Abstract
In contrast to external high energy photon or proton therapy, targeted radionuclide therapy (TRNT) is a systemic cancer treatment allowing targeted irradiation of a primary tumor and all its metastases, resulting in less collateral damage to normal tissues. The α-emitting radionuclide bismuth-213 (213Bi) has interesting properties and can be considered as a magic bullet for TRNT. The benefits and drawbacks of targeted alpha therapy with 213Bi are discussed in this review, covering the entire chain from radionuclide production to bedside. First, the radionuclide properties and production of 225Ac and its daughter 213Bi are discussed, followed by the fundamental chemical properties of bismuth. Next, an overview of available acyclic and macrocyclic bifunctional chelators for bismuth and general considerations for designing a 213Bi-radiopharmaceutical are provided. Finally, we provide an overview of preclinical and clinical studies involving 213Bi-radiopharmaceuticals, as well as the future perspectives of this promising cancer treatment option.
1. Introduction
In the diagnosis, monitoring, and treatment of cancer patients, nuclear medicine plays an important role. Radiopharmaceuticals consist generally out of three functional elements: (1) a vector molecule (or carrier) that shows high selectivity and affinity for a target; (2) a radionuclide; and (3) a linker or chelator to attach the former to the latter (Figure 1) [1,2,3,4]. A fundamental and critical component of a radiometal-based radiopharmaceutical is the chelator, the ligand that binds the radiometal ion in a stable coordination complex so that it can be properly directed to its molecular target in vivo [5,6,7]. The radiopharmaceutical is mostly distributed within the body by the vascular system and allows targeting of a primary tumor and all its metastases. The specific decay characteristics of the radionuclide attached to the vector molecule determine if the radiopharmaceutical can be used for diagnostic (molecular imaging) or therapeutic (TRNT, targeted radionuclide therapy) purposes. The goal of targeted radionuclide therapy is to deliver sufficiently high doses of ionizing radiation to specific disease sites for cure, disease control, or symptomatic (e.g., pain of hormonal secretion) palliation. The biological effect of radionuclide therapy is obtained by three mechanisms: (1) Interaction of ionizing radiation with water in which chemically active free radicals (reactive oxygen species, ROS) are formed that can react with biomolecules (phospholipids, proteins, RNA, DNA, etc.), thereby irreversibly damaging the cells. (2) Direct interaction of ionizing radiation with DNA in which single-strand (SSB), double-strand (DSB), or cluster breaks can occur. (3) During treatment, a phenomenon called the abscopal effect can occur when radiation reduces not only the targeted tumor but also leads to the shrinkage of untreated tumors elsewhere in the body. Though the exact biological mechanisms accountable for the abscopal effect are yet to be identified, the immune system is considered the major player in this significant role [1,8].
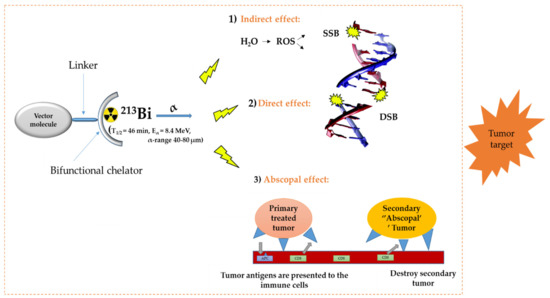
Figure 1.
General concept of TRNT. ROS = reactive oxygen species, SSB = single strand break, DSB = double strand break, APC = antigen presenting cells, CD6 = cluster of differentiation 6. The DNA structure was reproduced from Gill and Vallis 2019, with permission from the Royal Society of Chemistry [9], Royal Society of Chemistry, 2019.
Auger-, beta minus (β–-), and alpha (α-) emitters are the three main subgroups of therapeutic radionuclides. α-decay is a radioactive decay in which an α-particle, consisting of two protons and two neutrons, is emitted from the nucleus. Tumor cells can be selectively destroyed, and the healthy tissue is minimally damaged due to the short penetration depth of α-particles (40–80 µm, corresponding to 2–10 cell diameter). An α-particle is characterized by a high-energy linear transfer (LET, 100 kEV/µm) and thus a high relative biological effect (RBE). The high LET of α-particles results in a high rate of double-strand and cluster DNA breaks and consequently irreparable damage, producing the high kill rate of α-emitting radioisotopes both in normoxic as well as in a hypoxic tumor cell environment, which is known to be more resistant to photon and electron-based irradiation [10]. Therefore, targeted alpha therapy (TAT) can be considered as promising cancer treatment, especially suitable for the treatment of tumors with small diameters and which have a spatially homogeneous expression of the molecule targeted by the vector. Sufficient expression of the target in the malignant tissue must be confirmed before TAT can be initiated. This can be done by first performing a positron emission tomography (PET) or single photon emission computed tomography (SPECT) scan, after injection of the diagnostic sister of the therapeutic radiopharmaceutical. If the diagnostic radiopharmaceutical can truthfully predict the pharmacokinetics of its therapeutic sister, the diagnostic scan also allows accurate calculation of the dosimetry. This class of radiopharmaceuticals are called theranostic agents [11].
