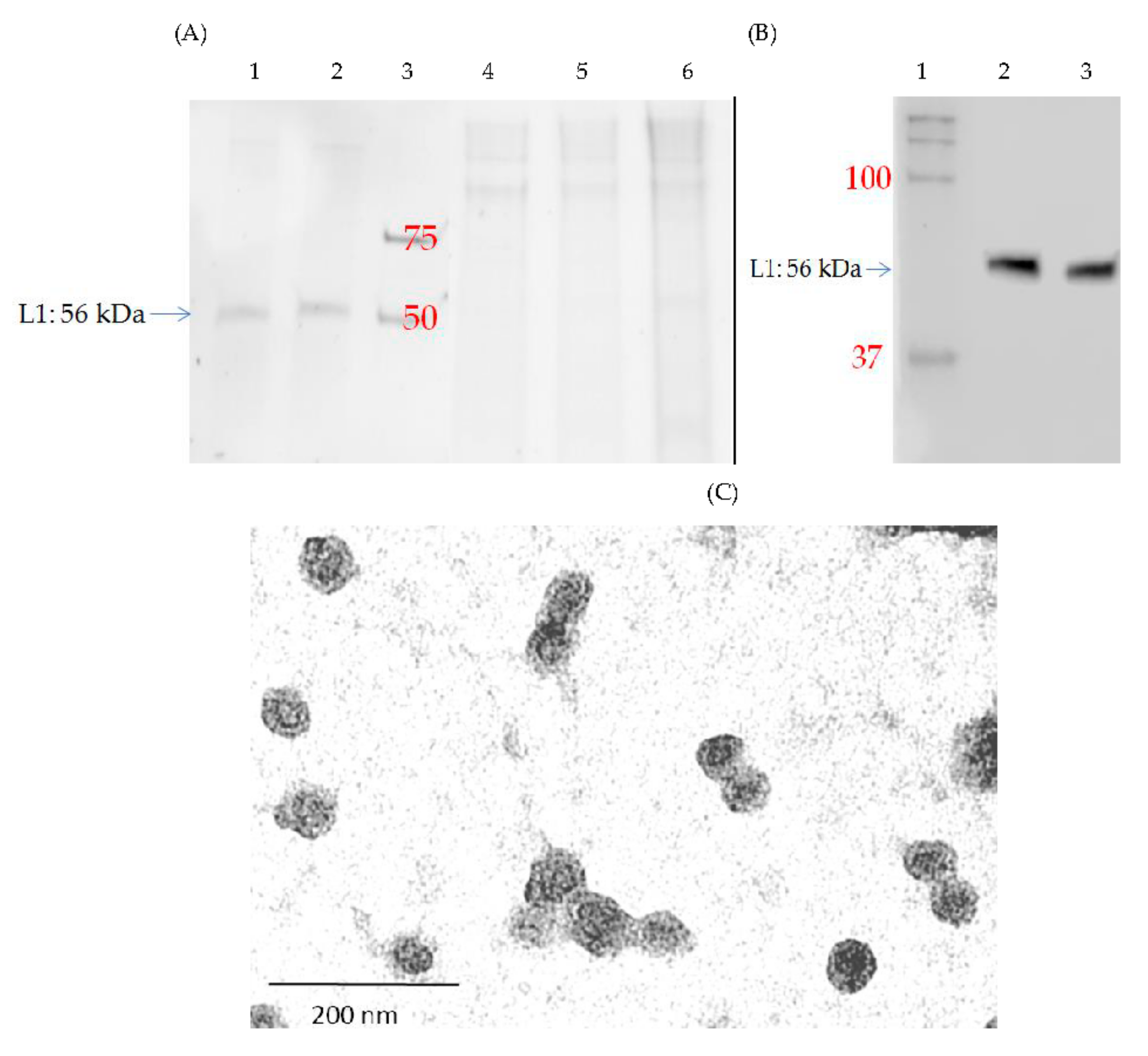
3.3. Production and Purification of Chimeric L1P18 VLPs
3.4. Characterization of Purified L1P18 VLPs
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bosch, F.X.; Manos, M.M.; Muñoz, N.; Sherman, M.; Jansen, A.M.; Peto, J.; Schiffman, M.H.; Moreno, V.; Kurman, R.; Shah, K.V. Prevalence of human papillomavirus in cervical cancer: A worldwide perspective. International biological study on cervical cancer (IBSCC) Study Group. J. Nat. Cancer Inst. 1995, 7, 796–802. [Google Scholar] [CrossRef]
- Zur Hausen, H. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 20, 260–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steben, M.; Duarte-Franco, E. Human papillomavirus infection: Epidemiology and pathophysiology. Gynecol. Oncol. 2007, 107, S2–S5. [Google Scholar] [CrossRef] [PubMed]
- Cervarix European Public Assessment Report. European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/cervarix (accessed on 26 August 2021).
