
Inhibitory Effects of Schisandra Lignans on Cytochrome P450s and Uridine 5′-Diphospho-Glucuronosyl Transferases in Human Liver Microsomes

 and
and 
Abstract
:1. Introduction
2. Materials and Methods
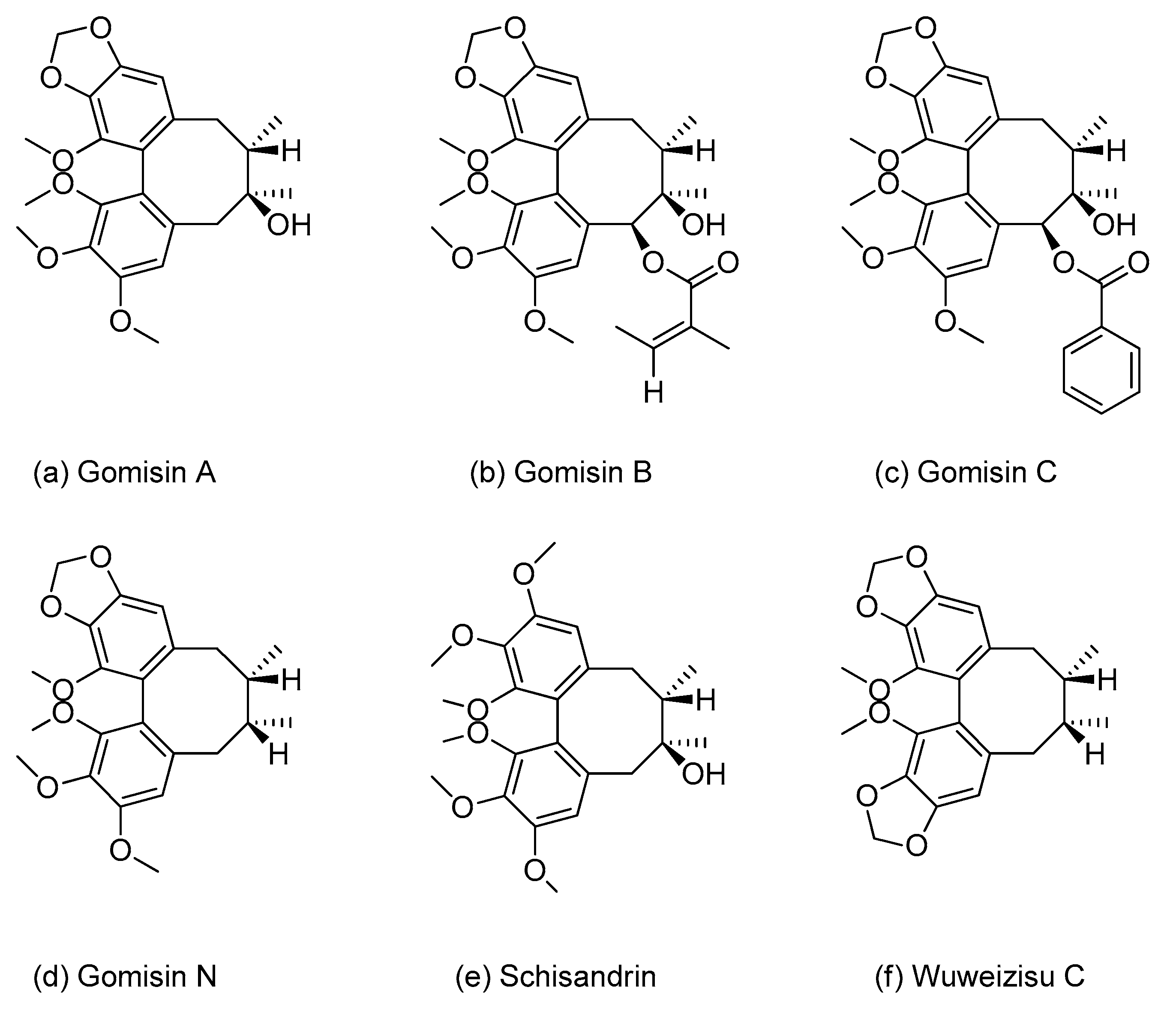
2.1. Chemicals and Reagents
2.2. Inhibitory Effects of Six Lignans against Human Cytochrome P450 Activity
2.3. Inhibitory Effects of Gomisin A against Recombianat CYP2C8, CYP2C19, and CYP3A4 Activity
2.4. Inhibitory Effects of Six Lignans against Human Uridine-5-Diphosphoglucuronosyl Transferase Activity
2.5. Characterization of Reactive Metabolites of Gomisin A in Recombinant P450 Isoforms
2.6. LC-MS/MS Analysis
2.7. Data Analysis
3. Results and Discussion
3.1. Inhibition of Cytochrome P450 Activities by Six Lignans
3.2. Inhibitory Effects of Gomisin A against Recombinant CYP2C8, CYP2C19, and CYP3A4
3.3. Inhibition of UGT Enzyme Activities by Six Lignans
3.4. Characterization of Reactive Metabolites of Gomisin A in Recombinant P450 Isoforms
3.5. Evaluation of Drug Interaction Potential of Six Lignans
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hao, M.; Zhao, Y.; Chen, P.; Huang, H.; Liu, H.; Jiang, H.; Zhang, R.; Wang, H. Structure-Activity Relationship and Substrate-Dependent Phenomena in Effects of Ginsenosides on Activities of Drug-Metabolizing P450 Enzymes. PLoS ONE 2008, 3, e2697. [Google Scholar] [CrossRef] [PubMed]
- Parvez, M.K.; Rishi, V. Herb-Drug Interactions and Hepatotoxicity. Curr. Drug Metab. 2019, 20, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.-E.F.; Frye, R.F. Effects of Herbal Supplements on Drug Glucuronidation. Review of Clinical, Animal, andIn VitroStudies. Planta Med. 2010, 77, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Adiwidjaja, J.; Boddy, A.V.; McLachlan, A.J. Physiologically Based Pharmacokinetic Modelling of Hyperforin to Predict Drug Interactions with St John’s Wort. Clin. Pharmacokinet. 2019, 58, 911–926. [Google Scholar] [CrossRef] [PubMed]
- Malati, C.Y.; Robertson, S.M.; Hunt, J.D.; Chairez, C.; Alfaro, R.M.; Kovacs, J.A.; Penzak, S.R. Influence of Panax ginseng on cytochrome P450 (CYP)3A and P-glycoprotein (P-gp) activity in healthy participants. J. Clin. Pharmacol. 2012, 52, 932–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderón, M.M.; Chairez, C.L.; Gordon, L.A.; Alfaro, R.M.; Kovacs, J.A.; Penzak, S.R. Influence of Panax ginseng on the steady state pharmacokinetic profile of lopinavir-ritonavir in healthy volunteers. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2014, 34, 1151–1158. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.; Tao, G.-Y.; Wang, G.; Chen, Y.; Zhang, W.; He, Y.-J.; Li, Q.; Lei, H.