It is also important to mention here that the theranostic approach could also involve a single radionuclide agent—this theory stipulates the use of a single radionuclide, producing both therapeutic radiation and a low-energy γ-ray for SPECT to deliver dosimetric data. Examples of some of these radionuclides are 47Sc (T½ = 3.35 d) and 67Cu (T½ = 2.58 d) [12].
The α-emitting radionuclide 213Bi (T1/2 = 45.6 min, Eα = 8.4 MeV, γ = 440 keV, α-particle range = 40–80 µm) has interesting properties and might be considered as a magic bullet for TRNT [5,9]. However, like every cancer treatment modality, 213Bi-based TAT has advantages and disadvantages. Indeed, 213Bi can be considered as powerful precision ammunition, but it needs to be handled with care. In function of the target, the most suited vector molecule needs to be selected and the biological half-life of the vector should be compatible with the 45.6 min physical half-life of 213Bi, which is substantially shorter than any therapeutic radionuclide in current routine clinical use. Further, an appropriate chelator needs to be carefully chosen to match the physical properties of 213Bi.
In this review, we will discuss advantages and disadvantages of TAT using 213Bi, covering the entire chain from radionuclide production to bedside. This includes production of the mother isotope 225Ac and the use of the 225Ac/213Bi generator, chemical and physical properties of 213Bi, an overview of acyclic and macrocyclic chelating agents that are available for 213Bi, general considerations for designing a 213Bi-radiopharmaceutical, and an in-depth overview of non-clinical and clinical studies that have been performed, discussing dosimetry and toxicity issues. Lastly, we will discuss the future perspectives of this intriguing potential cancer treatment.
2. Radionuclide Properties and Production of 225Ac and Its Daughter 213Bi
2.1. Decay Properties of 225Ac and 213Bi
225Ac is the parent radionuclide of 213Bi and is a relatively long-lived α-emitter with a half-life of 9.9 d. It decays via a cascade of six short-lived radionuclide daughters to stable 209Bi with four net alpha particles emitted per decay (Figure 2). 213Bi has a half-life of 45.6 min and shows branching decay (β– and α decay), and most of the α-particles emitted originate from the β– branch (see Figure 2). Indeed, it mostly decays via β– emission to the short-lived α-emitter 213Po (T1/2 = 4.2 µs, Eα = 8.375 MeV, 97.8%, Figure 2). The residual 2.2% of 213Bi decays leads to 209Tl (Eα = 5.549 MeV, 0.16%, Eα = 5.869 MeV, 2.0%). Finally, 213Po and 209Tl decay via 209Pb (T1/2 = 3.25 h, β–) to stable 209Bi [13].
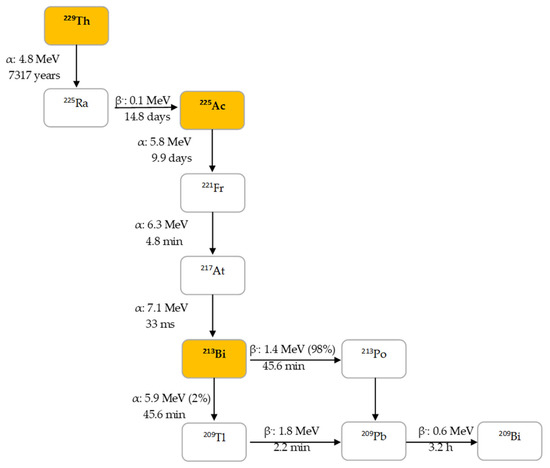
Figure 2.
Decay chain of thorium-229 to 225Ac and 213Bi.
In human tissue, the α-particle released by 213Po (Eα = 8.375 MeV) has a path length of 85 µm. It is this radionuclide that produces >98% of the α-particle energy released per disintegration of 213Bi and could be considered as the radionuclide that provides 213Bi cytotoxicity. The bulk of the total particle energy released per disintegration of 213Bi comes from α-decay, accounting for 92.7%, while 7.3% comes from β-particle emission, which includes the decay of 209Pb [14]. The decay of 213Bi is followed by the emission of a 435 keV-photon (98% abundance) that could potentially be used for SPECT with a gamma camera equipped with high energy collimators, permitting detailed evaluation of the biodistribution of 213Bi in vivo [15].
It is noteworthy to mention that high activity 225Ac/213Bi generators are required to allow production of clinical amounts of 213Bi. Optimal clinical injected activities depend on the vector molecules; for prostate-specific membrane antigen (PSMA)-targeting radiopharmaceuticals, this has been determined to be 4–8 MBq of 225Ac [16,17]. In contrast, to have the same number of alpha-particles emitted, 5–10 GBq is needed for 213Bi-labeled radiopharmaceuticals [18]. This was estimated based on the Equations (1) and (2) below and the fact that 225Ac delivers four α particles in its total decay scheme, while 213Bi delivers net only one α particle. However, this is a rough estimation that does not consider the biological clearance of the radioprobes. Definitely, the much shorter half-life of 213Bi compared to the half-life of 225Ac is the key reason for the high activity levels that are needed when using 213Bi for targeted radionuclide therapy (Equation (2)).
where A = activity (Bq, disintegrations per second), N = number of atoms, λ = decay constant.
