
A Physiologically-Based Pharmacokinetic Framework for Prediction of Drug Exposure in Malnourished Children


Abstract
:1. Introduction
2. Materials and Methods
2.1. Software
2.2. Physiologically Based Pharmacokinetics
2.3. Modeling Physiological Changes due to Malnutrition
2.3.1. Creation of Virtual Malnourished Pediatric Populations
2.3.2. Creation of Physiological Scaling Parameters
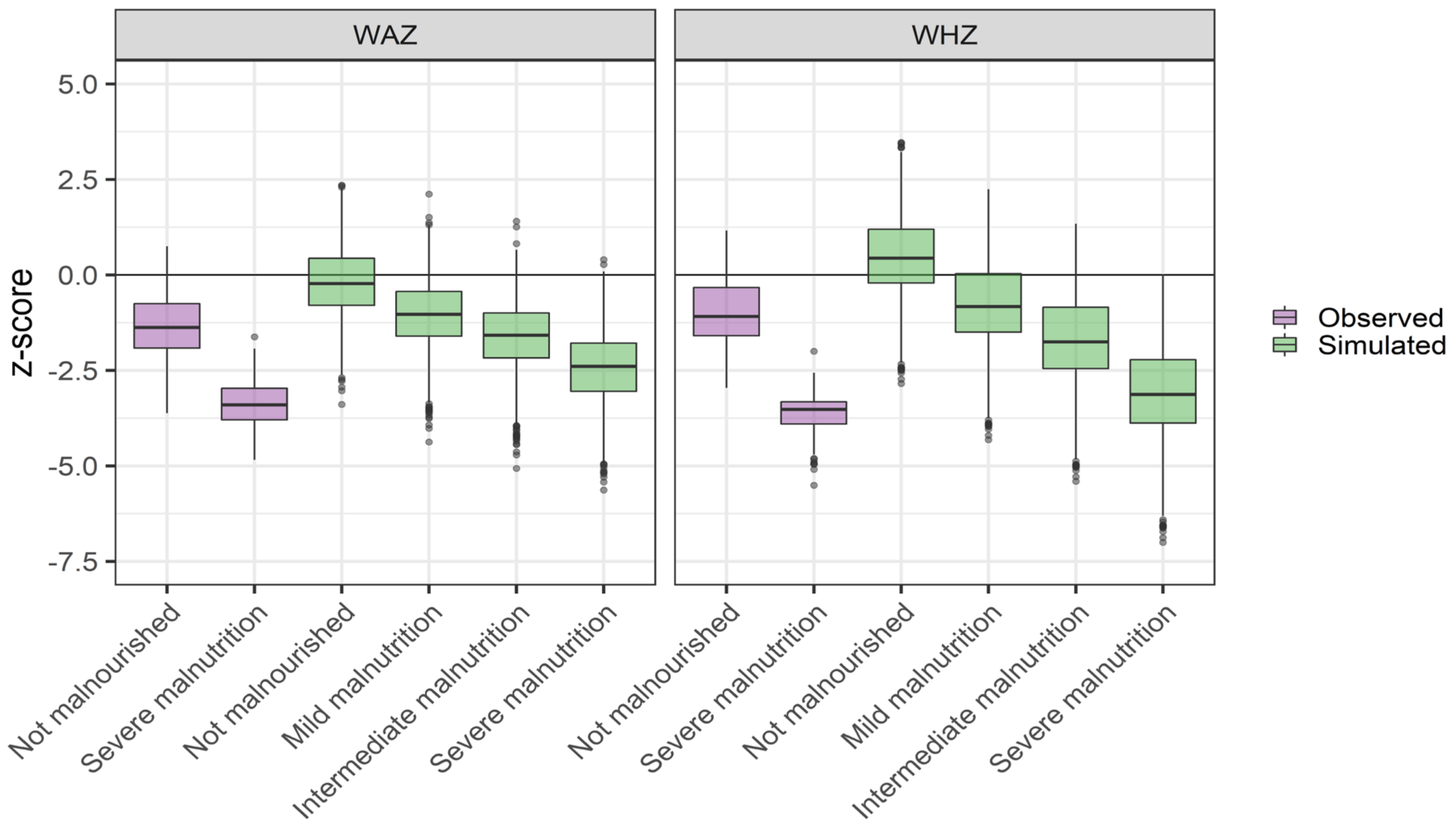
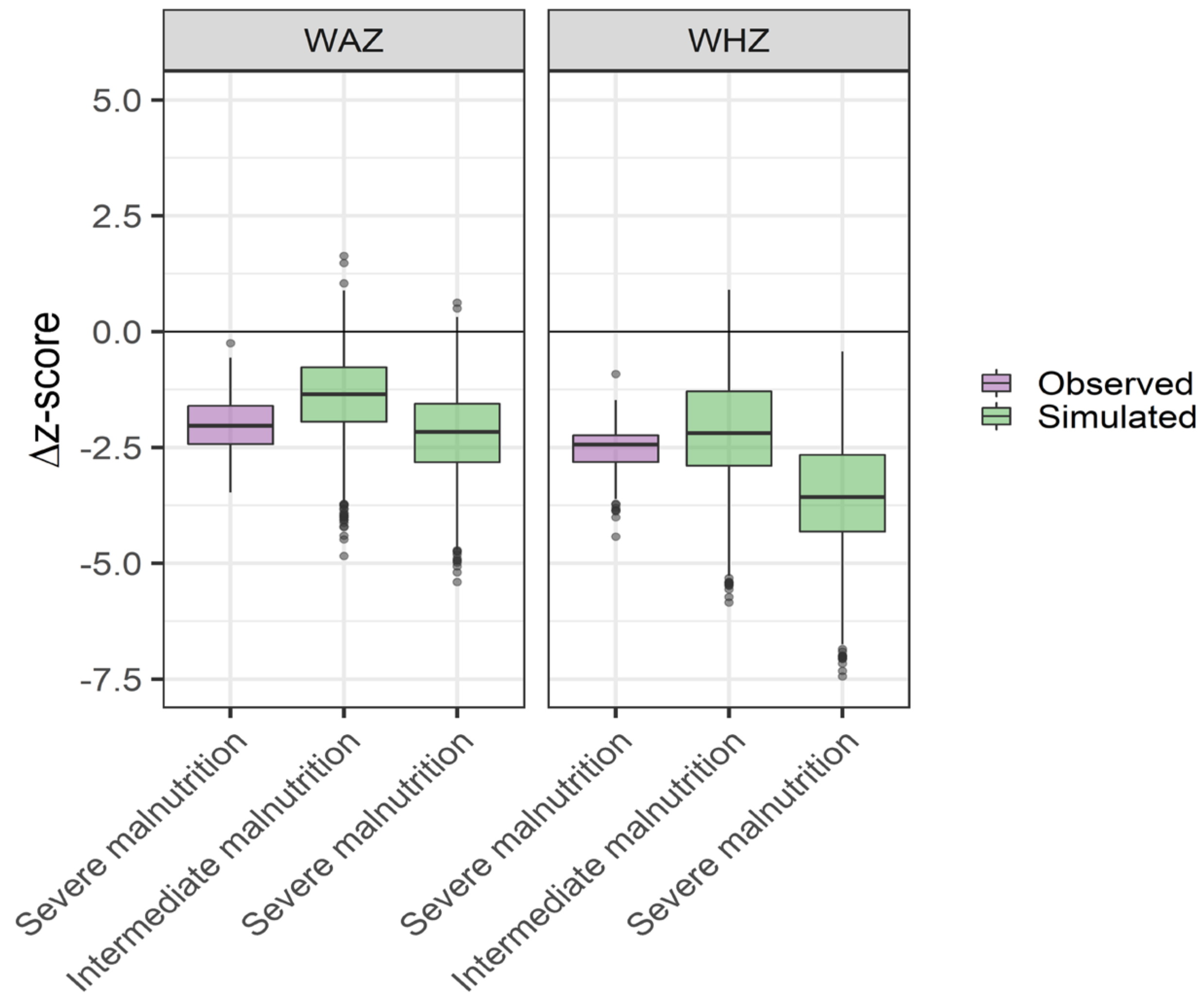
2.3.3. Evaluation of Physiological Scaling
2.4. PBPK Drug Models and Study Data Used in PK Evaluation
2.4.1. Caffeine
2.4.2. Cefoxitin
2.4.3. Ciprofloxacin
2.4.4. Lumefantrine
2.4.5. Pyrimethamine
2.4.6. Sulfadoxine
3. Results
3.1. Physiological Scaling Parameters
3.2. PBPK Drug Model Development
3.2.1. Caffeine
3.2.2. Cefoxitin
3.2.3. Ciprofloxacin
3.2.4. Lumefantrine

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Caffeine 1 | Cefoxitin | Ciprofloxacin 2 | Lumefantrine | Pyrimethamine | Sulfadoxine |
|---|---|---|---|---|---|---|
| logP | 0.87 | 0.84 3 | 0.95 | 3.09 3 | 3.14 3 | 3.74 3 |
| fu.p | 0.7 | 0.48 4 | 0.67 | 0.0029 3 | 0.095 5 | 0.036 5 |
| Mw | 194.2 | 427.45 | 331.3 | 528.9 | 248.71 | 310.33 |
