
Sirolimus Pharmacokinetics Variability Points to the Relevance of Therapeutic Drug Monitoring in Pediatric Oncology

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design: Drug Combinations and Administration
2.2. Blood Sampling and Analysis
2.3. Compartmental Pharmacokinetic Analysis
2.3.1. Base Model
2.3.2. Covariate’s Selection
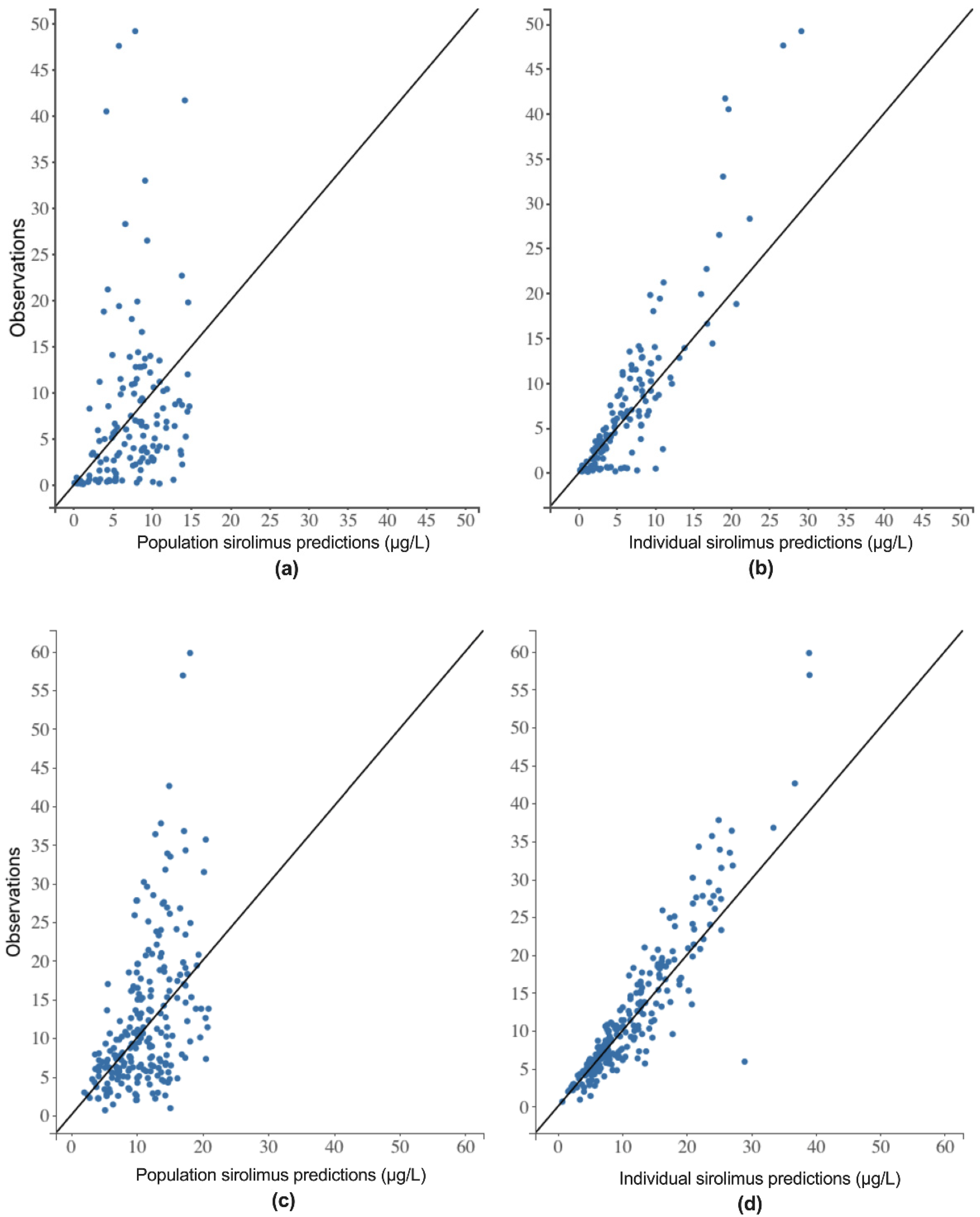
2.3.3. Model Validation
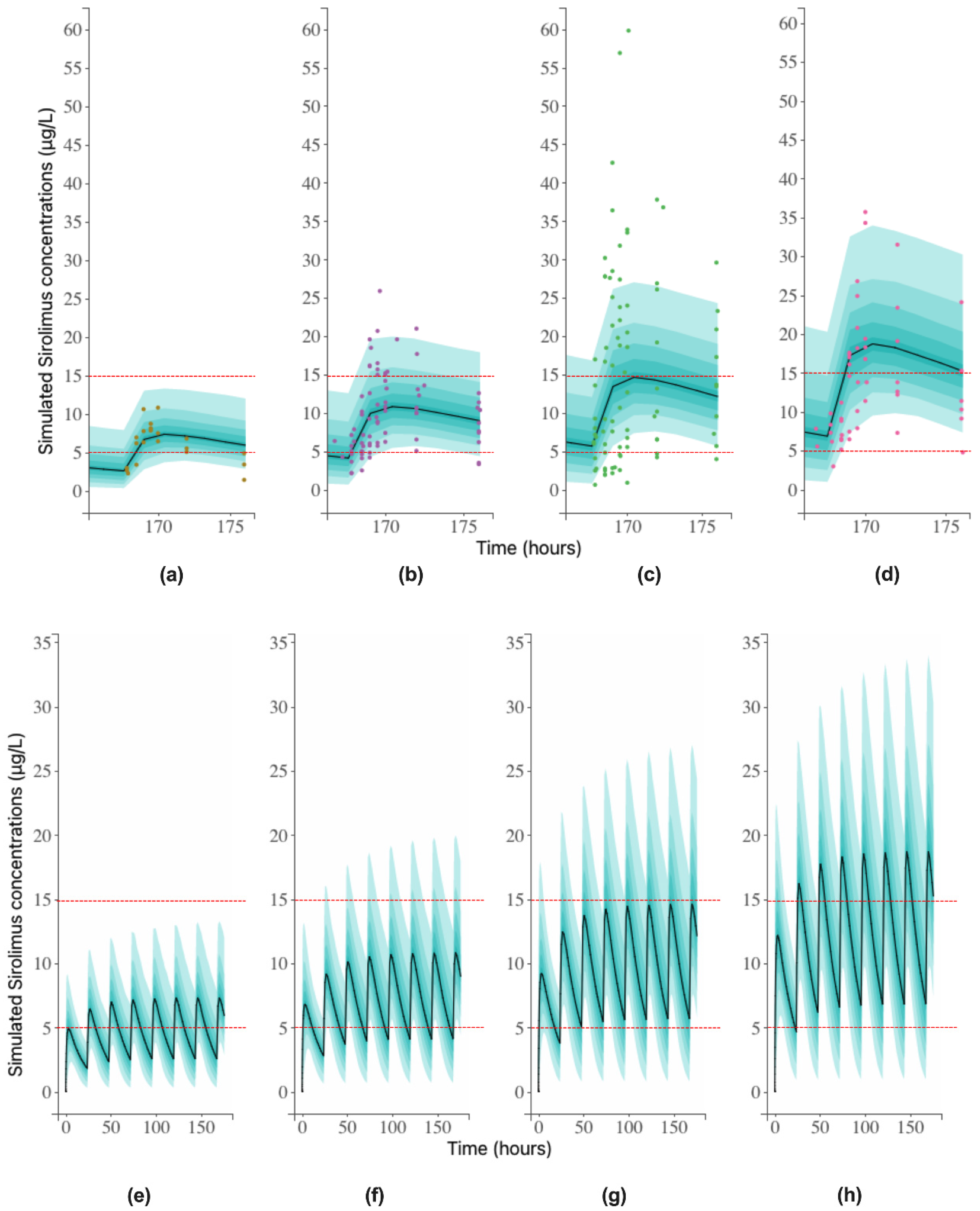
2.3.4. Simulation of Successive Concentrations
2.4. Statistical Analyses
3. Results
3.1. Population Characteristics
3.2. Pharmacokinetic Results
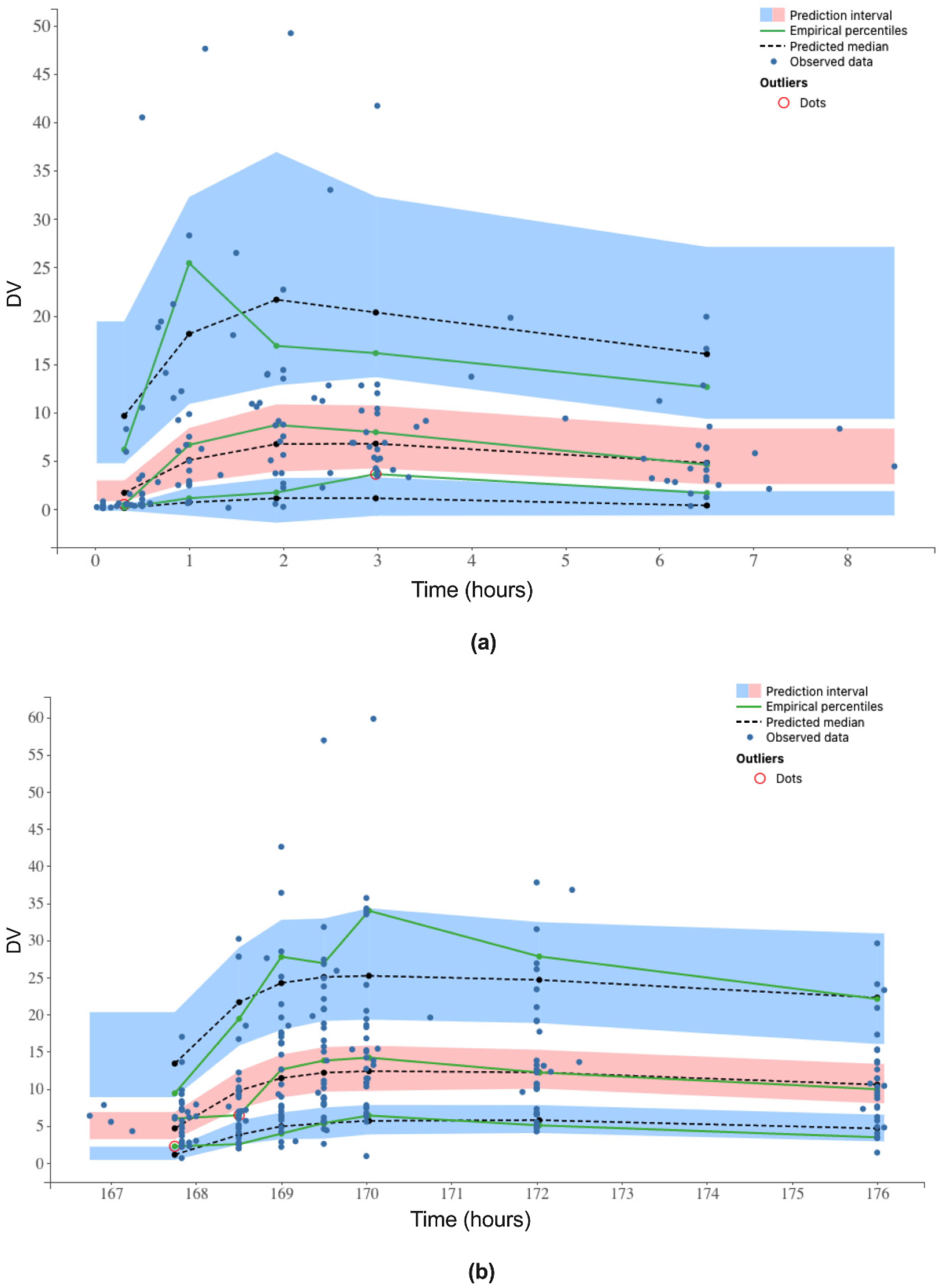
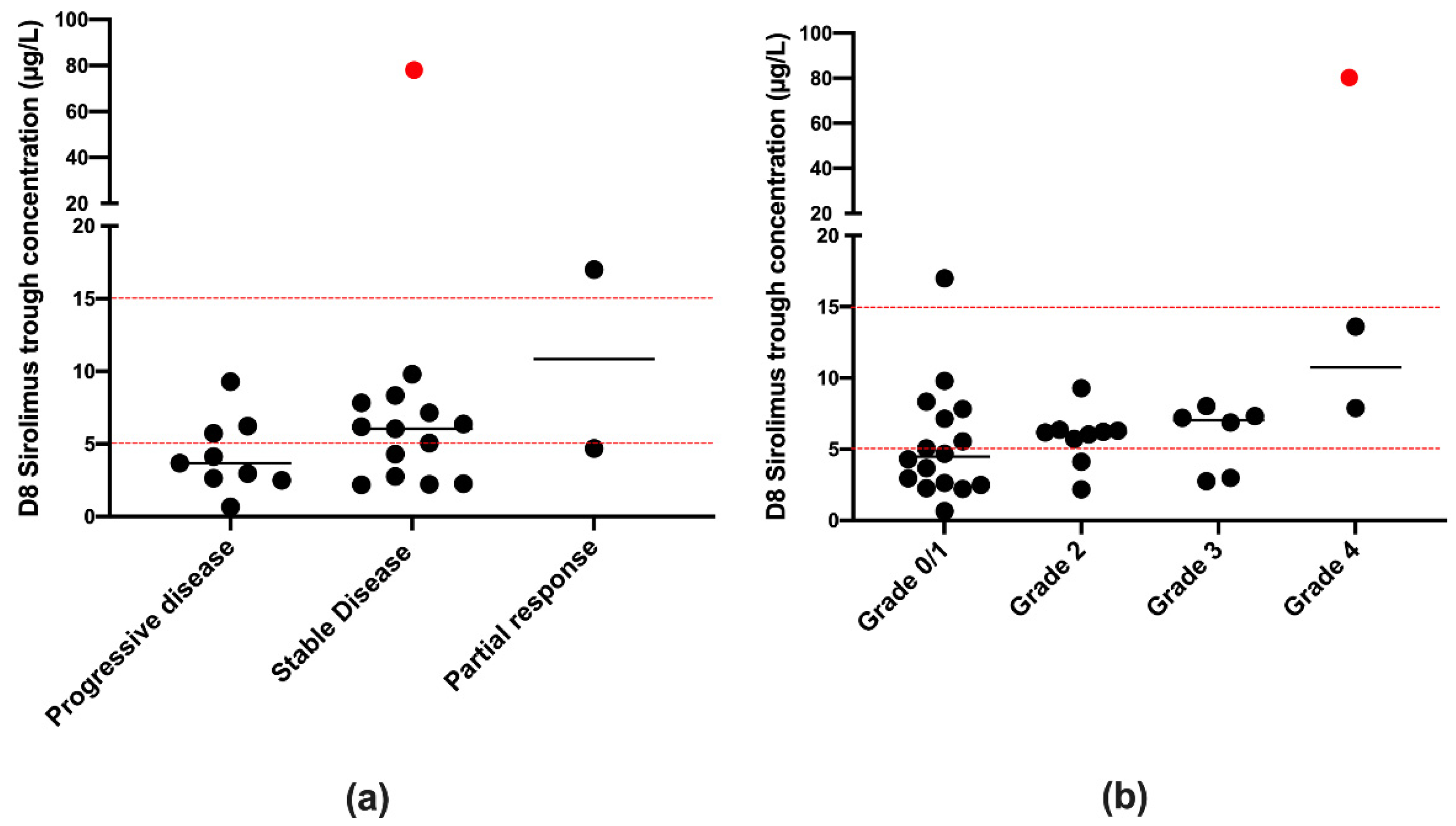
3.3. Simulated Concentrations and Therapeutic Range
3.4. Efficacy and Toxicities’ Assessment after Two Cycles of Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Diekmann, F.; Campistol, J.M. Conversion from Calcineurin Inhibitors to Sirolimus in Chronic Allograft Nephropathy: Benefits and Risks. Nephrol. Dial. Transplant. 2006, 21, 562–568. [Google Scholar] [CrossRef]
- Vergès, B.; Cariou, B. MTOR Inhibitors and Diabetes. Diabetes Res. Clin. Pract. 2015, 110, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Ekshyyan, O.; Ferdinandez, L.H.; Rong, X.; Caldito, G.; Nathan, C.-A.O. Efficacy and Comparative Effectiveness of Sirolimus as an Anticancer Drug. Laryngoscope 2011, 121, 978–982. [Google Scholar] [CrossRef]
- Vincenzi, B.; Napolitano, A.; D’Onofrio, L.; Frezza, A.M.; Silletta, M.; Venditti, O.; Santini, D.; Tonini, G. Targeted Therapy in Sarcomas: Mammalian Target of Rapamycin Inhibitors from Bench to Bedside. Expert Opin. Investig. Drugs 2011, 20, 1685–1705. [Google Scholar] [CrossRef]
- Mondesire, W.H.; Jian, W.; Zhang, H.; Ensor, J.; Hung, M.-C.; Mills, G.B.; Meric-Bernstam, F. Targeting Mammalian Target of Rapamycin Synergistically Enhances Chemotherapy-Induced Cytotoxicity in Breast Cancer Cells. Clin. Cancer Res. 2004, 10, 7031–7042. [Google Scholar] [CrossRef]