A = λ·N
λ = ln(2)/T1/2
2.2. Current Strategies for 225Ac Production
Several approaches for 225Ac and 213Bi production have been recently reviewed and discussed in detail (Figure 3) [7,13]. The most utilized strategy is the radiochemical extraction of 225Ac from 229Th (T1/2 = 7317 years) sources originating from the decay of fissile 233U. This results in “carrier free” 225Ac using the generator method, but the available 233U stocks and thus 229Th stocks are limited because 233U management is restricted by the requirements concerning non-proliferation of fissile materials. Most of the 213Bi and 225Ac used in clinical tests and research activities worldwide has so far been produced by this approach [19]. Two examples of 229Th sources that can produce clinically relevant activities of 225Ac are (1) the US Department of Energy, Oak Ridge National Laboratory (ORNL) in Oak Ridge, TN, United States of America and (2) the Directorate for Nuclear Safety and Security of the JRC of the European Commission in Karlsruhe, Germany. Additionally, at the Belgian Nuclear Research Centre (SCK CEN) in Mol, Belgium, very pure sources of 229Th were identified, processed, and used for pre-clinical studies [20]. The total global annual 225Ac production volume is approximately 55–65 GBq [21] and cannot meet the growing demand for 225Ac, but interestingly, it has been reported that stocks will increase by extraction of additional 229Th from US legacy wastes [7].
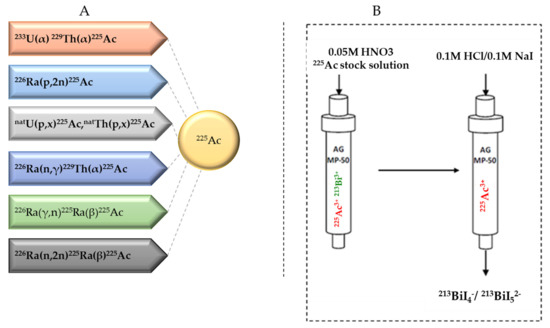
Figure 3.
(A) Production routes for 225Ac and (B) the 225Ac/213Bi generator.
Cyclotron production is an alternative method for 225Ac production. Medium-energy proton irradiation of 226Ra using the reaction 226Ra(p,2n)225Ac is considered to be a viable approach. The advantage of this method is the widespread availability of appropriate cyclotrons worldwide, especially in Europe [22,23], and high amounts of clinical doses of 225Ac could be produced in a reliable way. It is important to mention that medical cyclotrons used to produce radioisotopes (15–25 MeV) are considered to be feasible for basic and applied research [24]. The downside with this approach is that 226Ra is not an easy isotope to work with due to the presence of the highly radiotoxic noble gas 222Rn in its decay chain. Therefore, it is unlikely that this will ever enter a clinical cyclotron.
Further, high-energy proton irradiation (0.6–2 GeV) of uranium and thorium spallation targets via the reaction natU(p,x)225Ac is another approach for the production of 225Ac. The existence of several suitable high-energy proton facilities makes this an achievable prospect. However, such accelerator-produced 225Ac contains a low percentage (0.1–0.3%) 227Ac (T1/2 = 21.77y) at the end of bombardment, which might be a serious limitation in terms of clinical translation and waste management [25,26]. Nevertheless, 225Ac obtained through high-energy accelerators could be entirely appropriate for 225Ac/213Bi generator production (see 2.3), even with co-production of 227Ac, as all actinium species will be retained on the generator.
Another production path being explored is transmutation of 226Ra to 229Th by an intense neutron flux. This will lead to three successive neutron capture reactions: 226Ra(n,γ)227Ra, 227Ac(n,γ)228Ac, and 228Th(n,γ)229Th. However, production of many orders of magnitude of 228Th (T1/2 = 1.9 y) intermediate, and handling of the radium target remains a challenge [18].
226Ra(γ,n)225Ra is another reaction path for 225Ac production that has been determined experimentally [27,28]. This strategy explored irradiation of old radium needles with high-energy X-rays from electron linear accelerators (linacs).
Finally, transmutation of 226Ra to 225Ra by fast neutrons via the reaction 226Ra(n,2n)225Ra is also under consideration. It is noteworthy to point out that the limitation for all the strategies discussed is the handling of the radium targets. However, earlier results with these methods have been promising [22].
To conclude, all clinical studies with 225Ac/213Bi radiopharmaceuticals were performed to date with 225Ac originating from 229Th stocks, but other accelerator-based production routes were heavily investigated in the last decade. This progress will hopefully assure reliable production and delivery of 225Ac to radiopharmacy institutes in the near future, allowing more preclinical research and multicenter clinical trials with both 225Ac and 213Bi-radiopharmaceuticals.