| pKa | 0.8 B | 3.58 A 6 | 6.09 A, 8.62 B | 9.35 B 7 | 6.9 B 5 | 6.2 A 5 |
| Solubility 8 | 21.6 @ pH = 7 | 0.2 @ pH = 7 9 | 38.4 @ pH = 7 | fasted: 0.0097 3 @ pH = 6.5 fed—high fat: 0.18 3 @ pH = 5 fed—milk: 0.05 3 @ pH = 5 | 0.12 @ pH = 5 5 | 0.474 @ pH = 5 5 |
| Distribution | PK-Sim | RR | PK-Sim | PK-Sim | RR 10 | RR 11 |
| Pint | 223 3 | 0.161 12 | 1.57 3, 13 | 24.4 3 | 6370 3 | 1690 3 |
| Renal elimination | CLspec = 2.46 ×10−3 | CLspec = 3.8 3 + GFR | CLspec = 1.61 + GFR | GFR | GFR × 0.21 3 | |
| Hepatic elimination | Vmax.CYP1A2 = 73.1 3 Km.CYP1A2 = 14.7 | CLspec.CYP1A2 = 0.043 CLspec.bile = 0.096 | CLint.CYP3A4 = 93.7 3 | CLspec = 0.089 3 |
3.2.5. Pyrimethamine
3.2.6. Sulfadoxine
3.3. Pharmacokinetic Evaluation of Physiological Scaling Parameters
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Collins, S.; Dent, N.; Binns, P.; Bahwere, P.; Sadler, K.; Hallam, A. Management of Severe Acute Malnutrition in Children. Lancet 2006, 368, 1992–2000. [Google Scholar] [CrossRef]
- UNICEF—Progress for Children 2007—Introduction. Available online: https://www.unicef.org/progressforchildren/2007n6/index_41401.htm (accessed on 28 October 2020).
- De Onis, M.; Borghi, E.; Arimond, M.; Webb, P.; Croft, T.; Saha, K.; De-Regil, L.M.; Thuita, F.; Heidkamp, R.; Krasevec, J.; et al. Prevalence Thresholds for Wasting, Overweight and Stunting in Children under 5 Years. Public Health Nutr. 2019, 22, 175–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshikoya, K.A.; Sammons, H.M.; Choonara, I. A Systematic Review of Pharmacokinetics Studies in Children with Protein-Energy Malnutrition. Eur. J. Clin. Pharm. 2010, 66, 1025–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnaswamy, K. Drug Metabolism and Pharmacokinetics in Malnourished Children. Clin. Pharm. 1989, 17, 68–88. [Google Scholar] [CrossRef]
- European Medicines Agency Paediatric Investigation Plans. Available online: https://www.ema.europa.eu/en/human-regulatory/research-development/paediatric-medicines/paediatric-investigation-plans (accessed on 28 October 2020).
- U.S. Food and Drug Administration Pediatric Study Plans: Content of and Process for Submitting Initial Pediatric Study Plans and Amended Initial Pediatric Study Plans. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/pediatric-study-plans-content-and-process-submitting-initial-pediatric-study-plans-and-amended (accessed on 28 October 2020).