- Houghton, P.J.; Morton, C.L.; Kolb, E.A.; Gorlick, R.; Lock, R.; Carol, H.; Reynolds, C.P.; Maris, J.M.; Keir, S.T.; Billups, C.A.; et al. Initial Testing (Stage 1) of the MTOR Inhibitor Rapamycin by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2008, 50, 799–805. [Google Scholar] [CrossRef]
- Pencreach, E.; Guérin, E.; Nicolet, C.; Lelong-Rebel, I.; Voegeli, A.-C.; Oudet, P.; Larsen, A.K.; Gaub, M.-P.; Guenot, D. Marked Activity of Irinotecan and Rapamycin Combination toward Colon Cancer Cells In Vivo and In Vitro Is Mediated through Cooperative Modulation of the Mammalian Target of Rapamycin/Hypoxia-Inducible Factor-1α Axis. Clin. Cancer Res. 2009, 15, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Houghton, P.J.; Morton, C.L.; Gorlick, R.; Lock, R.B.; Carol, H.; Reynolds, C.P.; Kang, M.H.; Maris, J.M.; Keir, S.T.; Kolb, E.A.; et al. Stage 2 Combination Testing of Rapamycin with Cytotoxic Agents by the Pediatric Preclinical Testing Program. Mol. Cancer 2010, 9, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Martin-Liberal, J.; Tirado, O.M.; Muro, X.G. del Sirolimus plus Gemcitabine: A New Therapeutic Combination for Resistant Sarcomas? Expert Rev. Anticancer Ther. 2015, 15, 257–259. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nguyen, A.; Moussallieh, F.M.; Mackay, A.; Cicek, A.E.; Coca, A.; Chenard, M.P.; Weingertner, N.; Lhermitte, B.; Letouzé, E.; Guérin, E.; et al. Characterization of the Transcriptional and Metabolic Responses of Pediatric High Grade Gliomas to MTOR-HIF-1α Axis Inhibition. Oncotarget 2017, 8, 71597–71617. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Morgenstern, D.A.; Marzouki, M.; Bartels, U.; Irwin, M.S.; Sholler, G.L.S.; Gammon, J.; Yankanah, R.; Wu, B.; Samson, Y.; Baruchel, S. Phase I Study of Vinblastine and Sirolimus in Pediatric Patients with Recurrent or Refractory Solid Tumors. Pediatr. Blood Cancer 2014, 61, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Vo, K.T.; Karski, E.E.; Nasholm, N.M.; Allen, S.; Hollinger, F.; Gustafson, W.C.; Long-Boyle, J.R.; Shiboski, S.; Matthay, K.K.; DuBois, S.G. Phase 1 Study of Sirolimus in Combination with Oral Cyclophosphamide and Topotecan in Children and Young Adults with Relapsed and Refractory Solid Tumors. Oncotarget 2017, 8, 23851–23861. [Google Scholar] [CrossRef] [PubMed]
