
Assessing the Mechanism of Fluoxetine-Mediated CYP2D6 Inhibition

 , ,
, , 

{kind=link}
Abstract
:1. Introduction
2. Mechanisms of CYP450 Inhibition
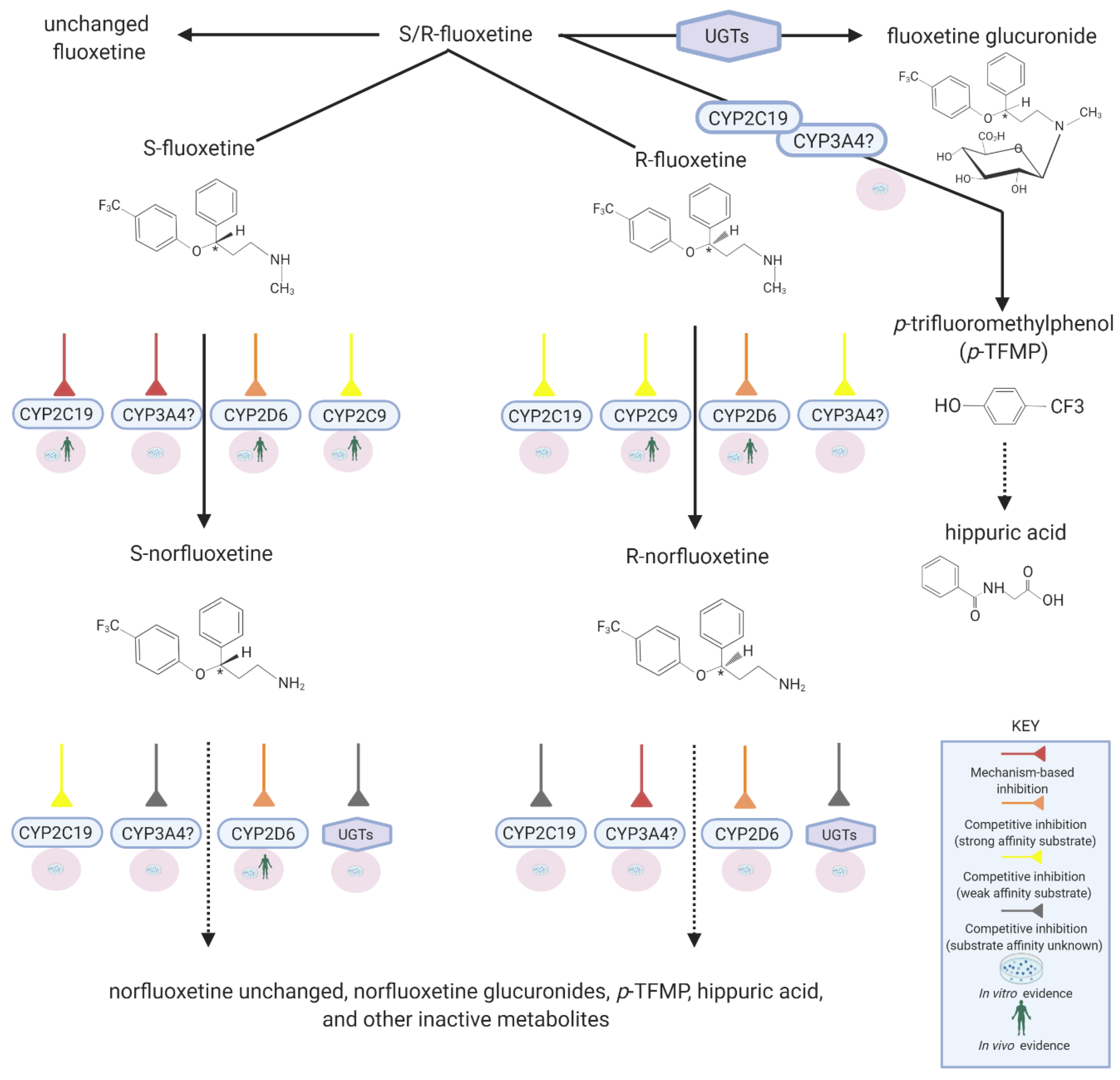
3. Fluoxetine Metabolism
4. Mechanism of CYP2D6 Inhibition by Fluoxetine
5. Time Course of CYP2D6 Inhibition
6. CYP2C19 and Fluoxetine
7. CYP2C9 and Fluoxetine
8. CYP3A4 and Fluoxetine
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rushton, C.A.; Strömberg, A.; Jaarsma, T.; Kadam, U.T. Multidrug and optimal heart failure therapy prescribing in older general practice populations: A clinical data linkage study. BMJ Open 2014, 4, e003698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.; Preskorn, S. Multiple Medication Use in General Practice and Psychiatry: So What? Psychiatr. Times 2005, 22, 8. [Google Scholar]
- Miccoli, R.; Penno, G.; del Prato, S. Multidrug Treatment of Type 2 Diabetes. Diabetes Care 2011, 34 (Suppl. 2), S231–S235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilsdon, T.D.; Hill, C.L. Managing the drug treatment of rheumatoid arthritis. Aust. Prescr. 2017, 40, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courlet, P.; Livio, F.; Guidi, M.; Cavassini, M.; Battegay, M.; Stoeckle, M.; Buclin, T.; Alves Saldanha, S.; Csajka, C.; Marzolini, C.; et al. Polypharmacy, Drug–Drug Interactions, and Inappropriate Drugs: New Challenges in the Aging Population With HIV. Open Forum Infect. Dis. 2019, 6. [Google Scholar] [CrossRef] [Green Version]
- Al-Musawe, L.; Torre, C.; Guerreiro, J.P.; Rodrigues, A.T.; Raposo, J.F.; Mota-Filipe, H.; Martins, A.P. Polypharmacy, potentially serious clinically relevant drug-drug interactions, and inappropriate medicines in elderly people with type 2 diabetes and their impact on quality of life. Pharmacol. Res. Perspect. 2020, 8, e00621. [Google Scholar] [CrossRef] [PubMed]