-P.; Jiang, F.; Hu, D.-L.; et al. Effects of Ginkgo biloba Extract Ingestion on the Pharmacokinetics of Talinolol in Healthy Chinese Volunteers. Ann. Pharmacother. 2009, 43, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-E.; Ha, N.; Kim, Y.; Kim, H.; Lee, J.W.; Jeon, J.-Y.; Kim, M.-G. Effect of epigallocatechin-3-gallate, major ingredient of green tea, on the pharmacokinetics of rosuvastatin in healthy volunteers. Drug Des. Dev. Ther. 2017, 11, 1409–1416. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Wang, X.; Xu, X.; Kong, L. Effect of Schisandra sphenanthera extract on the concentration of tacrolimus in the blood of liver transplant patients. Int. J. Clin. Pharmacol. Ther. 2010, 48, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Al-Jenoobi, F.I.; Al-Thukair, A.A.; Alam, M.A.; Abbas, F.A.; Al-Mohizea, A.M.; Alkharfy, K.M.; Al-Suwayeh, S.A. Effect of Curcuma longa on CYP2D6- and CYP3A4-mediated metabolism of dextromethorphan in human liver microsomes and healthy human subjects. Eur. J. Drug Metab. Pharmacokinet. 2014, 40, 61–66. [Google Scholar] [CrossRef]
- Szopa, A.; Ekiert, R.; Ekiert, H. Current knowledge of Schisandra chinensis (Turcz.) Baill. (Chinese magnolia vine) as a medicinal plant species: A review on the bioactive components, pharmacological properties, analytical and biotechnological studies. Phytochem. Rev. 2017, 16, 195–218. [Google Scholar] [CrossRef] [Green Version]
- Sowndhararajan, K.; Deepa, P.; Kim, M.; Park, S.J.; Kim, S. An overview of neuroprotective and cognitive enhancement properties of lignans from Schisandra chinensis. Biomed. Pharmacother. 2018, 97, 958–968. [Google Scholar] [CrossRef]
- Li, W.-L.; Xin, H.-W.; Yu, A.-R.; Wu, X.-C. In vivo effect of Schisandrin B on cytochrome P450 enzyme activity. Phytomedicine 2013, 20, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.-G.; Yang, B.-Y.; Liang, J.; Wang, J.-S.; Kuang, H.-X. Simultaneous quantification of five dibenzocyclooctadiene lignans in Schisandra chinensis by HPLC separation and fluorescence detection. Anal. Methods 2014, 6, 5981. [Google Scholar] [CrossRef]
- Sun, J.; Jing, S.; Jiang, R.; Wang, C.; Zhang, C.; Chen, J.; Li, H. Metabolomics study of the therapeutic mechanism of Schisandra chinensis lignans on aging rats induced by D-galactose. Clin. Interv. Aging 2018, 13, 829–841. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, K.; Taguchi, H.; Ikeya, Y.; Endo, T.; Yosioka, I. The Constituents of Schizandra chinensis BAILL. XIII. Quantitative Analysis of Lignans in the Fruits of Schizandra chinensis BAILL. by High Performance Liquid Chromatography. Yakugaku Zasshi 1983, 103, 743–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Sun, T.; Wu, J.