- Yellepeddi, V.; Rower, J.; Liu, X.; Kumar, S.; Rashid, J.; Sherwin, C.M.T. State-of-the-Art Review on Physiologically Based Pharmacokinetic Modeling in Pediatric Drug Development. Clin. Pharm. 2019, 58, 1–13. [Google Scholar] [CrossRef]
- Barrett, J.S.; Della Casa Alberighi, O.; Läer, S.; Meibohm, B. Physiologically Based Pharmacokinetic (PBPK) Modeling in Children. Clin. Pharm. Ther. 2012, 92, 40–49. [Google Scholar] [CrossRef]
- Michelet, R.; Bocxlaer, J.V.; Vermeulen, A. PBPK in Preterm and Term Neonates: A Review. Curr. Pharm. Des. 2017, 23, 5943–5954. [Google Scholar] [CrossRef]
- Johnson, T.N.; Rostami-Hodjegan, A. Resurgence in the Use of Physiologically Based Pharmacokinetic Models in Pediatric Clinical Pharmacology: Parallel Shift in Incorporating the Knowledge of Biological Elements and Increased Applicability to Drug Development and Clinical Practice. Paediatr. Anaesth. 2011, 21, 291–301. [Google Scholar] [CrossRef]
- Leong, R.; Vieira, M.L.T.; Zhao, P.; Mulugeta, Y.; Lee, C.S.; Huang, S.-M.; Burckart, G.J. Regulatory Experience with Physiologically Based Pharmacokinetic Modeling for Pediatric Drug Trials. Clin. Pharm. Ther. 2012, 91, 926–931. [Google Scholar] [CrossRef]
- Templeton, I.E.; Jones, N.S.; Musib, L. Pediatric Dose Selection and Utility of PBPK in Determining Dose. AAPS J. 2018, 20, 31. [Google Scholar] [CrossRef]
- Eissing, T.; Kuepfer, L.; Becker, C.; Block, M.; Coboeken, K.; Gaub, T.; Goerlitz, L.; Jaeger, J.; Loosen, R.; Ludewig, B.; et al. A Computational Systems Biology Software Platform for Multiscale Modeling and Simulation: Integrating Whole-Body Physiology, Disease Biology, and Molecular Reaction Networks. Front. Physiol. 2011, 2, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thelen, K.; Coboeken, K.; Willmann, S.; Burghaus, R.; Dressman, J.B.; Lippert, J. Evolution of a Detailed Physiological Model to Simulate the Gastrointestinal Transit and Absorption Process in Humans, Part I: Oral Solutions. J. Pharm. Sci. 2011, 100, 5324–5345. [Google Scholar] [CrossRef] [PubMed]
- Thelen, K.; Coboeken, K.; Willmann, S.; Dressman, J.B.; Lippert, J. Evolution of a Detailed Physiological Model to Simulate the Gastrointestinal Transit and Absorption Process in Humans, Part II: Extension to Describe Performance of Solid Dosage Forms. J. Pharm. Sci. 2012, 101, 1267–1280. [Google Scholar] [CrossRef] [PubMed]
- Willmann, S.; Höhn, K.; Edginton, A.; Sevestre, M.; Solodenko, J.; Weiss, W.; Lippert, J.; Schmitt, W. Development of a Physiology-Based Whole-Body Population Model for Assessing the Influence of Individual Variability on the Pharmacokinetics of Drugs. J. Pharm. Pharm. 2007, 34, 401–431. [Google Scholar] [CrossRef] [PubMed]
- Barac-Nieto, M.; Spurr, G.B.; Lotero, H.; Maksud, M.G. Body Composition in Chronic Undernutrition. Am. J. Clin. Nutr. 1978, 31, 23–40. [Google Scholar] [CrossRef]
- Bosy-Westphal, A.; Reinecke, U.; Schlörke, T.; Illner, K.; Kutzner, D.; Heller, M.; Müller, M.J. Effect of Organ and Tissue Masses on Resting Energy Expenditure in Underweight, Normal Weight and Obese Adults. Int. J. Obes. Relat. Metab. Disord. 2004, 28, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Berg, J.; Lane, V. Pathology Harmony; a Pragmatic and Scientific Approach to Unfounded Variation in the Clinical Laboratory. Ann. Clin. Biochem. 2011, 48, 195–197. [Google Scholar] [CrossRef]
- Du Bois, D.; Du Bois, E.F. A Formula to Estimate the Approximate Surface Area If Height and Weight Be Known. 1916. Nutrition 1989, 5, 303–311. [Google Scholar]
- WHO Global Database on Child Growth and Malnutrition. Available online: http://www.who.int/nutgrowthdb/en/ (accessed on 28 October 2020).