- Naing, A.; Lorusso, P.; Fu, S.; Hong, D.; Chen, H.X.; Doyle, L.A.; Phan, A.T.; Habra, M.A.; Kurzrock, R. Insulin Growth Factor Receptor (IGF-1R) Antibody Cixutumumab Combined with the MTOR Inhibitor Temsirolimus in Patients with Metastatic Adrenocortical Carcinoma. Br. J. Cancer 2013, 108, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Fouladi, M.; Perentesis, J.P.; Wagner, L.M.; Vinks, A.A.; Reid, J.M.; Ahern, C.; Thomas, G.; Mercer, C.A.; Krueger, D.A.; Houghton, P.J.; et al. A Phase I Study of Cixutumumab (IMC-A12) in Combination with Temsirolimus (CCI-779) in Children with Recurrent Solid Tumors: A Children’s Oncology Group Phase I Consortium Report. Clin. Cancer Res. 2015, 21, 1558–1565. [Google Scholar] [CrossRef]
- Wagner, L.M.; Fouladi, M.; Ahmed, A.; Krailo, M.D.; Weigel, B.; DuBois, S.G.; Doyle, L.A.; Chen, H.; Blaney, S.M. Phase II Study of Cixutumumab in Combination with Temsirolimus in Pediatric Patients and Young Adults with Recurrent or Refractory Sarcoma: A Report from the Children’s Oncology Group. Pediatr. Blood Cancer 2015, 62, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Bagatell, R.; Norris, R.; Ingle, A.M.; Ahern, C.; Voss, S.; Fox, E.; Little, A.R.; Weigel, B.J.; Adamson, P.C.; Blaney, S. Phase 1 Trial of Temsirolimus in Combination with Irinotecan and Temozolomide in Children, Adolescents and Young Adults with Relapsed or Refractory Solid Tumors: A Children’s Oncology Group Study. Pediatr. Blood Cancer 2014, 61, 833–839. [Google Scholar] [CrossRef]
- Mody, R.; Naranjo, A.; Van Ryn, C.; Yu, A.L.; London, W.B.; Shulkin, B.L.; Parisi, M.T.; Servaes, S.-E.-N.; Diccianni, M.B.; Sondel, P.M.; et al. Irinotecan-Temozolomide with Temsirolimus or Dinutuximab in Children with Refractory or Relapsed Neuroblastoma (COG ANBL1221): An Open-Label, Randomised, Phase 2 Trial. Lancet Oncol. 2017, 18, 946–957. [Google Scholar] [CrossRef]