-J.; Cao, Y.-F.; Fang, Z.-Z.; Sun, H.-Z.; Zhu, Z.-T.; Yang, K.; Liu, Y.-Z.; Gonzalez, F.J.; et al. Inhibition of human CYP3A4 and CYP3A5 enzymes by gomisin C and gomisin G, two lignan analogs derived from Schisandra chinensis. Fitoterapia 2017, 119, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-J.; Ge, G.-B.; He, Y.-Q.; Wang, P.; Dai, Z.-R.; Ning, J.; Hu, L.-H.; Yang, L. Gomisin A is a Novel Isoform-Specific Probe for the Selective Sensing of Human Cytochrome P450 3A4 in Liver Microsomes and Living Cells. AAPS J. 2015, 18, 134–145. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.M.; Xu, S.Y.; Hu, H.H.; Zhou, H.; Jiang, H.D.; Yu, L.S.; Zeng, S. Identification of CYP2C19 inhibitors from phytochemicals using the recombinant human enzyme model. Die Pharm. 2014, 69, 362–366. [Google Scholar]
- Wan, C.-K.; Tse, A.; Yu, Z.-L.; Zhu, G.-Y.; Wang, H.; Fong, D. Inhibition of cytochrome P450 3A4 activity by schisandrol A and gomisin A isolated from Fructus Schisandrae chinensis. Phytomedicine 2010, 17, 702–705. [Google Scholar] [CrossRef]
- Iwata, H.; Tezuka, Y.; Kadota, S.; Hiratsuka, A.; Watabe, T. Identification and Characterization of Potent Cyp3a4 Inhibitors in Schisandra Fruit Extract. Drug Metab. Dispos. 2004, 32, 1351–1358. [Google Scholar] [CrossRef]
- Kim, M.J.; Kim, H.; Cha, I.J.; Park, J.S.; Shon, J.H.; Liu, K.H.; Shin, J.G. High-throughput screening of inhibitory potential of nine cytochrome P450 enzymes in vitro using liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 2651–2658. [Google Scholar] [CrossRef]
- Itkonen, M.K.; Tornio, A.; Filppula, A.M.; Neuvonen, M.; Neuvonen, P.J.; Niemi, M.; Backman, J.T. Clopidogrel but Not Prasugrel Significantly Inhibits the CYP2C8-Mediated Metabolism of Montelukast in Humans. Clin. Pharmacol. Ther. 2018, 104, 495–504. [Google Scholar] [CrossRef]
- Bedada, S.K.; Boga, P.K. Effect of piperine on CYP2E1 enzyme activity of chlorzoxazone in healthy volunteers. Xenobiotica 2016, 47, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.-Y.; Shen, J.H.; Lemler, S.M.; Skaar, T.C.; Li, L.; Blievernicht, J.; Zanger, U.M.; Kim, K.-B.; Shin, J.-G.; Flockhart, D.A.; et al. Rifampin enhances cytochrome P450 (CYP) 2B6-mediated efavirenz 8-hydroxylation in healthy volunteers. Drug Metab. Pharmacokinet. 2016, 31, 107–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Zhang, L.; Duan, L.X.; Wu, J.J.; Hu, M.; Liu, Z.Q.; Wang, C.Y. Potential of herb-drug / herb interactions between substrates and inhibitors of UGTs derived from herbal medicines. Pharmacol. Res. 2019, 150, 104510. [Google Scholar] [CrossRef]