- Chotsiri, P.; Denoeud-Ndam, L.; Baudin, E.; Guindo, O.; Diawara, H.; Attaher, O.; Smit, M.; Guerin, P.J.; Doumbo, O.K.; Wiesner, L.; et al. Severe Acute Malnutrition Results in Lower Lumefantrine Exposure in Children Treated with Artemether-Lumefantrine for Uncomplicated Malaria. Clin. Pharm. Ther. 2019, 106, 1299–1309. [Google Scholar] [CrossRef] [Green Version]
- Akinyinka, O.O.; Sowunmi, A.; Honeywell, R.; Renwick, A.G. The Effects of Acute Falciparum Malaria on the Disposition of Caffeine and the Comparison of Saliva and Plasma-Derived Pharmacokinetic Parameters in Adult Nigerians. Eur. J. Clin. Pharm. 2000, 56, 159–165. [Google Scholar] [CrossRef]
- Akinyinka, O.O.; Sowunmi, A.; Honeywell, R.; Renwick, A.G. The Pharmacokinetics of Caffeine in Nigerian Children Suffering from Malaria and Kwashiorkor. Eur. J. Clin. Pharm. 2000, 56, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Kampf, D.; Schurig, R.; Korsukewitz, I.; Brückner, O. Cefoxitin Pharmacokinetics: Relation to Three Different Renal Clearance Studies in Patients with Various Degrees of Renal Insufficiency. Antimicrob. Agents Chemother. 1981, 20, 741–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carver, P.L.; Nightingale, C.H.; Quintiliani, R. Pharmacokinetics and Pharmacodynamics of Total and Unbound Cefoxitin and Cefotetan in Healthy Volunteers. J. Antimicrob. Chemother. 1989, 23, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, N.; Mithal, Y.; Witcomb, M. Cefoxitin: Intravenous Pharmacokinetics and Intramuscular Bioavailability in Kwashiorkor. Br. J. Clin. Pharm. 1980, 9, 623–627. [Google Scholar] [CrossRef] [Green Version]
- Schlender, J.-F.; Teutonico, D.; Coboeken, K.; Schnizler, K.; Eissing, T.; Willmann, S.; Jaehde, U.; Stass, H. A Physiologically-Based Pharmacokinetic Model to Describe Ciprofloxacin Pharmacokinetics Over the Entire Span of Life. Clin. Pharm. 2018, 57, 1613–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallicano, K.; Sahai, J. Lack of Gender Effect on Ciprofloxacin Pharmacokinetics in Humans. Br. J. Clin. Pharm. 1996, 42, 632–634. [Google Scholar] [CrossRef] [PubMed]
- Thuo, N.; Ungphakorn, W.; Karisa, J.; Muchohi, S.; Muturi, A.; Kokwaro, G.; Thomson, A.H.; Maitland, K. Dosing Regimens of Oral Ciprofloxacin for Children with Severe Malnutrition: A Population Pharmacokinetic Study with Monte Carlo Simulation. J. Antimicrob. Chemother. 2011, 66, 2336–2345. [Google Scholar] [CrossRef] [PubMed]
- Alison, T.; Wanchana, U. Development of a Physiologically Based Pharmacokinetic Model for Children with Severe Malnutrition. PAGE. Abstr. Annu. Meet. Popul. Approach Group Eur. 2013, 22, 2711. [Google Scholar]
- Lefèvre, G.; Thomsen, M.S. Clinical Pharmacokinetics of Artemether and Lumefantrine (Riamet®). Clin. Drug Investig. 1999, 18, 467–480. [Google Scholar] [CrossRef]
- Heimbach, T. Formulation-Dependent Pediatric Physiologically Based Pharmacokinetic (PPBPK) Modeling to Aid Drug Development. Presentation at Challenges and Strategies to Facilitate Formulation Development of Pediatric Drug Products, a Symposium Sponsored by the University of Maryland Center of Excellence in Regulatory Science and Innovation (M-CERSI). 2016. Available online: https://www.pharmacy.umaryland.edu/media/SOP/wwwpharmacyumarylandedu/centers/cersievents/pedsformulation/wen-presentation-notes.pdf (accessed on 28 October 2020).