- Qayed, M.; Cash, T.; Tighiouart, M.; MacDonald, T.J.; Goldsmith, K.C.; Tanos, R.; Kean, L.; Watkins, B.; Suessmuth, Y.; Wetmore, C.; et al. A Phase I Study of Sirolimus in Combination with Metronomic Therapy (CHOAnome) in Children with Recurrent or Refractory Solid and Brain Tumors. Pediatr. Blood Cancer 2020, 67, e28134. [Google Scholar] [CrossRef]
- Jannier, S.; Kemmel, V.; Sebastia Sancho, C.; Chammas, A.; Sabo, A.-N.; Pencreach, E.; Farace, F.; Chenard, M.P.; Lhermitte, B.; Geoerger, B.; et al. SFCE-RAPIRI Phase I Study of Rapamycin Plus Irinotecan: A New Way to Target Intra-Tumor Hypoxia in Pediatric Refractory Cancers. Cancers 2020, 12, 3051. [Google Scholar] [CrossRef]
- Mahalati, K.; Kahan, B.D. Clinical Pharmacokinetics of Sirolimus. Clin. Pharmacokinet. 2001, 40, 573–585. [Google Scholar] [CrossRef]
- Stenton, S.B.; Partovi, N.; Ensom, M.H.H. Sirolimus: The Evidence for Clinical Pharmacokinetic Monitoring. Clin. Pharmacokinet. 2005, 44, 769–786. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Hu, L.; Han, Z.; Hou, K.; Fang, H.; Zhang, G.; Feng, Y.; Huang, L. The Effect of ABCB1 Polymorphism on Sirolimus in Renal Transplant Recipients: A Meta-Analysis. Transl. Urol. 2020, 9, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Emoto, C.; Fukuda, T.; Venkatasubramanian, R.; Vinks, A.A. The Impact of CYP3A5*3 Polymorphism on Sirolimus Pharmacokinetics: Insights from Predictions with a Physiologically-Based Pharmacokinetic Model. Br. J. Clin. Pharmacol. 2015, 80, 1438–1446. [Google Scholar] [CrossRef] [PubMed]
- Golubović, B.; Vučićević, K.; Radivojević, D.; Kovačević, S.V.; Prostran, M.; Miljković, B. Exploring Sirolimus Pharmacokinetic Variability Using Data Available from the Routine Clinical Care of Renal Transplant Patients—Population Pharmacokinetic Approach. J. Med. Biochem. 2019, 38, 323–331. [Google Scholar] [CrossRef]
- Ribbing, J.; Jonsson, E.N. Power, Selection Bias and Predictive Performance of the Population Pharmacokinetic Covariate Model. J Pharmacokinet. Pharmacodyn. 2004, 31, 109–134. [Google Scholar] [CrossRef]