2.3. 225Ac/213Bi Radionuclide Generators
225Ac can be loaded on 225Ac/213Bi generators to deliver 213Bi on site, but it can also be used directly as a therapeutic radionuclide. 225Ac/213Bi generators are well explored and discussed [29,30,31]. The most established strategy is based on the direct generator method, in which the parent 225Ac in acidic solution (e.g., 0.05M HNO3) is strongly retained by the sorbent (e.g., AG MP-50 cation exchange resin) and 213Bi is eluted. Elution is performed generally with a mixture of 0.1M HCl/0.1M NaI to obtain 213Bi in the form of 213BiI4- and 213BiI52- that can be directly used for radiochemistry purposes. (Figure 3) [7]. These generators can potentially be used clinically due to the relatively long parent half-life, which allows shipment of the generator to radiopharmacy facilities at long distance. Additionally, the transient equilibrium of the 225Ac-213Bi permits elution at approximately every 3 h [32]. These generators can provide weeks of reliable in-house generation of 213Bi for radiolabeling purposes [33]. High activity (up to 4 GBq 225Ac) generator systems, developed at JRC Karlsruhe, have been reported with yields of 213Bi elution exceeding 80% and low breakthrough of 225Ac of less than 0.2 ppm [32].
3. Fundamental Chemistry of Bi
Bi is an element that in comparison to the lighter main group metals has received only limited attention in the past. However, Bi compounds are or were used in a wide spectrum of applications ranging from non-toxic pigments and catalysts to biocompatible additives in dental materials and remedies in human and veterinary medicine [34,35]. To select a good chelator for 213Bi to be used in radiopharmaceutical applications, it is important to have an in-depth knowledge about the chemical properties of Bi.
The Bi atom has 83 electrons distributed among different energy levels. Electrons occupying the highest and outermost energy levels are responsible for its chemical reactivity [34]. Bi has a [Xe] 4f145d106s26p3 electron configuration and tends to form trivalent bismuth (Bi3+) with 6s2 valence configuration. For this tendency of the 6s2 electron pair to remain formally unoxidized in Bi compounds (i.e., core-like nature of the 6s electrons), the term “inert pair effect” [36], or “nonhybridization effect” has often been used [37]. Bi has two major oxidation states (III and V), with Bi(III) being the most common and stable oxidation state. However, rare occurrences of Bi(II) and Bi(IV) have been reported [38].
The aqueous Bi chemistry is predominantly dominated by Bi(III) and, unlike the d-block metals, it forms complex and clustered structures across the pH scale [39,40]. At low pH (e.g., pKa1 = 1.5), Bi(III) readily undergoes hydrolysis to form Bi(III) hydroxide [41]. In dilute aqueous solutions ([Bi] ˂ 1 × 10−5 molarity), mononuclear complexes predominate considerably over a wide range of pH values [42], and these mononuclear complexes of Bi largely follow a stepwise addition of OH– such that Bi(OH)x(3−x) (where x ≤ 4) complexes are formed sequentially with increasing pH.
Bi(III) is a borderline metal ion, according to Pearson’s hard–soft acid–base (HSAB) theory [43]. Nevertheless, with considerable affinity for nitrogen and oxygen donor atoms [44,45,46], some researchers have reported Bi(III) as a strong Lewis acid [39]. This property makes complexation with amine donor groups possible even at low pH [47]. The complexation kinetics with N- and O-donor atoms is often fast, with equilibrium achieved in a few minutes [47]. Some amino ligand binding constants have predictive nature, which can be expressed as logK1(polyamine) = 1.152 logβn(NH3) + (n−1) × 1.744, where βn refers to the stability of the complex containing primary amine analogues and n is the number of primary amine analogues of the polyamine [47]. As observed with aqua ligands, small O-donor ligands bridge multiple Bi atoms to form cluster species [48].
In Bi coordination chemistry, Bi(III) complexes exhibit a variety of coordination numbers, with values between 3–10 [41]. Low coordination numbers can be well-predicted using valence shell electron pair repulsion theory (VSEPR, see Table 1) [41]. Nonetheless, higher coordination numbers (>6) require more complex levels of theory to account for structural influence on geometry (e.g., vibrational coupling, etc.).

Table 1.
Summary of geometries for each coordination number. Table was adapted from Sun H. et al., 1997, with permission from [41], John Wiley and Sons, 2006.
4. Bifunctional Chelating Ligands for 213Bi
When a radiometal is used for targeted radionuclide therapy, a chelator with an extra reactive functional group is required. This moiety, also called a bifunctional chelator (BF), guarantees a covalent connection with the vector molecule and forms a stable radiometal complex. Because of the high cost of the α-emitting radionuclides, short physical half-life in the case of 213Bi, and radioprotection reasons, quantitative yields using fast and mild radiolabeling conditions (e.g., room temperature, 5 min) are desired to facilitate efficient good manufacturing practice (GMP)-compliant on-site production of 213Bi radiopharmaceuticals. Further, if the vector molecule is a heat-sensitive biomolecule or peptide, mild aqueous labelling conditions should be applied to avoid degradation of the vector molecule. Importantly, the radiometal–chelator complex should have a high thermodynamic stability and kinetic inertness to avoid the in vivo dissociation that typically occurs via transchelation to serum proteins and enzymes. Several papers have reported that naked 213Bi, dissociated from its chelating ligands, tends to accumulate in the kidney [5,49,50]. In this chapter, we will give an overview of most important bifunctional chelators currently used for Bi3+.