- Lefèvre, G.; Carpenter, P.; Souppart, C.; Schmidli, H.; McClean, M.; Stypinski, D. Pharmacokinetics and Electrocardiographic Pharmacodynamics of Artemether-Lumefantrine (Riamet) with Concomitant Administration of Ketoconazole in Healthy Subjects. Br. J. Clin. Pharm. 2002, 54, 485–492. [Google Scholar] [CrossRef] [Green Version]
- Ashley, E.A.; Stepniewska, K.; Lindegårdh, N.; Annerberg, A.; Kham, A.; Brockman, A.; Singhasivanon, P.; White, N.J.; Nosten, F. How Much Fat Is Necessary to Optimize Lumefantrine Oral Bioavailability? Trop. Med. Int. Health 2007, 12, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Edstein, M.D. Pharmacokinetics of Sulfadoxine and Pyrimethamine after Fansidar Administration in Man. Chemotherapy 1987, 33, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.N.; Little, F.; Camba, T.; Cassam, Y.; Raman, J.; Boulle, A.; Barnes, K.I. Efficacy of Sulphadoxine-Pyrimethamine with or without Artesunate for the Treatment of Uncomplicated Plasmodium Falciparum Malaria in Southern Mozambique: A Randomized Controlled Trial. Malar. J. 2009, 8, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, K.I.; Little, F.; Smith, P.J.; Evans, A.; Watkins, W.M.; White, N.J. Sulfadoxine-Pyrimethamine Pharmacokinetics in Malaria: Pediatric Dosing Implications. Clin. Pharm. Ther. 2006, 80, 582–596. [Google Scholar] [CrossRef]
- De Kock, M.; Tarning, J.; Workman, L.; Allen, E.N.; Tekete, M.M.; Djimde, A.A.; Bell, D.J.; Ward, S.A.; Barnes, K.I.; Denti, P. Population Pharmacokinetic Properties of Sulfadoxine and Pyrimethamine: A Pooled Analysis to Inform Optimal Dosing in African Children with Uncomplicated Malaria. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [Green Version]
- Valentin, J. Basic Anatomical and Physiological Data for Use in Radiological Protection: Reference Values: ICRP Publication 89. Ann. ICRP 2002, 32, 1–277. [Google Scholar] [CrossRef]
- Kotila, O.A.; Olaniyi, O.O.; Adegoke, A.O.; Babalola, C.P. Experimental Determination of the Physicochemical Properties of Lumefantrine. Afr. J. Med. Med. Sci. 2013, 42, 209–214. [Google Scholar]
- Colussi, D.; Parisot, C.; Legay, F.; Lefèvre, G. Binding of Artemether and Lumefantrine to Plasma Proteins and Erythrocytes. Eur. J. Pharm. Sci. 1999, 9, 9–16. [Google Scholar] [CrossRef]
- Wolff, J.A.; Margolis, S.; Bujdoso-Wolff, K.; Matusick, E.; MacLean, W.C. Plasma and Red Blood Cell Fatty Acid Composition in Children with Protein-Calorie Malnutrition. Pediatr. Res. 1984, 18, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Leichsenring, M.; Sütterlin, N.; Less, S.; Bäumann, K.; Anninos, A.; Becker, K. Polyunsaturated Fatty Acids in Erythrocyte and Plasma Lipids of Children with Severe Protein-Energy Malnutrition. Acta Paediatr. 1995, 84, 516–520. [Google Scholar] [CrossRef]
- Etukudo, M.H.; Agbedana, E.O.; Akinyinka, O.O.; Osifo, B.O. Plasma Electrolytes, Total Cholesterol, Liver Enzymes, and Selected Antioxidant Status in Protein Energy Malnutrition. Afr. J. Med. Med. Sci. 1999, 28, 81–85. [Google Scholar] [PubMed]
- Charman, S.A.; Andreu, A.; Barker, H.; Blundell, S.; Campbell, A.; Campbell, M.; Chen, G.; Chiu, F.C.K.; Crighton, E.; Katneni, K.; et al. An In Vitro Toolbox to Accelerate Anti-Malarial Drug Discovery and Development. Malar. J. 2020, 19, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouankie, J.B.; Senczuk, W.; Florek, E. Urinary Elimination Kinetics of Pyrimethamine. Eur. J. Drug Metab. Pharm. 2009, 34, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Charpiat, B.; Thiébaut, R.; Salmi, L.R. Systematic Search and Analysis of Published Pharmacokinetic Data Related to Sulfadoxine. European TOXO PREVENTION Project. 2005. Available online: https://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.495.3346&rep=rep1&type=pdf (accessed on 28 October 2020).