- Liu, C.; Cao, Y.-F.; Fang, Z.-Z.; Zhang, Y.-Y.; Hu, C.-M.; Sun, X.-Y.; Huang, T.; Zeng, J.; Fan, X.-R.; Hong, M. Strong inhibition of deoxyschizandrin and schisantherin A toward UDP-glucuronosyltransferase (UGT) 1A3 indicating UGT inhibition-based herb–drug interaction. Fitoterapia 2012, 83, 1415–1419. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Ryu, B.; Lee, J.S.; Choi, J.-H.; Jang, D.S. Schisandrosides A–D, Dibenzocyclooctadiene Lignan Glucosides from the Roots of Schisandra chinensis. Chem. Pharm. Bull. 2015, 63, 746–751. [Google Scholar] [CrossRef] [Green Version]
- Opletal, L.; Sovova, H.; Bartlova, M. Dibenzo[a,c]cyclooctadiene lignans of the genus Schisandra: Importance, isolation and determination. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2004, 812, 357–371. [Google Scholar] [CrossRef]
- Kim, H.-J.; Lee, H.; Ji, H.-K.; Lee, T.; Liu, K.-H. Screening of ten cytochrome P450 enzyme activities with 12 probe substrates in human liver microsomes using cocktail incubation and liquid chromatography–tandem mass spectrometry. Biopharm. Drug Dispos. 2019, 40, 101–111. [Google Scholar] [CrossRef]
- Perloff, E.S.; Mason, A.K.; Dehal, S.S.; Blanchard, A.P.; Morgan, L.; Ho, T.; Dandeneau, A.; Crocker, R.M.; Chandler, C.M.; Boily, N.; et al. Validation of cytochrome P450 time-dependent inhibition assays: A two-time point IC50 shift approach facilitates kinact assay design. Xenobiotica 2009, 39, 99–112. [Google Scholar] [CrossRef]
- Joo, J.; Lee, B.; Lee, T.; Liu, K.-H. Screening of six UGT enzyme activities in human liver microsomes using liquid chromatography/triple quadrupole mass spectrometry. Rapid Commun. Mass Spectrom. 2014, 28, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Qiu, B.; Zou, L.; Liu, K.; Xu, X.; Zhu, H. Schisandrin B elicits the Keap1-Nrf2 defense system via carbene reactive metabolite which is less harmful to mice liver. Drug Des. Dev. Ther. 2018, 12, 4033–4046. [Google Scholar] [CrossRef] [Green Version]
- Rauniyar, N. Parallel Reaction Monitoring: A Targeted Experiment Performed Using High Resolution and High Mass Accuracy Mass Spectrometry. Int. J. Mol. Sci. 2015, 16, 28566–28581. [Google Scholar] [CrossRef] [Green Version]
- Zhai, J.; Zhang, F.; Gao, S.; Chen, L.; Feng, G.; Yin, J.; Chen, W. Time- and NADPH-Dependent Inhibition on CYP3A by Gomisin A and the Pharmacokinetic Interactions between Gomisin A and Cyclophosphamide in Rats. Molecules 2017, 22, 1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Xin, H.; Su, M.; Xiong, L. Inhibitory effects of schisandrin A and schisandrin B on CYP3A activity. Methods Find. Exp. Clin. Pharmacol. 2010, 32, 163–169. [Google Scholar] [CrossRef]