- Gardasil European Public Assessment Report. European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/gardasil (accessed on 26 August 2021).
- Gardasil 9 European Public Assessment Report. European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/gardasil-9 (accessed on 26 August 2021).
- Centers for Disease Control and Prevention (CDC). MMWR. Morbidity and Mortality Weekly Report; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2010; Volume 59, pp. 626–629. [Google Scholar]
- D’Andrilli, G.; Bovicelli, A.; Giordano, A. HPV vaccines: State of the art. J. Cell Physiol. 2010, 224, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Cladel, N.M.; Budgeon, L.R.; Balogh, K.K.; Cooper, T.K.; Hu, J.; Christensen, N.D. A novel pre-clinical murine model to study the life cycle and progression of cervical and anal papillomavirus infections. PLoS ONE 2015, 10, e0120128. [Google Scholar] [CrossRef]
- Harper, D.M.; DeMars, L.R. HPV vaccines—A review of the first decade. Gynecol. Oncol. 2017, 146, 196–204, Erratum in 2017, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Global HIV & AIDS Statistics—2020 Fact Sheet. Available online: http://unaids.mio.guru/en/resources/fact-sheet (accessed on 26 August 2021).
- Unicef Press Release on 25 November 2020. 320,000 Children and Adolescents Newly Infected with HIV in 2019, 1 Every 100 Seconds—UNICEF. UNICEF Warns of COVID-19 Disruptions to HIV Service Delivery in One Third of High Burden Countries. Available online: https://www.unicef.org/press-releases/320000-children-and-adolescents-newly-infected-hiv-2019-1-every-100-seconds-unicef (accessed on 26 August 2021).
- Van de Perre, P.; Goga, A.; Ngandu, N.; Nagot, N.; Moodley, D.; King, R.; Molès, J.P.; Mosqueira, B.; Chirinda, W.; Scarlatti, G.; et al. Eliminating postnatal HIV transmission in high incidence areas: Need for complementary biomedical interventions. Lancet 2021, 3, 1316–1324. [Google Scholar] [CrossRef]
- Musumari, P.M.; Techasrivichien, T.; Srithanaviboonchai, K.; Wanyenze, R.K.; Matovu, J.K.B.; Poudyal, H.; Suguimoto, S.P.; Zamani, S.; Tangmunkongvorakul, A.; Ono-Kihara, M.; et al. HIV epidemic in fishing communities in Uganda: A scoping review. PLoS ONE 2021, 16, e0249465. [Google Scholar] [CrossRef] [PubMed]
- Esteban, I.; Pastor-Quiñones, C.; Usero, L.; Plana, M.; García, F.; Leal, L. In the Era of mRNA Vaccines, Is There Any Hope for HIV Functional Cure? Viruses 2021, 13, 3790. [Google Scholar] [CrossRef] [PubMed]
- He, X.Q.; Huang, Y.Q.; Zeng, Y.M.; Qin, Y.Y.; Tang, S.Q.; Xu, X.L.; Harypursat, V.; Lu, Y.Q.; Liu, M.; Yuan, J.; et al. Timing of antiretroviral therapy for HIV-infected patients with cytomegalovirus retinitis: Study protocol of a multi-center prospective randomized controlled trial. Trials 2021, 22, 218. [Google Scholar] [CrossRef] [PubMed]
- Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayaphan, S.; Kaewkungwal, J.; Chiu, J.; Paris, R.; Premsri, N.; Namwat, C.; de Souza, M.; Adams, E.; et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 2009, 361, 2209–2220. [Google Scholar] [CrossRef]
- Barouch, D.H. Challenges in the development of an HIV-1 vaccine. Nature 2008, 455, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Malagón, T.; MacCosham, A.; Burchell, A.N.; El-Zein, M.; Tellier, P.P.; Coutlée, F.; Franco, E.L. HITCH Study Group. Sex- and Type-specific Genital Human Papillomavirus Transmission Rates between Heterosexual Partners: A Bayesian Reanalysis of the HITCH Cohort. Epidemiology 2021, 32, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Cromwell, I.; Smith, L.W.; van der Hoek, K.; Hedden, L.; Coldman, A.J.; Cook, D.; Franco, E.L.; Krajden, M.; Martin, R.; Lee, M.H.; et al. Cost-effectiveness analysis of primary human papillomavirus testing in cervical cancer screening: Results from the HPV FOCAL Trial. Cancer Med. 2021, 10, 2996–3003. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Qin, C.; Chen, R.; Huang, Y.; Xu, Y.; Tang, Q.; Liang, X.; Peng, B.; Shao, Y.; Yang, Y.; et al. Epidemiological profile and molecular genetic characterization of HIV-1 among female sex workers and elderly male clients in Guangxi, China. Emerg. Microbes Infect. 2021, 10, 384–395. [Google Scholar] [CrossRef]