- Bell, D.J.; Nyirongo, S.K.; Mukaka, M.; Molyneux, M.E.; Winstanley, P.A.; Ward, S.A. Population Pharmacokinetics of Sulfadoxine and Pyrimethamine in Malawian Children with Malaria. Clin. Pharm. Ther. 2011, 89, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Lippert, J.; Burghaus, R.; Edginton, A.; Frechen, S.; Karlsson, M.; Kovar, A.; Lehr, T.; Milligan, P.; Nock, V.; Ramusovic, S.; et al. Open Systems Pharmacology Community-An Open Access, Open Source, Open Science Approach to Modeling and Simulation in Pharmaceutical Sciences. CPT Pharmacomet. Syst. Pharm. 2019, 8, 878–882. [Google Scholar] [CrossRef] [Green Version]
- Keys, A.; Brozek, J.; Hensckel, A.; Mickelsen, O.; Taylor, H.L. The Biology of Human Starvation; University of Minnesota Press: Minneapolis, MN, USA, 1950; Volume 1, ISBN 978-0-8166-7234-9. [Google Scholar]
- Lazzerini, M.; Tickell, D. Antibiotics in Severely Malnourished Children: Systematic Review of Efficacy, Safety and Pharmacokinetics. Bull. World Health Organ. 2011, 89, 594–607. [Google Scholar] [CrossRef]
- Freerks, L.; Papadatou Soulou, E.; Batchelor, H.; Klein, S. A Review of GI Conditions Critical to Oral Drug Absorption in Malnourished Children. Eur J. Pharm. Biopharm. 2019, 137, 9–22. [Google Scholar] [CrossRef]
- Klahr, S.; Alleyne, G.A. Effects of Chronic Protein-Calorie Malnutrition on the Kidney. Kidney Int. 1973, 3, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Pukrittayakamee, S.; Looareesuwan, S.; Keeratithakul, D.; Davis, T.M.E.; Teja-Isavadharm, P.; Nagachinta, B.; Weber, A.; Smith, A.L.; Kyle, D.; White, N.J. A Study of the Factors Affecting the Metabolic Clearance of Quinine in Malaria. Eur. J. Clin. Pharm. 1997, 52, 487–493. [Google Scholar] [CrossRef]
- Das, D.; Grais, R.F.; Okiro, E.A.; Stepniewska, K.; Mansoor, R.; van der Kam, S.; Terlouw, D.J.; Tarning, J.; Barnes, K.I.; Guerin, P.J. Complex Interactions between Malaria and Malnutrition: A Systematic Literature Review. BMC Med. 2018, 16, 186. [Google Scholar] [CrossRef] [Green Version]







| Study | Measurement | ||||
|---|---|---|---|---|---|
| Barac-Nieto 1 | Bosy-Westphal 2 | ||||
| Nutritional category 3 | M | I | S | IW | UW |
| Weight (kg) | 52.03 | 48.24 | 42.52 | 70.9 | 46.3 |
| Height (cm) | 156 | 157 | 156 | 178 | 165 |
| BWT/HT (kg/m) | 33.3 | 30.8 | 27.4 | 39.8 | 28.1 |
| % of standard BWT/HT | 89.5 | 82.7 | 73.9 | ||
| Serum albumin (g/100 mL) | 3.8 | 3 | 2.1 | ||
| Hematocrit | 44.4 | 37.2 | 32 | ||