- Richter, T.; Mürdter, T.E.; Heinkele, G.; Pleiss, J.; Tatzel, S.; Schwab, M.; Eichelbaum, M.; Zanger, U.M. Potent Mechanism-Based Inhibition of Human CYP2B6 by Clopidogrel and Ticlopidine. J. Pharmacol. Exp. Ther. 2003, 308, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Fairman, D.A.; Collins, C.; Chapple, S. Progress Curve Analysis of CYP1A2 Inhibition: A More Informative Approach to the Assessment of Mechanism-Based Inactivation? Drug Metab. Dispos. 2007, 35, 2159–2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awortwe, C.; Manda, V.K.; Avonto, C.; Khan, S.I.; Khan, I.A.; Walker, L.A.; Bouic, P.J.; Rosenkranz, B. In Vitro Evaluation of Reversible and Time-Dependent Inhibitory Effects of Kalanchoe crenata on CYP2C19 and CYP3A4 Activities. Drug Metab. Lett. 2015, 9, 48–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertelsen, K.M.; Venkatakrishnan, K.; Von Moltke, L.L.; Obach, R.S.; Greenblatt, D.J. Apparent Mechanism-based Inhibition of Human CYP2D6 in Vitro by Paroxetine: Comparison with Fluoxetine and Quinidine. Drug Metab. Dispos. 2003, 31, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Fang, Z.-Z.; Zhang, Y.-Y.; Ge, G.-B.; Huo, H.; Liang, S.-C.; Yang, L. Time-dependent inhibition (TDI) of CYP3A4 and CYP2C9 by noscapine potentially explains clinical noscapine-warfarin interaction. Br. J. Clin. Pharmacol. 2010, 69, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, J.T.; Davydova, N.Y.; Paragas, E.M.; Jones, J.P.; Davydov, D.R. Kinetic mechanism of time-dependent inhibition of CYP2D6 by 3,4-methylenedioxymethamphetamine (MDMA): Functional heterogeneity of the enzyme and the reversibility of its inactivation. Biochem. Pharmacol. 2018, 156, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Franklin, M.R. Human cytochrome p450 inhibition and metabolic-intermediate complex formation by goldenseal extract and its methylenedioxyphenyl components. Drug Metab. Dispos. 2003, 31, 1391–1397. [Google Scholar] [CrossRef]
- Ma, S.; Subramanian, R. Detecting and characterizing reactive metabolites by liquid chromatography/tandem mass spectrometry. J. Mass Spectrom. 2006, 41, 1121–1139. [Google Scholar] [CrossRef]
- Li, F.; Lu, J.; Ma, X. Profiling the Reactive Metabolites of Xenobiotics Using Metabolomic Technologies. Chem. Res. Toxicol. 2011, 24, 744–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, B.; Fitch, W.L. Screening and characterization of reactive metabolites using glutathione ethyl ester in combination with Q-trap mass spectrometry. J. Mass Spectrom. 2009, 44, 90–100. [Google Scholar] [CrossRef]
- Fang, Z.-Z.; Krausz, K.W.; Li, F.; Cheng, J.; Tanaka, N.; Gonzalez, F.J. Metabolic map and bioactivation of the anti-tumour drug noscapine. Br. J. Pharmacol. 2012, 167, 1271–1286. [Google Scholar] [CrossRef]