- Khandeparkar, A.; Rataboli, P.V. A study of harmful drug-drug interactions due to polypharmacy in hospitalized patients in Goa Medical College. Perspect. Clin. Res. 2017, 8, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Nachega, J.B.; Hsu, A.J.; Uthman, O.A.; Spinewine, A.; Pham, P.A. Antiretroviral therapy adherence and drug-drug interactions in the aging HIV population. AIDS 2012, 26 (Suppl. 1), S39–S53. [Google Scholar] [CrossRef]
- Campbell, N.L.; Dexter, P.; Perkins, A.J.; Gao, S.; Li, L.; Skaar, T.C.; Frame, A.; Hendrie, H.C.; Callahan, C.M.; Boustani, M.A. Medication adherence and tolerability of Alzheimer’s disease medications: Study protocol for a randomized controlled trial. Trials 2013, 14, 125. [Google Scholar] [CrossRef] [Green Version]
- McDonnell, A.M.; Dang, C.H. Basic review of the cytochrome p450 system. J. Adv. Pract. Oncol. 2013, 4, 263–268. [Google Scholar] [CrossRef]
- Estabrook, R.W. A passion for P450s (rememberances of the early history of research on cytochrome P450). Drug Metab. Dispos. 2003, 31, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Cytochrome p450 and chemical toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, A.H.; Gbedemah, M.; Martinez, A.M.; Nash, D.; Galea, S.; Goodwin, R.D. Trends in depression prevalence in the USA from 2005 to 2015: Widening disparities in vulnerable groups. Psychol. Med. 2018, 48, 1308–1315. [Google Scholar] [CrossRef] [PubMed]
- Mojtabai, R.; Olfson, M.; Han, B. National Trends in the Prevalence and Treatment of Depression in Adolescents and Young Adults. Pediatrics 2016, 138, 6. [Google Scholar] [CrossRef] [Green Version]
- Case, A.; Deaton, A. Rising morbidity and mortality in midlife among white non-Hispanic Americans in the 21st century. Proc. Natl. Acad. Sci. USA 2015, 112, 15078–15083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brody, D.J.; Gu, Q. Antidepressant Use Among Adults: United States, 2015–2018. Hyattsville, 2020. Available online: https://www.cdc.gov/nchs/data/databriefs/db377-H.pdf (accessed on 23 November 2020).