| Fat mass (%) | 17.7 | 19.8 | 15.2 | ||
| Brain (kg) | 1.51 | 1.13 | |||
| Heart (kg) | 0.33 | 0.22 | |||
| Liver (kg) | 1.64 | 0.94 | |||
| Spleen (kg) | 0.23 | 0.11 | |||
| Kidney (kg) | 0.36 | 0.21 | |||
| BMC (kg) | 2.68 | 1.93 | |||
| LBMtrunk (kg) | 24.1 | 16.9 | |||
| MM (kg) | 27.6 | 16.6 | |||
| Component | Not Malnourished | Malnutrition Level | |||
|---|---|---|---|---|---|
| Mild | Intermediate | Severe | |||
| Bone | 1 | 0.947 | 0.913 | 0.869 | |
| Brain | 1 | 0.918 | 0.865 | 0.797 | |
| Fat | 1 | 0.817 | 0.822 | 0.624 | |
| Gonads, intestines, lung, stomach | 1 | 0.936 | 0.894 | 0.84 | |
| Heart | 1 | 0.902 | 0.839 | 0.758 | |
| Kidney | 1 | 0.874 | 0.792 | 0.686 | |
| Liver | 1 | 0.872 | 0.789 | 0.682 | |
| Muscle | 1 | 0.893 | 0.771 | 0.715 | |
| Pancreas | 1 | 0.936 | 0.894 | 0.84 | |
| Skin | 1 | 0.954 | 0.922 | 0.879 | |
| Spleen | 1 | 0.844 | 0.743 | 0.612 | |
| arterial | 1 | 1.03 | 0.992 | 0.833 | |
| Blood | venous | 1 | 1.02 | 0.979 | 0.822 |
| portal vein | 1 | 1.02 | 0.982 | 0.825 | |
| Plasma proteins | 1 | 0.894 | 0.706 | 0.494 | |
| Hematocrit | 1 | 0.945 | 0.791 | 0.681 | |
| Biometric | Not Malnourished | Malnutrition Level | ||
|---|---|---|---|---|
| Mild | Intermediate | Severe | ||
| BWT (kg) | 73.0 | 65.3 | 60.3 | 53.9 |
| HT (cm) | 176 | 176 | 176 | 176 |
| BMI (kg/m2) | 23.6 | 21.1 | 19.5 | 17.4 |
| BWT/HT (kg/m) | 41.4 | 37.1 | 34.3 | 30.6 |
| BSA (m2) | 1.9 | 1.8 | 1.7 | 1.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sjögren, E.; Tarning, J.; Barnes, K.I.; Jonsson, E.N. A Physiologically-Based Pharmacokinetic Framework for Prediction of Drug Exposure in Malnourished Children. Pharmaceutics 2021, 13, 204. https://doi.org/10.3390/pharmaceutics13020204
Sjögren E, Tarning J, Barnes KI, Jonsson EN. A Physiologically-Based Pharmacokinetic Framework for Prediction of Drug Exposure in Malnourished Children. Pharmaceutics. 2021; 13(2):204. https://doi.org/10.3390/pharmaceutics13020204
Chicago/Turabian StyleSjögren, Erik, Joel Tarning, Karen I. Barnes, and E. Niclas Jonsson. 2021. "A Physiologically-Based Pharmacokinetic Framework for Prediction of Drug Exposure in Malnourished Children" Pharmaceutics 13, no. 2: 204. https://doi.org/10.3390/pharmaceutics13020204
APA StyleSjögren, E., Tarning, J., Barnes, K. I., & Jonsson, E. N. (2021). A Physiologically-Based Pharmacokinetic Framework for Prediction of Drug Exposure in Malnourished Children. Pharmaceutics, 13(2), 204. https://doi.org/10.3390/pharmaceutics13020204