- Harding, B.N.; Avoundjian, T.; Heckbert, S.R.; Whitney, B.M.; Nance, R.M.; Ruderman, S.A.; Kalani, R.; Tirschwell, D.L.; Ho, E.L.; Becker, K.J.; et al. HIV Viremia and Risk of Stroke Among People Living with HIV Who Are Using Antiretroviral Therapy. Epidemiology 2021, 32, 457–464. [Google Scholar] [CrossRef]
- Barrero, J.J.; Pagazartaundua, A.; Glick, B.S.; Valero, F.; Ferrer, P. Bioreactor-scale cell performance and protein production can be substantially increased by using a secretion signal that drives co-translational translocation in Pichia pastoris. N. Biotechnol. 2021, 60, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Oyama, K.; Ohkuri, T.; Inoue, M.; Caaveiro, J.M.M.; Ueda, T. High-level expression of human CH2 domain from the Fc region in Pichia pastoris and preparation of anti-CH2 antibodies. J. Biochem. 2021, 27, mvab039. [Google Scholar] [CrossRef] [PubMed]
- Garrigós-Martínez, J.; Vuoristo, K.; Nieto-Taype, M.A.; Tähtiharju, J.; Uusitalo, J.; Tukiainen, P.; Schmid, C.; Tolstorukov, I.; Madden, K.; Penttilä, M.; et al. Bioprocess performance analysis of novel methanol-independent promoters for recombinant protein production with Pichia pastoris. Microb. Cell Fact. 2021, 20, 74. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Yang, N.; Teng, D.; Hao, Y.; Ma, X.; Mao, R.; Wang, J. Design and High Expression of Non-glycosylated Lysostaphins in Pichia pastoris and Their Pharmacodynamic Study. Front. Microbiol. 2021, 12, 637662. [Google Scholar] [CrossRef] [PubMed]
- Vidyasagar, P.; Sridevi, V.N.; Rajan, S.; Praveen, A.; Srikanth, A.; Abhinay, G.; Siva Kumar, V.; Verma, R.R.; Rajendra, L. Generation and characterization of neutralizing monoclonal antibodies against baculo-expressed HPV 16 VLPs. Eur. J. Microbiol. Immunol. 2014, 4, 56–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Liu, H.; Zheng, J.; Chen, H.; Yang, J.; Wang, L.; Yan, X.; Wang, Y.; Si, L. Carboxyl terminus truncated human papillomavirus type L1 protein maintains its bioactivity and ability to form virus-like particles. J. Huazhong Univ. Sci. Technol. Med. Sci. 2004, 24, 537–539. [Google Scholar] [PubMed]
- Rose, R.C.; Bonnez, W.; Reichman, R.C.; Garcea, R.L. Expression of human papillomavirus type 11 L1 protein in insect cells: In vivo and in vitro assembly of viruslike particles. J. Virol. 1993, 67, 1936–1944. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Potter, C.S.; Carragher, B.; Lander, G.; Sworen, J.; Towne, V.; Abraham, D.; Duncan, P.; Washabaugh, M.W.; Sitrin, R.D. Characterization of virus-like particles in GARDASIL® by cryo transmission electron microscopy. Hum. Vaccines Immunother. 2014, 10, 734–739. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.S.; Casini, G.; Harrison, S.C.; Garcea, R.L. Papillomavirus capsid protein expression in Escherichia coli: Purification and assembly of HPV11 and HPV16 L. J. Mol. Biol. 2001, 16, 173–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Castro, R.; Acero Galindo, G.; García Salcedo, Y.; Uribe Campero, L.; Vazquez Perez, V.; Carrillo-Tripp, M.; Gevorkian, G.; Gomez Lim, M.A. Plant-based chimeric HPV-virus-like particles bearing amyloid-β epitopes elicit antibodies able to recognize amyloid plaques in APP-tg mouse and Alzheimer’s disease brains. Inflammopharmacology 2018, 26, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Olczak, P.; Roden, R.B.S. Progress in L2-Based Prophylactic Vaccine Development for Protection against Diverse Human Papillomavirus Genotypes and Associated Diseases. Vaccines 2020, 8, 568. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.; Ault, K.; Rouphael, N.; Beck, A.; Domjahn, B.; Xu, Y.; Anderson, E.J.; Cheng, A.; Nakamura, A.; Hoagland, R.J.; et al. Duration of Cellular and Humoral Responses after Quadrivalent Human Papillomavirus Vaccination in Healthy Female Adults with or without Prior Type 16 and/or 18 Exposure. Vaccines 2020, 30, 348. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Biswas, M.; Jose, T. HPV vaccine: Current status and future directions. Med. J. Armed Forces India 2015, 71, 171–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, H.; Cohen, J.; Hosmalin, A.; Cease, K.B.; Houghten, R.; Cornette, J.L.; DeLisi, C.; Moss, B.; Germain, R.N.; Berzofsky, J.A. An immunodominant epitope of the human immunodeficiency virus envelope glycoprotein gp160 recognized by class I major histocompatibility moleculerestricted murine cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA 1988, 85, 3105–3109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazan, S.B.; de Alencar Muniz Chaves, A.; Aires, K.A.; Cianciarullo, A.M.; Garcea, R.L.; Ho, P.L. Expression and characterization of HPV-16 L1 capsid protein in Pichia pastoris. Arch. Virol. 2009, 154, 1609–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanumantha Rao, N.; Baji Babu, P.; Rajendra, L.; Sriraman, R.; Pang, Y.Y.; Schiller, J.T.; Srinivasan, V.A. Expression of codon optimized major capsid protein (L1) of human papillomavirus type 16 and 18 in Pichia pastoris; purification and characterization of the virus-like particles. Vaccine 2011, 29, 7326–7334. [Google Scholar] [CrossRef] [Green Version]
- Sadeyen, J.R.; Tourne, S.; Shkreli, M.; Sizaret, P.Y.; Coursaget, P. Insertion of a foreign sequence on capsid surface loops of human papillomavirus type 16 virus-like particles reduces their capacity to induce neutralizing antibodies and delineates a conformational neutralizing epitope. Virology 2003, 309, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.S.; Garcea, R.L.; Goldberg, I.; Casini, G.; Harrison, S.C. Structure of small virus-like particles assembled from the L1 protein of human papillomavirus. Mol. Cell. 2000, 5, 557–567. [Google Scholar] [CrossRef]
- Buck, C.B.; Day, P.M.; Trus, B.L. The papillomavirus major capsid protein L. Virology 2013, 445, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Fuenmayor, J.; Gòdia, F.; Cervera, L. Production of virus-like particles for vaccines. N. Biotechnol. 2017, 39, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Eto, Y.; Saubi, N.; Ferrer, P.; Joseph, J. Designing Chimeric Virus-like Particle-based Vaccines for Human Papillomavirus and HIV: Lessons Learned. AIDS Rev. 2019, 21, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Doan, L.X.; Li, M.; Chen, C.; Yao, Q. Virus-like particles as HIV-1 vaccines. Rev. Med. Virol. 2005, 15, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, S.Y.; Lim, S.J.; Kim, J.Y.; Lee, S.J.; Kim, H.J. One-step chromatographic purification of human papillomavirus type 16 L1 protein from Saccharomyces cerevisiae. Protein Expr. Purif. 2010, 70, 68–74. [Google Scholar] [CrossRef]
- Park, M.A.; Kim, H.J.; Kim, H.J. Optimum conditions for production and purification of human papillomavirus type 16 L1 protein from Saccharomyces cerevisiae. Protein Expr. Purif. 2008, 59, 175–181. [Google Scholar] [CrossRef] [PubMed]
- GE Healthcare. The Use of Capto Core 700 and Capto Q ImpRes in the Purification of Human Papilloma Virus Like Particles. Application Note 29-0983-01 AB (Oct 2014). Available online: https://cdn.cytivalifesciences.com/dmm3bwsv3/AssetStream.aspx?mediaformatid=10061&destinationid=10016&assetid=17140 (accessed on 26 August 2021).