- Kane, S. Fluoxetine Hydrochloride, ClinCalc DrugStats Database, Version 21.0. Available online: https://clincalc.com/DrugStats/Drugs/FluoxetineHydrochloride (accessed on 23 November 2020).
- Prozac (Fluoxetine Capsules), US Food and Drug Administration. Revised 2017. 1987. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/018936s108lbl.pdf. (accessed on 23 November 2020).
- Wong, D.T.; Perry, K.W.; Bymaster, F.P. Case history: The discovery of fluoxetine hydrochloride (Prozac). Nat. Rev. Drug Discov. 2005, 4, 764–774. [Google Scholar] [CrossRef]
- Pigott, T.A.; Seay, S.M. A review of the efficacy of selective serotonin reuptake inhibitors in obsessive-compulsive disorder. J. Clin. Psychiatry 1999, 60, 101–106. [Google Scholar] [CrossRef]
- Wong, D.T.; Bymaster, F.P.; Engleman, E.A. Prozac (fluoxetine, lilly 110140), the first selective serotonin uptake inhibitor and an antidepressant drug: Twenty years since its first publication. Life Sci. 1995, 57, 411–441. [Google Scholar] [CrossRef]
- Mandrioli, R.; Forti, G.C.; Raggi, M.A. Fluoxetine metabolism and pharmacological interactions: The role of cytochrome p450. Curr. Drug Metab. 2006, 7, 127–133. [Google Scholar] [CrossRef]
- Dean, L. Amitriptyline Therapy and CYP2D6 and CYP2C19 Genotype; Pratt, V.M., McLeod, H.L., Rubinstein, W.S., Scott, S.A., Dean, L.C., Kattman, B.L., Malheiro, A.J., Eds.; National Center for Biotechnology Information: Bethesd, MD, USA, 2012. [Google Scholar]
- Smith, H.S. Opioid metabolism. Mayo Clin. Proc. 2009, 84, 613–624. [Google Scholar] [CrossRef] [Green Version]
- Jazwinska-Tarnawska, E.; Orzechowska-Juzwenko, K.; Niewinski, P.; Rzemislawska, Z.; Loboz-Grudzien, K.; Dmochowska-Perz, M.; Slawin, J. The influence of CYP2D6 polymorphism on the antiarrhythmic efficacy of propafenone in patients with paroxysmal atrial fibrillation during 3 months propafenone prophylactic treatment. Int. J. Clin. Pharmacol. Ther. 2001, 39, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Amchin, J.; Ereshefsky, L.; Zarycranski, W.; Taylor, K.; Albano, D.; Klockowski, P.M. Effect of venlafaxine versus fluoxetine on metabolism of dextromethorphan, a CYP2D6 probe. J. Clin. Pharmacol. 2001, 41, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Liston, H.L.; DeVane, C.L.; Boulton, D.W.; Risch, S.C.; Markowitz, J.S.; Goldman, J. Differential time course of cytochrome P450 2D6 enzyme inhibition by fluoxetine, sertraline, and paroxetine in healthy volunteers. J. Clin. Psychopharmacol. 2002, 22, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Deodhar, M.; Al Rihani, S.B.; Arwood, M.J.; Darakjian, L.; Dow, P.; Turgeon, J.; Michaud, V. Mechanisms of CYP450 Inhibition: Understanding Drug-Drug Interactions Due to Mechanism-Based Inhibition in Clinical Practice. Pharmaceutics 2020, 12, 846. [Google Scholar] [CrossRef] [PubMed]
- Shou, M.; Lin, Y.; Lu, P.; Tang, C.; Mei, Q.; Cui, D.; Tang, W.; Ngui, J.S.; Lin, C.C.; Singh, R.; et al. Enzyme kinetics of cytochrome P450-mediated reactions. Curr. Drug Metab. 2001, 2, 17–36. [Google Scholar] [CrossRef]