- Mizuno, T.; Fukuda, T.; Emoto, C.; Mobberley-Schuman, P.S.; Hammill, A.M.; Adams, D.M.; Vinks, A.A. Developmental Pharmacokinetics of Sirolimus: Implications for Precision Dosing in Neonates and Infants with Complicated Vascular Anomalies. Pediatr. Blood Cancer 2017, 64, e26470. [Google Scholar] [CrossRef]
- Mizuno, T.; Fukuda, T.; Christians, U.; Perentesis, J.P.; Fouladi, M.; Vinks, A.A. Population Pharmacokinetics of Temsirolimus and Sirolimus in Children with Recurrent Solid Tumours: A Report from the Children’s Oncology Group. Br. J. Clin. Pharmacol. 2017, 83, 1097–1107. [Google Scholar] [CrossRef]
- Dansirikul, C.; Morris, R.G.; Tett, S.E.; Duffull, S.B. A Bayesian Approach for Population Pharmacokinetic Modelling of Sirolimus. Br. J. Clin. Pharmacol. 2006, 62, 420–434. [Google Scholar] [CrossRef]
- Scott, J.R.; Courter, J.D.; Saldaña, S.N.; Widemann, B.C.; Fisher, M.; Weiss, B.; Perentesis, J.; Vinks, A.A. Population Pharmacokinetics of Sirolimus in Pediatric Patients With Neurofibromatosis Type 1. Ther. Drug Monit. 2013, 35, 332–337. [Google Scholar] [CrossRef]
- Schachter, A.D.; Benfield, M.R.; Wyatt, R.J.; Grimm, P.C.; Fennell, R.S.; Herrin, J.T.; Lirenman, D.S.; McDonald, R.A.; Munoz-Arizpe, R.; Harmon, W.E. Sirolimus Pharmacokinetics in Pediatric Renal Transplant Recipients Receiving Calcineurin Inhibitor Co-Therapy. Pediatr. Transplant. 2006, 10, 914–919. [Google Scholar] [CrossRef]
- Goyal, R.K.; Han, K.; Wall, D.A.; Pulsipher, M.A.; Bunin, N.; Grupp, S.A.; Mada, S.R.; Venkataramanan, R. Sirolimus Pharmacokinetics in Early Postmyeloablative Pediatric Blood and Marrow Transplantation. Biol. Blood Marrow Transplant. 2013, 19, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.; Sparidans, R.W.; Cheung, K.L.; Fukami, T.; Durmus, S.; Wagenaar, E.; Yokoi, T.; van Vlijmen, B.J.M.; Beijnen, J.H.; Schinkel, A.H. P-Glycoprotein, CYP3A, and Plasma Carboxylesterase Determine Brain and Blood Disposition of the MTOR Inhibitor Everolimus (Afinitor) in Mice. Clin. Cancer Res. 2014, 20, 3133–3145. [Google Scholar] [CrossRef] [PubMed]