- Law, K.H.; Tsang, M.W.; Wong, Y.K.; Tsang, M.S.; Lau, P.Y.; Wong, K.Y.; Ho, K.P.; Leung, Y.C. Efficient production of secretory Streptomyces clavuligerus β-lactamase inhibitory protein (BLIP) in Pichia pastoris. AMB Express 2018, 8, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahrl, R.J.; Gasser, B.; Mattanovich, D.; Ferrer, P. Detection and Elimination of Cellular Bottlenecks in Protein-Producing Yeasts. Methods Mol. Biol. 1923, 75–95. [Google Scholar] [CrossRef]
- Coimbra, E.C.; Gomes, F.B.; Campos, J.F.; D’arc, M.; Carvalho, J.C.; Mariz, F.C.; Jesus, A.L.; Stocco, R.C.; Beçak, W.; Freitas, A.C. Production of L1 protein from different types of HPV in Pichia pastoris using an integrative vector. Braz. J. Med. Biol. Res. 2011, 44, 1209–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrero, J.J.; Casler, J.C.; Valero, F.; Ferrer, P.; Glick, B.S. An improved secretion signal enhances the secretion of model proteins from Pichia pastoris. Microb. Cell Fact. 2018, 17, 161. [Google Scholar] [CrossRef]
- Cook, J.C.; Joyce, J.G.; George, H.A.; Schultz, L.D.; Hurni, W.M.; Jansen, K.U.; Hepler, R.W.; Ip, C.; Lowe, R.S.; Keller, P.M.; et al. Purification of virus-like particles of recombinant human papillomavirus type 11 major capsid protein L1 from Saccharomyces cerevisiae. Protein Expr. Purif. 1999, 17, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, D.; Rolf, T.; Suckow, M.; Kranz, A.; Barbian, A.; Chan, J.A.; Leitsch, J.; Weniger, M.; Jenzelewski, V.; Kouskousis, B.; et al. Establishment of a yeast-based VLP platform for antigen presentation. Microb. Cell Fact. 2018, 17, 17. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step | Total Protein (µg) a | Total L1P18 (µg) | Recovery Yield (%) | Purity (%) c |
|---|---|---|---|---|
| Cell lysate supernatants | - | 81.3 b | 100 | - |
| Ammonium sulfate precipitation/ removal of precipitated contaminants | - | 46.2 b | 56.8 | - |
| Size exclusion chromatography | - | 12.4 b | 15.3 | - |
| Ultracentrifugation | - | 9.57 b | 11.8 | - |
| Ultrafiltration | 7.81 | 7.50 d | 9.23 | 96% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eto, Y.; Saubi, N.; Ferrer, P.; Joseph-Munné, J. Expression of Chimeric HPV-HIV Protein L1P18 in Pichia pastoris; Purification and Characterization of the Virus-like Particles. Pharmaceutics 2021, 13, 1967. https://doi.org/10.3390/pharmaceutics13111967
Eto Y, Saubi N, Ferrer P, Joseph-Munné J. Expression of Chimeric HPV-HIV Protein L1P18 in Pichia pastoris; Purification and Characterization of the Virus-like Particles. Pharmaceutics. 2021; 13(11):1967. https://doi.org/10.3390/pharmaceutics13111967
Chicago/Turabian StyleEto, Yoshiki, Narcís Saubi, Pau Ferrer, and Joan Joseph-Munné. 2021. "Expression of Chimeric HPV-HIV Protein L1P18 in Pichia pastoris; Purification and Characterization of the Virus-like Particles" Pharmaceutics 13, no. 11: 1967. https://doi.org/10.3390/pharmaceutics13111967
APA StyleEto, Y., Saubi, N., Ferrer, P., & Joseph-Munné, J. (2021). Expression of Chimeric HPV-HIV Protein L1P18 in Pichia pastoris; Purification and Characterization of the Virus-like Particles. Pharmaceutics, 13(11), 1967. https://doi.org/10.3390/pharmaceutics13111967