- Zheng, J.; Ma, L.; Xin, B.; Olah, T.; Humphreys, W.G.; Zhu, M. Screening and Identification of GSH-Trapped Reactive Metabolites Using Hybrid Triple Quadruple Linear Ion Trap Mass Spectrometry. Chem. Res. Toxicol. 2007, 20, 757–766. [Google Scholar] [CrossRef]
- Zhuo, X.; Huang, X.S.; Degnan, A.P.; Snyder, L.B.; Yang, F.; Huang, H.; Shu, Y.-Z.; Johnson, B.M. Identification of Glutathione Conjugates of Acetylene-Containing Positive Allosteric Modulators of Metabotropic Glutamate Receptor Subtype 5. Drug Metab. Dispos. 2015, 43, 578–589. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Tao, X.; Di, P.; Yang, Y.; Li, J.; Qian, X.; Feng, J.; Chen, W. Effects of Traditional Chinese Medicine Wuzhi Capsule on Pharmacokinetics of Tacrolimus in Rats. Drug Metab. Dispos. 2013, 41, 1398–1403. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, S.; Hu, J.; Li, Y. Multifaceted interaction of the traditional Chinese medicinal herb Schisandra chinensis with cytochrome P450-mediated drug metabolism in rats. J. Ethnopharmacol. 2014, 155, 1473–1482. [Google Scholar] [CrossRef]
- Li, W.-L.; Xin, H.-W.; Su, M.-W. Inhibitory Effects of Continuous Ingestion of Schisandrin A on CYP3A in the Rat. Basic Clin. Pharmacol. Toxicol. 2011, 110, 187–192. [Google Scholar] [CrossRef]
- Li, X.-Q.; Björkman, A.; Andersson, T.B.; Ridderström, M.; Masimirembwa, C.M. Amodiaquine clearance and its metabolism to N-desethylamodiaquine is mediated by CYP2C8: A new high affinity and turnover enzyme-specific probe substrate. J. Pharmacol. Exp. Ther. 2002, 300, 399–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Václavíková, R.; Horský, S.; Simek, P.; Gut, I. Paclitaxel metabolism in rat and human liver microsomes is inhibited by phenolic antioxidants. Naunyn-Schmiedeberg Arch. Pharmacol. 2003, 368, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Bidstrup, T.B.; Bjørnsdottir, I.; Sidelmann, U.G.; Thomsen, M.S.; Hansen, K.T. CYP2C8 and CYP3A4 are the principal enzymes involved in the human in vitro biotransformation of the insulin secretagogue repaglinide. Br. J. Clin. Pharmacol. 2003, 56, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.A.; Park, P.W.; Hong, S.J.; Park, J.-Y. The Effect of CYP2C19 Polymorphism on the Pharmacokinetics and Pharmacodynamics of Clopidogrel: A Possible Mechanism for Clopidogrel Resistance. Clin. Pharmacol. Ther. 2008, 84, 236–242. [Google Scholar] [CrossRef]
- Andersson, T.; Cederberg, C.; Edvardsson, G.; Heggelund, A.; Lundborg, P. Effect of omeprazole treatment on diazepam plasma levels in slow versus normal rapid metabolizers of omeprazole. Clin. Pharmacol. Ther. 1990, 47, 79–85. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| P450 Enzyme | Substrate | Concentration (μM) | Metabolite | SRM Transition (m/z) | Polarity * | Collision Energy (eV) |