- Arimori, K.; Kuroki, N.; Hidaka, M.; Iwakiri, T.; Yamsaki, K.; Okumura, M.; Ono, H.; Takamura, N.; Kikuchi, M.; Nakano, M. Effect of P-Glycoprotein Modulator, Cyclosporin A, on the Gastrointestinal Excretion of Irinotecan and Its Metabolite SN-38 in Rats. Pharm. Res. 2003, 20, 910–917. [Google Scholar] [CrossRef]
- Riera, P.; Artigas-Baleri, A.; Salazar, J.; Sebio, A.; Virgili, A.C.; Arranz, M.J.; Páez, D. ABCB1 Genetic Variants as Predictors of Irinotecan-Induced Severe Gastrointestinal Toxicity in Metastatic Colorectal Cancer Patients. Front. Pharmacol. 2020, 11, 973. [Google Scholar] [CrossRef]
- El-Sankary, W.; Bombail, V.; Gibson, G.G.; Plant, N. Glucocorticoid-Mediated Induction of CYP3A4 Is Decreased by Disruption of a Protein: DNA Interaction Distinct from the Pregnane X Receptor Response Element. Drug Metab. Dispos. 2002, 30, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Jusko, W.J.; Ferron, G.M.; Mis, S.M.; Kahan, B.D.; Zimmerman, J.J. Pharmacokinetics of Prednisolone during Administration of Sirolimus in Patients with Renal Transplants. J. Clin. Pharmacol. 1996, 36, 1100–1106. [Google Scholar] [CrossRef]
- Bäckman, L.; Kreis, H.; Morales, J.M.; Wilczek, H.; Taylor, R.; Burke, J.T. Sirolimus Steady-State Trough Concentrations Are Not Affected by Bolus Methylprednisolone Therapy in Renal Allograft Recipients. Br. J. Clin. Pharmacol. 2002, 54, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Wang, J.-S.; Backman, J.T.; Laitila, J.; Neuvonen, P.J. Trimethoprim and Sulfamethoxazole Are Selective Inhibitors of CYP2C8 and CYP2C9, Respectively. Drug Metab. Dispos. 2002, 30, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Böttiger, Y.; Brattström, C.; Bäckman, L.; Claesson, K.; Burke, J.T. Trimethoprim-Sulphamethoxazole Does Not Affect the Pharmacokinetics of Sirolimus in Renal Transplant Recipients. Br. J. Clin. Pharmacol. 2005, 60, 566–569. [Google Scholar] [CrossRef][Green Version]
- Zimmerman, J.J.; Kahan, B.D. Pharmacokinetics of Sirolimus in Stable Renal Transplant Patients after Multiple Oral Dose Administration. J. Clin. Pharmacol. 1997, 37, 405–415. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Levels | Irinotecan Dose (mg/m2, D1) | Sirolimus Dose (mg/m2/day) | Included Patients |