4.1. DTPA and DTPA-Derivatives
The discovery of acyclic chelators with rapid radiometal ion coordination kinetics, high in vivo stability, and kinetic inertness has become a requirement for TRNT due to the emergence of antibody vectors.
As one of the oldest chelators used in radiopharmaceuticals, BF diethylenetriamine-N,N,N’,N’’,N’’-pentaacetic acid (DTPA, Figure 4A) can rapidly radiolabel different radiometal ions, even at room temperature [3]. However, in vivo stability remains the major limitation, and radiometal DTPA complexes are generally less stable as the corresponding complexes with DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid, Figure 4C). BF-DTPA has been used successfully with the FDA-approved SPECT agent 111In-DTPA-octreotide (OctreoScanTM), a somatostatin receptor targeting peptide-conjugate, used for imaging neuroendocrine tumors [3,51]. Additionally, it has been successfully radiolabeled with different radiometals, such as 213Bi, 111In, 177Lu, 64Cu, and 86/90Y, but has been made superfluous by the introduction of more stable DTPA-derivatives such as 1B4M-DTPA and BF-CHX-A’’-DTPA (Figure 4B) [2,3]. BF-1B4M-DTPA (Figure 4A), a DTPA derivative that contains a single methyl group on one of its ethylene backbones, has been successfully used for the FDA-approved 90Y therapeutic immunoconjugate ibritumomab tiuxetan (BF-1B4M-DTPA, Zevalin®) [52]. The presence of the cyclohexyl moiety of CHX-A’’-DTPA provides the chelator with additional rigidity and imposes a significant degree of preorganization on the metal ion binding site, augmenting kinetic inertness, but impeding radiolabeling kinetics as compared with DTPA [3].
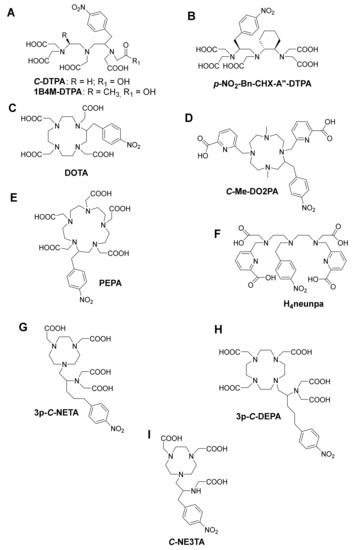
Figure 4.
Some structures of bifunctional chelators currently used for 213Bi (A) DTPA and 1B4M-DTPA (B) CHX-A’’-DTPA, (C) DOTA, (D) Me-DO2PA, (E) PEPA, (F) H4neunpa, (G) 3p-C-NETA (H) 3p-C-DEPA and (I) C-NE3TA.
While kidney uptake caused by in vivo dissociation of 213Bi from the chelator is problematic with 213Bi-DTPA constructs, [53] a slight improvement is recorded with 213Bi(1B4M-DTPA). Current 213Bi TAT research mostly uses CHX-A”-DTPA (logKML = 34.9–35.6) [53,54], as the Bi complex has shown significantly improved inertness compared with Bi-DTPA. A clear backbone rigidity effect on 206Bi release was observed after a study comparing renal uptake of B72.3-mAb radiolabeled with 206Bi using DTPA, CHX-A’’-DTPA, and 1B4M-DTPA as chelating ligands. Kidney uptake amounted to 27.2%ID/g for DTPA, 7.8%ID/g for CHX-A’’-DTPA, and 13.2%ID/g for BF-1B4M-DTPA, respectively [55]. CHX-A’’-DTPA displayed considerably superior stability over DTPA; however, its stability could still not match that of DOTA.
4.2. DOTA and DOTA-Derivatives
The current “gold standard” BF chelator for 213Bi is the amino carboxylate macrocycle DOTA, and 213Bi-DOTA bioconjugates have been reported to be stable in vitro and in vivo for at least two hours [56,57]. Bi3+ adopts a square antiprism geometry with DOTA in the [Bi-DOTA]-complex (see Figure 5) [58]. Despite the relatively high thermodynamic stability of the 213Bi-DOTA complex (logKML = 30.3), DOTA has several drawbacks as a 213Bi chelator. DOTA’s typical radiolabeling conditions require heating at high temperatures (e.g., 30–60 min at 95 °C) at pH 4–9, depending on the type of buffer used. Additionally, it has been demonstrated that a high concentration of DOTA (10 µM) is required to achieve quantitative yields for 213Bi-labeling. In contrast, for CHX-A’’-DTPA, a concentration of 1 µM is in general sufficient to achieve quantitative yields [59]. The relatively short half-life of 213Bi requires fast radiolabeling, which is not the case for DOTA and results in significant loss of radioactivity due to decay. Additionally, the high temperatures are unsuitable for heat-sensitive proteins of interest for TRNT, as this may cause the proteins to denature.