|---|---|---|---|---|---|---|
| 1A2 | Phenacetin | 20 | Acetaminophen | 152 > 110 | ESI+ | 25 |
| 2A6 | Coumarin | 1 | 7-Hydroxycoumarin | 163 > 107 | ESI+ | 17 |
| 2B6 | Bupropion | 3 | 6-Hydroxybupropion | 256 > 238 | ESI+ | 10 |
| 2C8 | Amodiaquine | 0.1 | N-Desethylamodiaquine | 328 > 283 | ESI+ | 13 |
| 2C9 | Diclofenac | 1 | 4-Hydroxydiclofenac | 312 > 231 | ESI+ | 15 |
| 2C19 | S-Mephenytoin | 40 | 4-Hydroxymethenytoin | 235 > 150 | ESI+ | 15 |
| 2D6 | Dextromethorphan | 2 | Dextrorphan | 258 > 157 | ESI+ | 30 |
| 2E1 | Chlorzoxazone | 5 | 6-Hydroxychlorzoxazone | 184 > 120 | ESI− | 18 |
| 3A4 | Midazolam | 0.1 | 1′-Hydroxymidazolam | 342 > 203 | ESI+ | 28 |
| Nifedipine | 0.2 | Dehydronifedipine | 345 > 284 | ESI+ | 30 | |
| Testosterone | 2 | 6β-Hydroxytestosterone | 305 > 269 | ESI+ | 15 | |
| IS | Trimipramine | 0.007 | - | 295 > 100 | ESI+ | 17 |
| UGT Enzyme | Substrate | Concentration (μM) | Metabolite | SRM Transition (m/z) | Polarity | Collision Energy (eV) |
|---|---|---|---|---|---|---|
| 1A1 | SN-38 * | 0.5 | SN-38 glucuronide | 569 > 393 | ESI+ | 30 |
| 1A3 | Chenodeoxycholic acid | 2 | CDCA-24 glucuronide | 567 > 391 | ESI- | 20 |
| 1A4 | Trifluoperazine | 0.5 | TFP N-glucuronide | 584 > 408 | ESI+ | 30 |
| 1A6 | N-Acetylserotonin | 1 | N-SER glucuronide | 395 > 219 | ESI+ | 10 |
| 1A9 | Mycophenolic acid | 0.2 | MPA 7-O-glucuronide | 495 > 319 | ESI- | 25 |
| 2B7 | Naloxone | 0.2 | NX 3-glucuronide | 504 > 310 | ESI+ | 30 |
| IS | Estrone-β-D-glucuronide | 0.25 | 445 > 269 | ESI- | 35 |
| P450 Enzyme | Probe Substrate | IC50 (µM) | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gomisin A | Gomisin B | Gomisin C | Gomisin N | Schisandrin | Wuweizisu C | ||||||||||||||
| RI * | TDI ** | IC50 Shift | RI | TDI | IC50 Shift | RI | TDI | IC50 Shift | RI | TDI | IC50 Shiaft | RI | TDI | IC50 Shift | RI | TDI | IC50 Shift | ||
| 1A2 | Phenacetin | 37.4 | >50 | - | >50 | >50 | - | >50 | >50 | - | >50 | >50 | - | 4.0 | >50 | - | 25.6 | 22.4 | <1.5 |
| 2A6 | Coumarin | >50 | >50 | - | >50 | >50 | - | >50 | >50 | - | 38.2 | >50 | - | 40.9 | >50 | - | >50 | >50 | - |
| 2B6 | Bupropion | >50 | >50 | - | 42.6 | >50 | - | 33.1 | >50 | - | 38.3 | >50 | - | 14.7 | >50 | - | 2.9 | 1.4 | 2.1 |
| 2C8 | Amodiaquine | 29.3 | 2.8 | 10.5 | 16.5 | 4.9 | 3.4 | 10.9 | 5.8 | 1.9 | 31.7 | 9.9 | 3.2 | 22.0 | >50 | - | 21.0 | 14.6 | <1.5 |
| 2C9 | Diclofenac | 45.4 | 22.0 | 2.1 | 45.7 | >50 | - | >50 | >50 | - | 36.2 | 10.9 | 3.3 | 43.0 | >50 | - | 8.9 | 3.6 | 2.5 |