|---|---|---|---|
| 1 | 125 | 1 | 3 |
| 2 | 125 | 1.5 | 6 |
| 3 | 125 | 2 | 6 |
| 4 | 125 | 2.5 | 3 |
| 5 | 200 | 1.5 | 3 |
| 6 | 200 | 2 | 3 |
| 7 | 200 | 2.5 | 3 |
| 8 | 240 | 1.5 | 3 |
| 9 | 240 | 2 | 6 |
| 10 | 240 | 2.5 | 6 |
| Variable | D1 | D8 |
|---|---|---|
| Demographics | ||
| Number of pharmacokinetic profiles | 27 | 34 |
| Number of observations | 127 | 229 |
| Age (years) (mean ± SD) | 11.7 ± 5.9 | 12.6 ± 5.6 |
| <12 years | 14 | 14 |
| ≥12 years | 13 | 20 |
| Male/Female | 16/11 | 21/13 |
| Body weight (kg) (mean ± SD) | 38.5 ± 18.6 | 40.7 ± 17.4 |
| Height (cm) (mean ± SD) | 140.9 ± 31.2 | 146.0 ± 29.25 |
| Body surface area (m2) (mean ± SD) | 1.2 ± 0.4 | 1.3 ± 0.4 |
| Body mass index (kg/m2) (mean ± SD) | 18.1 ± 2.9 | 18.1 ± 2.8 |
| Central nervous system tumor/other diagnostics | 12/15 | 14/20 |
| Sirolimus information | ||
| Real administered dose (mean ± SD, (n)) | ||
| Dose level group 1 mg/m2 | 1.22 ± 0.81 (2) | 1.14 ± 0.59 (3) |
| Dose level group 1.5 mg/m2 | 2.03 ± 0.67 (10) | 2.04 ± 0.63 (11) |
| Dose level group 2 mg/m2 | 2.26 ± 0.88 (9) | 2.54 ± 0.78 (13) |
| Dose level group 2.5 mg/m2 | 2.78 ± 0.92 (6) | 2.93 ± 0.94 (7) |
| Irinotecan administration | ||
| Irinotecan infusion before sirolimus intake | Yes | No |
| Real administered dose (mean ± SD, (n)) | ||
| Dose level group 125 mg/m2 | 150 ± 52.3 (14) | - |
| Dose level group 200 mg/m2 | 307 ± 80.5 (5) | - |
| Dose level group 240 mg/m2 | 247 ± 102 (8) | - |
| Comedication | ||
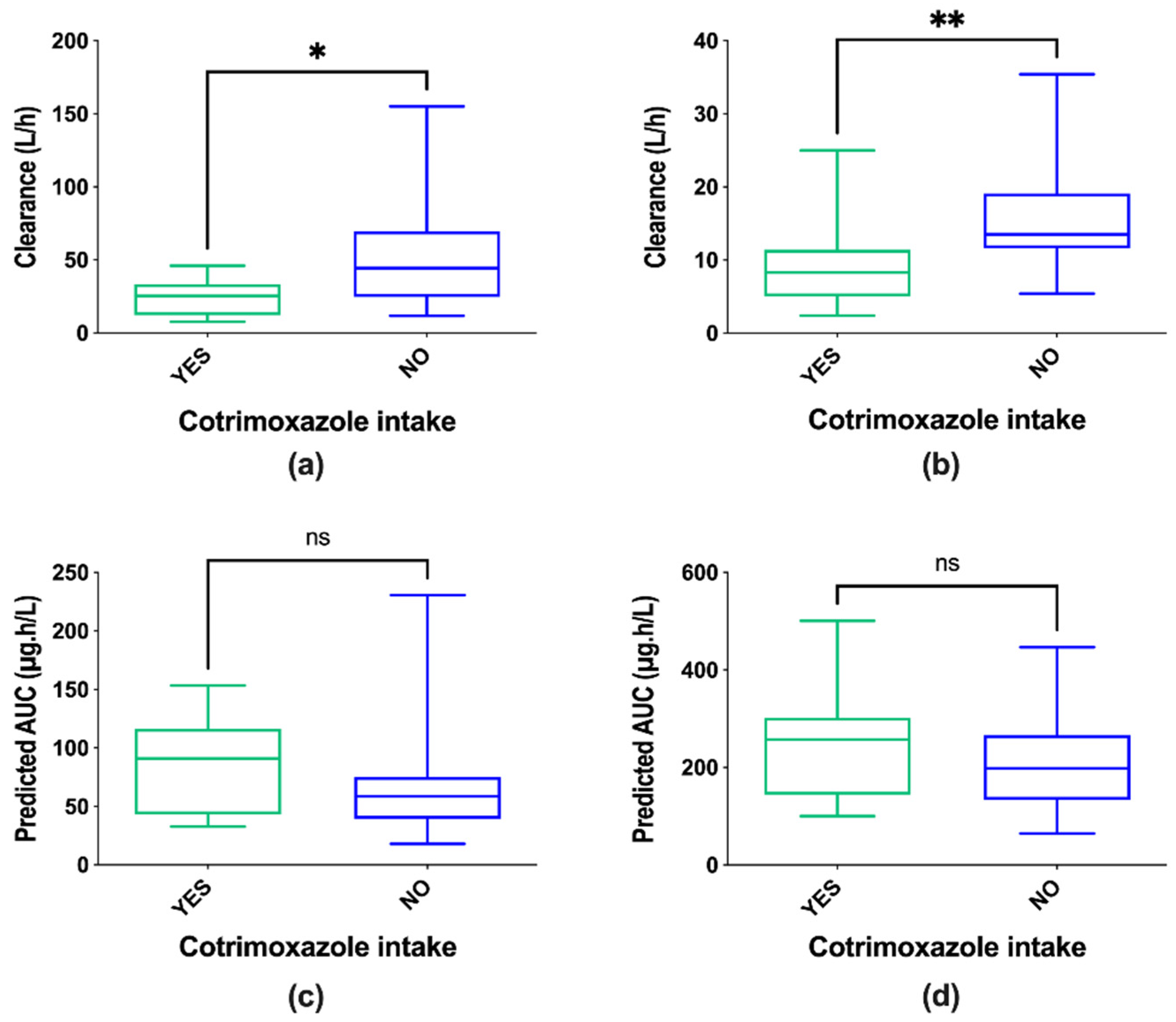
| Number of patients on cotrimoxazole Median doses 400/80 mg Range from 200/40 to 800/160 mg | 11 | 15 |
| Number of patients on glucocorticoids | 3 | 3 |
| Parameter | Definition | Base Model Estimation (RSE) | Final Model Estimation (RSE) | ||
|---|---|---|---|---|---|
| Structural model | D1 | D8 | D1 | D8 | |
| ka (h−1) | Absorption rate constant | 0.52 (41.6) | 1.02 (27.1) | 0.46 (39.2) | 0.97 (27.1) |
| Cl/F (L.h−1) | Apparent sirolimus clearance | 21.5 (22.8) | 10.0 (11.5) | 23.9 (21.8) | 11.9 (9.2) |