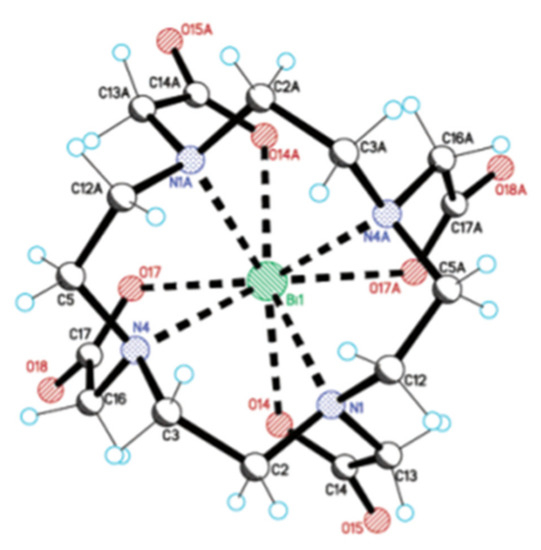
Figure 5.
Crystal structure of NaBiDOTA·H2O. Reprinted (adapted) from E. Brücher et al., 2003, with permission from [58]; American Chemical Society, 2003.
BF-PEPA (Figure 4E), a chelator that is an expanded version of DOTA, has also been studied with 205/206Bi. This chelator was developed to improve the association kinetics of Bi-DOTA radiocomplexes. Unfortunately, this chelator was discovered not to be ideal for Bi because a lower tumor uptake and increased kidney uptake of 205,206Bi-B3-PEPA was observed compared with the corresponding CHX-A’’-DTPA complex [50].
Me-DO2PA (Figure 4D), a [12] aneN4 bearing two picolinic acid arms and two methyl-capped amines, has also been reported to be stable in vivo when radiolabeled with 213Bi [60,61].
4.3. NETA and DEPA-Derivatives
NETA ({4-[2-(bis-carboxymethyl-amino)-ethyl]-7-carboxymethyl- [1,4,7] triazonan-1-yl}-acetic acid, Figure 4 G) and DEPA (7-[2-(bis-carboxymethyl-amino)-ethyl]-4,10-bis-carboxymethyl-1,4,7,10-tetraazacyclododec-1-yl-acetic acid, Figure 4H) are promising chelators for radionuclide therapy of α- and β–-emitters, including 90Y, 177Lu (T1/2 = 6.7 days), 206Bi, 207Bi, 213Bi, and 212Pb. NETA possesses both a parent macrocyclic NOTA (1,4,7-triazacyclononane-N,N’,N’’-triacetic acid) backbone and a flexible acyclic tridentate pendant arm (iminodiacetic acid). DEPA on the other hand is made up of a donor system incorporating both macrocyclic DOTA and an acyclic tridentate pendant arm (iminodiacetic acid). The idea of designing NETA/DEPA was to integrate the advantage of both the macrocyclic and acyclic frameworks, i.e., rapid complexation (favorable formation kinetics) at ambient temperatures with radionuclides and high thermodynamic stability [62,63].
A 205/206Bi-NETA analogue displayed a lower degree of degradation compared with 205/206Bi-CHX-A′′-DTPA after in vitro challenge experiments. Biodistribution experiments of unconjugated [205/206Bi(C-NETA)]- in non-tumor-bearing mice showed a high level of kidney retention (24.63 ± 2.79%ID/g) after one hour. However, a biodistribution study of the seven-coordinate analogue [205/206Bi(C-NE3TA)] (Figure 4I) showed reduced kidney retention (4.69 ± 0.55%ID/g), even though having a seemingly non-coordinatively saturated Bi3+ metal center [62,64]. A version of NETA with a prolonged linker between the coordinating functional groups and pendant isothiocyanate group (p-SCN-Bn) in the subsequent experiments stipulates that the instability could originate from coupling group interference. 3p-C-NETA-trastuzumab showed faster labeling of 205/206Bi compared with DOTA-trastuzumab, and an in vivo experiment on mice bearing subcutaneous tumors (LS-174T, which expresses HER-2, the target of trastuzumab) demonstrated significant tumor accumulation without increasing kidney uptake (after 24 h) [65]. This study showed that 3p-C-NETA could be radiolabeled at room temperature in quantitative yields and have high stability in vitro and in vivo, which was not the case with DOTA.
The 205/206Bi-DEPA complex has also been studied to show similarly promising results with DTPA as benchmark [66,67]. After 72 h p.i., 205/206Bi-C-DEPA-trastuzumab remained 100% intact in human serum. However, in the case of 205/206Bi-DTPA-trastuzumab, only 77% was intact after 72 h [66]. 205/206Bi-3p-C-DEPA-trastuzumab also showed significant tumor uptake in tumor-bearing (LS-174T) mice. As suggested by Price and Orvig, an in-depth assessment of NETA versus DEPA for Bi-based bioconjugates might be useful to identify the “gold standard” of Bi chelators [3]. Finally, a promising bifunctional picolinic acid-based scaffold chelator, H4neunpa (Figure 4F), has also been studied. The only concern is that the stability of the Bi3+ complex (log KML = 28.8) was lower than that of DOTA and DTPA; however, the pM value is the similar to that of DOTA (pM = 27), providing potential for in vivo use [68]. Complex geometry and thermodynamic parameters of 213Bi with different chelators are summarized in Table 2.
Table 2.
Overview of Bi3+ Chelators, Complex Geometry, and Thermodynamic Parameters. Table was adapted from Kostelnik et al., 2019, with permission from [2], American Chemical Society, 2019.