- Kalgutkar, A.S.; Obach, R.S.; Maurer, T.S. Mechanism-based inactivation of cytochrome P450 enzymes: Chemical mechanisms, structure-activity relationships and relationship to clinical drug-drug interactions and idiosyncratic adverse drug reactions. Curr. Drug Metab. 2007, 8, 407–447. [Google Scholar] [CrossRef]
- Fontana, E.; Dansette, P.M.; Poli, S.M. Cytochrome p450 enzymes mechanism based inhibitors: Common sub-structures and reactivity. Curr. Drug Metab. 2005, 6, 413–454. [Google Scholar] [CrossRef]
- Benfield, P.; Heel, R.C.; Lewis, S.P. Fluoxetine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy in depressive illness. Drugs 1986, 32, 481–508. [Google Scholar] [CrossRef]
- Liu, Z.-Q.; Tan, Z.-R.; Wang, D.; Huang, S.-L.; Wang, L.-S.; Zhou, H.-H. Simultaneous determination of fluoxetine and its metabolite p-trifluoromethylphenol in human liver microsomes using a gas chromatographic–electron-capture detection procedure. J. Chromatogr. B 2002, 769, 305–311. [Google Scholar] [CrossRef]
- Liu, Z.-Q.; Zhu, B.; Tan, Y.F.; Tan, Z.R.; Wang, L.S.; Huang, S.L.; Shu, Y.; Zhou, H.H. O-Dealkylation of fluoxetine in relation to CYP2C19 gene dose and involvement of CYP3A4 in human liver microsomes. J. Pharmacol. Exp. Ther. 2002, 300, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Urichuk, L.J.; Aspeslet, L.J.; Holt, A.; Silverstone, P.H.; Coutts, R.T.; Baker, G.B. Determination of p-trifluoromethylphenol, a metabolite of fluoxetine, in tissues and body fluids using an electron-capture gas chromatographic procedure. J. Chromatogr. B. Biomed. Sci. Appl. 1997, 698, 103–109. [Google Scholar] [CrossRef]
- Lerena, A.L.; Dorado, P.; Berecz, R.; González, A.P.; Ledó, E.M.P. Effect of CYP2D6 and CYP2C9 genotypes on fluoxetine and norfluoxetine plasma concentrations during steady-state conditions. Eur. J. Clin. Pharmacol. 2004, 59, 869–873. [Google Scholar] [CrossRef]
- Margolis, J.M.; Donnell, J.P.; Mankowski, D.C.; Ekins, S.; Obach, R.S. (R)-, (S)-, and Racemic Fluoxetine N-Demethylation by Human Cytochrome P450 Enzymes. Drug Metab. Dispos. 2000, 28, 1187–1191. [Google Scholar]
- Fjordside, L.; Jeppesen, U.; Eap, C.B.; Powell, K.; Baumann, P.; Brossen, K. The stereoselective metabolism of fluoxetine in poor and extensive metabolizers of sparteine. Pharmacogenet. Genom. 1999, 9. [Google Scholar] [CrossRef] [PubMed]
- Sager, J.E.; Lutz, J.D.; Foti, R.S.; Davis, C.; Kunze, K.L.; Isoherranen, N. Fluoxetine- and norfluoxetine-mediated complex drug-drug interactions: In vitro to in vivo correlation of effects on CYP2D6, CYP2C19, and CYP3A4. Clin. Pharmacol. Ther. 2014, 95, 653–662. [Google Scholar] [CrossRef] [Green Version]
- Alfaro, C.L.; Lam, Y.W.F.; Simpson, J.; Ereshefsky, L. CYP2D6 Inhibition by Fluoxetine, Paroxetine, Sertraline, and Venlafaxine in a Crossover Study: Intraindividual Variability and Plasma Concentration Correlations. J. Clin. Pharmacol. 2000, 40, 58–66. [Google Scholar] [CrossRef]
- Greenblatt, D.J.; Zhao, Y.; Venkatakrishnan, K.; Duan, S.X.; Harmatz, J.S.; Parent, S.J.; Court, M.H.; von Moltke, L.L. Mechanism of cytochrome P450-3A inhibition by ketoconazole. J. Pharm. Pharmacol. 2011, 63, 214–221. [Google Scholar] [CrossRef]
- Stevens, J.C.; Wrighton, S.A. Interaction of the enantiomers of fluoxetine and norfluoxetine with human liver cytochromes P450. J. Pharmacol. Exp. Ther. 1993, 266, 964–971. [Google Scholar]