| Vd/F (L) | Apparent volume of distribution | 92.8 (37.2) | 214.0 (15.7) | 88.9 (35.5) | 238.0 (9.9) |
| Covariates | |||||
| β_Vd_BS | Effect of body surface on Vd/F | - | - | 1.35 (31.0) | 1.41 (15.5) |
| β_Cl_BS | Effect of body surface on Cl/F | - | - | - | 1.09 (21.2) |
| IIV (CV) | |||||
| IIV-ka (%) | ka interindividual variability | 110.4 (26.9) | 141.8 (32.2) | 80.0 (25.3) | 168.5 (21.2) |
| IIV-Cl/F (%) | Cl/F interindividual variability | 95.6 (19.5) | 71.8 (13.4) | 92.9 (19.2) | 50.1 (13.5) |
| IIV-Vd/F (%) | Vd/F interindividual variability | 97.6 (24.6) | 69.8 (16.0) | 62.5 (38.3) | 29.4 (25.1) |
| Residual error | |||||
| b (%) | Proportional residual error | 0.549 (8.97) | 0.312 (6.25) | 0.538 (8.90) | 0.306 (6.19) |
| OFV | −2 log-likelihood value | 755.97 | 1361.87 | 747.66 | 1320.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabo, A.-N.; Jannier, S.; Becker, G.; Lessinger, J.-M.; Entz-Werlé, N.; Kemmel, V. Sirolimus Pharmacokinetics Variability Points to the Relevance of Therapeutic Drug Monitoring in Pediatric Oncology. Pharmaceutics 2021, 13, 470. https://doi.org/10.3390/pharmaceutics13040470
Sabo A-N, Jannier S, Becker G, Lessinger J-M, Entz-Werlé N, Kemmel V. Sirolimus Pharmacokinetics Variability Points to the Relevance of Therapeutic Drug Monitoring in Pediatric Oncology. Pharmaceutics. 2021; 13(4):470. https://doi.org/10.3390/pharmaceutics13040470
Chicago/Turabian StyleSabo, Amelia-Naomi, Sarah Jannier, Guillaume Becker, Jean-Marc Lessinger, Natacha Entz-Werlé, and Véronique Kemmel. 2021. "Sirolimus Pharmacokinetics Variability Points to the Relevance of Therapeutic Drug Monitoring in Pediatric Oncology" Pharmaceutics 13, no. 4: 470. https://doi.org/10.3390/pharmaceutics13040470
APA StyleSabo, A.-N., Jannier, S., Becker, G., Lessinger, J.-M., Entz-Werlé, N., & Kemmel, V. (2021). Sirolimus Pharmacokinetics Variability Points to the Relevance of Therapeutic Drug Monitoring in Pediatric Oncology. Pharmaceutics, 13(4), 470. https://doi.org/10.3390/pharmaceutics13040470