5. General Considerations for Designing a 213Bi-radiopharmaceutical
In order to realize the potential and favorable properties of 213Bi, specifically targeted carriers need to be developed [1,3,4]. Indeed, the vector molecule is an essential part of the therapeutic radiopharmaceutical because it is responsible for the selective interaction with the target, leading to a high concentration of the radionuclide in the target tissue. The target of interest should have sufficiently high differential expression in the target tissue versus the background tissue, combined with sufficiently high absolute expression (Bmax). Several targets are currently being investigated for TAT with 213Bi-labeled probes, such as human epidermal growth factor receptor 2 (HER2) [69], cluster of differentiation 20 (CD20) [70], CD33 [71,72], CD45 [73], substance P [74,75,76], somatostatin receptors [19], prostate-specific membrane antigen (PSMA) [77], and epidermal growth factor receptor (EGFR) [78].
Due to reversible interactions, such as an affinity-based receptor or a transporter binding, the retention in the target tissue can be governed by equilibrium association and dissociation. However, the tracer can be internalized and kept in the cell after binding to the target. This internalization can maximize selective irradiation of the tumor since it leads to pseudo-irreversible kinetics.
Radiopharmaceuticals are usually administered intravenously, distributed quickly over the body, and are concentrated in the target tissue or cells due to physical or chemical interactions. In the surrounding tissue, which lacks the interaction mechanism, the concentration of the radiopharmaceutical is in equilibrium with the plasma concentration that is reduced in function of time because of clearance from plasma by excretory organs such as kidneys and liver. Renal clearance and urinary excretion are preferred over hepatobiliary clearance for oncological applications for TAT with 213Bi since hepatobiliary clearance results in a slow transfer through the gastro-intestinal tract, resulting in undesired high levels of abdominal activity.
The vector molecule can consist of a small organic molecule, a peptide, or a protein including antibodies and antibody fragments (Figure 6) [4]. It is important that the vector molecule has a high affinity and selectivity for the target and maintains this affinity and selectivity after conjugation with the radionuclide. As 213Bi is a radiometal, derivatization of small organic molecules with a bulky chelator usually significantly alters its binding properties. Therefore, mostly peptides and antibody fragments are used as vector molecule in 213Bi-labeled radiopharmaceuticals. Their large size and the presence of non-binding pockets usually make them less sensitive to changes in target affinity when being derivatized with bulky bifunctional ligands.
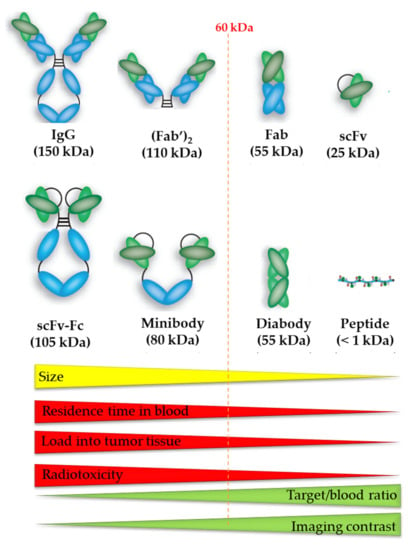
Figure 6.
The size of the vector molecule determines the residence time of blood, tumor accumulation, radiotoxicity, target-to-blood ratio, and imaging contrast of the radiopharmaceutical. Image was adapted from Hong H. et al., 2008 and modified with permission from [79], SAGE Journals, 2008.
An important factor when assessing the therapeutic potential of vectors is the kinetic profile of the carrier. In general, vector molecules with longer circulation time in blood have the highest tumor accumulation, which is beneficial for the efficacy of TAT treatment. On the other hand, longer residence time in blood also involves unavoidable higher radiation dose to healthy tissues. Therefore, the half-life of the radionuclide should be compatible with the plasma half-life of the vector to ensure a sufficient high tumor/background ratio. One should note that the selectivity for radiation damage to malignant tissue is potentially higher for longer-lived radionuclides, which is beneficial in a therapeutic setting. In this respect, the short physical half-life of 213Bi is a disadvantage when combined with a carrier molecule with a long plasma half-life. Accumulation of such a carrier in the tumor tissue is too slow in comparison with the half-life of 213Bi, resulting in less selective irradiation of the tumor tissue. In contrast, 213Bi is a much better match with vector molecules that have a short biological half-life, such as some small molecules, several peptides, and antibody fragments (including nanobodies). These molecules accumulate rapidly in the tumor tissue, and therefore allow 213Bi to deposit its dose to the tumor before it is fully decayed. The TAT studies with 213Bi-PSMA-617 in a patient with metastasized castration-resistant prostate cancer that was refractory to 177Lu-radiotherapy illustrates the potential of the combination of 213Bi with a vector molecule with short biological half-life [80]. Further, an in vitro and in vivo preclinical study with a 213Bi-labeled nanobody for TAT showed promising results (see Table 3) [5].
Table 3.
Overview of some preclinical studies with 213Bi.