- Bertelsen, K.M.; Venkatakrishnan, K.; von Moltke, L.L.; Obach, R.S.; Greenblatt, D.J. Apparent mechanism-based inhibition of human CYP2D6 in vitro by paroxetine: Comparison with fluoxetine and quinidine. Drug Metab. Dispos. 2003, 31, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Bertilsson, L.; Dahl, M.-L.; Dalén, P.; Al-Shurbaji, A. Molecular genetics of CYP2D6: Clinical relevance with focus on psychotropic drugs. Br. J. Clin. Pharmacol. 2002, 53, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Scordo, M.G.; Spina, E.; Dahl, M.-L.; Gatti, G.; Perucca, E. Influence of CYP2C9, 2C19 and 2D6 genetic polymorphisms on the steady-state plasma concentrations of the enantiomers of fluoxetine and norfluoxetine. Basic Clin. Pharmacol. Toxicol. 2005, 97, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, S.; Huang, M.; Hu, H.; Yu, L.; Zeng, S. Characterizing the Effect of Cytochrome P450 (CYP) 2C8, CYP2C9, and CYP2D6 Genetic Polymorphisms on Stereoselective N-demethylation of Fluoxetine. Chirality 2014, 26, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Gassó, P.; Rodríguez, N.; Mas, S.; Pagerols, M.; Blázquez, A.; Plana, M.T.; Torra, M.; Lázaro, L.; Lafuente, A. Effect of CYP2D6, CYP2C9 and ABCB1 genotypes on fluoxetine plasma concentrations and clinical improvement in children and adolescent patients. Pharmacogenomics J. 2014, 14, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Eap, C.B.; Bondolfi, G.; Zullino, D.; Savary-Cosendai, L.; Powell-Golay, K.; Kosel, M.; Baumann, P. Concentrations of the Enantiomers of Fluoxetine and Norfluoxetine After Multiple Doses of Fluoxetine in Cytochrome P4502D6 Poor and Extensive Metabolizers. J. Clin. Psychopharmacol. 2001, 21, 330–334. [Google Scholar] [CrossRef]
- Zajecka, J.; Fawcett, J.; Amsterdam, J.; Quitkin, F.; Reimherr, F.; Rosenbaum, J.; Michelson, D.; Beasley, C. Safety of Abrupt Discontinuation of Fluoxetine: A Randomized, Placebo-Controlled Study. J. Clin. Psychopharmacol. 1998, 18, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Bogetto, F.; Bellino, S.; Revello, R.B.; Patria, L. Discontinuation syndrome in dysthymic patients treated with selective serotonin reuptake inhibitors: A clinical investigation. CNS Drugs 2002, 16, 273–283. [Google Scholar] [CrossRef]
- Hicks, J.K.; Swen, J.J.; Thorn, C.F.; Sangkuhl, K.; Kharasch, E.D.; Ellingrod, V.L.; Skaar, T.C.; Müller, D.J.; Gaedigk, A.; Stingl, J.C. Clinical Pharmacogenetics Implementation Consortium guideline for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants. Clin. Pharmacol. Ther. 2013, 93, 402–408. [Google Scholar] [CrossRef] [Green Version]
- Preskorn, S.H.; Alderman, J.; Chung, M.; Harrison, W.; Messig, M.; Harris, S. Pharmacokinetics of desipramine coadministered with sertraline or fluoxetine. J. Clin. Psychopharmacol. 1994, 14, 90–98. [Google Scholar] [CrossRef]
- Pato, M.T.; Murphy, D.L.; Devane, L.C.P. Sustained Plasma Concentrations of Fluoxetine and/or Norfluoxetine Four and Eight Weeks After Fluoxetine Discontinuation. J. Clin. Psychopharmacol. 1991, 11, 224–225. [Google Scholar] [CrossRef]