| 2C19 | S-Mephenytoin | 11.2 | 4.8 | 2.3 | >50 | 37.8 | - | 16.3 | 20.4 | - | 10.4 | 3.5 | 3.0 | 5.3 | 46.1 | - | 2.7 | 1.5 | 1.8 |
| 2D6 | Dextromethorphan | 45.7 | >50 | - | 45.5 | >50 | - | >50 | >50 | - | 42.0 | >50 | - | 40.2 | >50 | - | 20.3 | 28.2 | - |
| 2E1 | Chlorzoxazone | >50 | 15.6 | >3.2 | >50 | 20.6 | >2.4 | >50 | 24.4 | >2.0 | >50 | 23.6 | >2.1 | 4.2 | 36.0 | - | >50 | 25.2 | >2.0 |
| 3A | Midazolam | 3.1 | 1.2 | 2.6 | 0.42 | 0.12 | 3.5 | 0.30 | 0.10 | 3.0 | 4.5 | 1.7 | 2.7 | 10.5 | 35.0 | - | 25.9 | 2.5 | 10.4 |
| Nifedipine | 1.8 | 0.77 | 2.3 | 0.32 | 0.10 | 3.2 | 0.26 | 0.09 | 2.9 | 1.4 | 0.61 | 2.4 | 16.0 | 43.3 | - | 5.6 | 1.2 | 4.7 | |
| Testosterone | 2.3 | 0.77 | 3.0 | 0.28 | 0.09 | 3.1 | 0.19 | 0.09 | 2.1 | 1.3 | 0.55 | 2.4 | 5.8 | 20.6 | - | 3.6 | 1.2 | 3.0 | |
| Recombinant P450 Enzyme | Probe Substrate | IC50 (μM) * | ||
|---|---|---|---|---|
| Gomisin A | ||||
| RI ** | TDI ** | IC50 shift | ||
| rCYP2C8 | Amodiaquine | 30.4 ± 8.1 | 3.32 ± 1.05 | 9.2 |
| rCYP2C19 | S-Mephenytoin | 11.3 ± 3.3 | 4.98 ± 0.49 | 2.3 |
| rCYP3A4 | Midazolam | 1.51 ± 0.20 | 0.51 ± 0.07 | 3.0 |
| UGT Enzyme | Substrate | IC50 (μM) * | |||||
|---|---|---|---|---|---|---|---|
| Gomisin A | Gomisin B | Gomisin C | Gomisin N | Schisandrin | Wuweizisu C | ||
| 1A1 | SN-38 ** | >50 | 20.7 | 24.0 | >50 | >50 | >50 |
| 1A3 | Chenodeoxycholic acid | >50 | 16.5 | 15.0 | 26.9 | >50 | >50 |
| 1A4 | Trifluoperazine | >50 | >50 | >50 | >50 | >50 | >50 |
| 1A6 | N-Acetylserotonin | >50 | >50 | >50 | >50 | >50 | >50 |
| 1A9 | Mycophenolic acid | >50 | >50 | >50 | >50 | >50 | >50 |
| 2B6 | Naloxone | >50 | >50 | >50 | >50 | >50 | >50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seo, H.-J.; Ji, S.-B.; Kim, S.-E.; Lee, G.-M.; Park, S.-Y.; Wu, Z.; Jang, D.S.; Liu, K.-H. Inhibitory Effects of Schisandra Lignans on Cytochrome P450s and Uridine 5′-Diphospho-Glucuronosyl Transferases in Human Liver Microsomes. Pharmaceutics 2021, 13, 371. https://doi.org/10.3390/pharmaceutics13030371
Seo H-J, Ji S-B, Kim S-E, Lee G-M, Park S-Y, Wu Z, Jang DS, Liu K-H. Inhibitory Effects of Schisandra Lignans on Cytochrome P450s and Uridine 5′-Diphospho-Glucuronosyl Transferases in Human Liver Microsomes. Pharmaceutics. 2021; 13(3):371. https://doi.org/10.3390/pharmaceutics13030371
Chicago/Turabian StyleSeo, Hyung-Ju, Seung-Bae Ji, Sin-Eun Kim, Gyung-Min Lee, So-Young Park, Zhexue Wu, Dae Sik Jang, and Kwang-Hyeon Liu. 2021. "Inhibitory Effects of Schisandra Lignans on Cytochrome P450s and Uridine 5′-Diphospho-Glucuronosyl Transferases in Human Liver Microsomes" Pharmaceutics 13, no. 3: 371. https://doi.org/10.3390/pharmaceutics13030371