- Iwasaki, S.; Hirabayashi, H.; Amano, N. Quantitative prediction of the extent of drug–drug interaction using a physiologically based pharmacokinetic model that includes inhibition of drug metabolism determined in cryopreserved hepatocytes. Xenobiotica 2018, 48, 770–780. [Google Scholar] [CrossRef]
- Stresser, D.; Mason, A.; Perloff, E.; Ho, T.; Crespi, C.; Dandeneau, A.; Morgan, L.; Dehal, S. Differential Time- and NADPH-dependent Inhibition of CYP2C19 by Enantiomers of Fluoxetine. Drug Metab. Dispos. 2009, 37, 695–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ring, B.J.; Eckstein, J.A.; Gillespie, J.S.; Binkley, S.N.; VandenBranden, M.; Wrighton, S.A. Identification of the human cytochromes p450 responsible for in vitro formation of R- and S-norfluoxetine. J. Pharmacol. Exp. Ther. 2001, 297, 1044–1050. [Google Scholar] [PubMed]
- Delavenne, X.; Magnin, M.; Basset, T.; Piot, M.; Mallouk, N.; Ressnikoff, D.; Garcin, A.; Laporte, S.; Garnier, P.; Mismetti, P. Investigation of drug-drug interactions between clopidogrel and fluoxetine. Fundam. Clin. Pharmacol. 2013, 27, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Bykov, K.; Schneeweiss, S.; Donneyong, M.M.; Dong, Y.-H.; Choudhry, N.K.; Gagne, J.J. Impact of an Interaction Between Clopidogrel and Selective Serotonin Reuptake Inhibitors. Am. J. Cardiol. 2017, 119, 651–657. [Google Scholar] [CrossRef]
- Schmider, J.; Greenblatt, D.J.; von Moltke, L.L.; Karsov, D.; Shader, R.I. Inhibition of CYP2C9 by selective serotonin reuptake inhibitors in vitro: Studies of phenytoin p-hydroxylation. Br. J. Clin. Pharmacol. 1997, 44, 495–498. [Google Scholar] [CrossRef] [Green Version]
- Lutz, J.D.; VandenBrink, B.M.; Babu, K.N.; Nelson, W.L.; Kunze, K.L.; Isoherranen, N. Stereoselective inhibition of CYP2C19 and CYP3A4 by fluoxetine and its metabolite: Implications for risk assessment of multiple time-dependent inhibitor systems. Drug Metab. Dispos. 2013, 41, 2056–2065. [Google Scholar] [CrossRef] [Green Version]
- Lam, Y.W.F.; Alfaro, C.L.; Ereshefsky, L.; Miller, M. Pharmacokinetic and pharmacodynamic interactions of oral midazolam with ketoconazole, fluoxetine, fluvoxamine, and nefazodone. J. Clin. Pharmacol. 2003, 43, 1274–1282. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deodhar, M.; Rihani, S.B.A.; Darakjian, L.; Turgeon, J.; Michaud, V. Assessing the Mechanism of Fluoxetine-Mediated CYP2D6 Inhibition. Pharmaceutics 2021, 13, 148. https://doi.org/10.3390/pharmaceutics13020148
Deodhar M, Rihani SBA, Darakjian L, Turgeon J, Michaud V. Assessing the Mechanism of Fluoxetine-Mediated CYP2D6 Inhibition. Pharmaceutics. 2021; 13(2):148. https://doi.org/10.3390/pharmaceutics13020148
Chicago/Turabian StyleDeodhar, Malavika, Sweilem B. Al Rihani, Lucy Darakjian, Jacques Turgeon, and Veronique Michaud. 2021. "Assessing the Mechanism of Fluoxetine-Mediated CYP2D6 Inhibition" Pharmaceutics 13, no. 2: 148. https://doi.org/10.3390/pharmaceutics13020148
APA StyleDeodhar, M., Rihani, S. B. A., Darakjian, L., Turgeon, J., & Michaud, V. (2021). Assessing the Mechanism of Fluoxetine-Mediated CYP2D6 Inhibition. Pharmaceutics, 13(2), 148. https://doi.org/10.3390/pharmaceutics13020148