Although carriers with a short plasma half-life are preferred, several 213Bi-labeled vectors with slow kinetics have been reported. One way to bypass the kinetic incompatibility is local administration. Locoregional delivery of 213Bi-radiopharmaceuticals compared to systematic administration has the potential to significantly increase efficacy, while minimizing systemic toxicity to non-targeted tissues. A disadvantage is that it is no longer a systemic treatment and that not all metastases will be treated, except if after locoregional injection there is substantial spill-over to the systemic circulation (e.g., injection in the hepatic artery). With locoregional delivery, the short-lived radionuclide 213Bi can be combined with vector molecules with a long biological half-life, such as mAbs, as only high binding affinity to the target is important and not the plasma pharmacokinetic properties of the radiopharmaceutical.
Pretargeted radiotherapy is another approach for combining a vector with long plasma half-life with a radionuclide with a short half-life, such as 213Bi [99]. First, a (slow) tumor-accumulating vector molecule carrying a tag is administered systemically. Once accumulated at the target sites and largely cleared from the blood, a radiolabeled agent that rapidly recognizes the tag of the tumor-bound vector in vivo and is cleared fast from plasma is injected. Upon encountering the targeting vector, fast and efficient bio-orthogonal ligation will take place between the two molecules, which leads to the in vivo formation of the final radioconjugate, resulting in specific irradiation of target tissue and low radiation burden of healthy tissue.
In the following two chapters non-clinical and clinical experience with 213Bi-labeled probes will be discussed, organized by the type of vector molecule.
6. Preclinical TAT Studies with 213Bi-labeled Probes
6.1. Antibodies
Monoclonal antibodies (mAbs), with their impeccable affinity for tumor antigens, have become powerful tools in the diagnosis and treatment of cancer, particularly when combined with therapeutic agents such as radionuclides and cytotoxic drugs, thanks to advances in hybridoma technology in the 1980s [87,100]. A mAb has the potential to bind antigen epitopes with high affinity, including tumor-related antigens, and are interesting vector molecules for TRNT because of high tumor accumulation (Figure 6) [4]. A variety of preclinical investigations have been conducted using mAbs labeled with 213Bi as radiometal (Table 3).
TRNT performed with 3.7 MBq of 213Bi-labeled 9E7.4 anti-CD138 mAb increased median survival to 80 days, compared with 37 days for the untreated control, and resulted in 45% cure in a multiple myeloma mice model. β–-TRNT performed with 18.5 MBq of 177Lu-labeled 9E7.4 mAb was well tolerated and increased mouse survival significantly (54 vs. 37 days in the control group); however, the authors reported that no mice were cured with this treatment [87].
Another study has shown that fractionated intravesical TRNT with 213Bi-anti-EGFR-mAb is a promising approach in advanced bladder carcinoma. Therapeutic efficacy was evaluated via overall survival and toxicity toward normal urothelium by histopathological analysis. Mice without treatment and those treated with the native anti-EGFR-mAb showed median survivals of 65.4 and 57.6 d, respectively. After fractionated treatment with 0.93 MBq, 213Bi-anti-EGFR-MAb animals lived for an average of 141.5 d, with 33% survival for at least 268 d. The animals survived for an average of 131.8 d after fractionated treatment with 0.46 MBq 213Bi-anti-EGFR-mAb, with 30% survival for more than 300 d. It was concluded that no toxic side effects on the normal urothelium were observed, even after treatment with 3.7 MBq of 213Bi-anti-EGFR-mAb [81].
6.2. Antibody Fragments
Recent advances in bioengineering have led to the development of antibody fragments—such as Fab (50 kDa), F(ab’)2 (110kDa), 25-kDa single-chain Fv (scFv), diabodies (55 kDa), nanobodies (15kDa), and minibodies (80 kDa)—without affecting their affinity and specificity [101]. Smaller mAb derivatives are transported more rapidly to the tumor site and can penetrate the tumor more effectively. Because of their smaller size and lack of an Fc region, they are swiftly cleared from circulation, resulting in high tumor-to-background ratios at early time-points (Figure 6). Successful conjugation of 213Bi to anti-HER2 C6.5 scFv and diabody molecules has been reported. However, there was no tumor-specific therapeutic effect, which was most likely due to the in vivo instability of the scFv and diabody molecules. The physical half-life of 213Bi, 45.6 min, was found to be too short for the systemically administered diabody to accumulate sufficiently in the solid tumor [102].
For the first time, a 213Bi-labeled HER2-sdAb nanobody was successfully produced and characterized in vitro and in vivo in a preclinical setting. 213Bi-DTPA-2Rs15d sdAb exhibited high in vivo stability and specific accumulation in target tissue after i.v. administration in mice. When administered in therapeutic doses, 213Bi-DTPA-2Rs15d sdAb increased the median survival of mice (80 d compared with 56 d in the control group), particularly when used in combination with trastuzumab (140 d). These findings suggest that 213Bi-DTPA-sdAb could be used as a new radioconjugate for TAT, both alone and in combination with trastuzumab, to treat HER2+ metastatic cancer. Authors indicate it might be interesting to evaluate 213Bi-DTPA-2Rs15d sdAb in a trastuzumab-resistant tumor model. To conclude, the rapid accumulation of 213Bi-labeled 2Rs15d sdAb in HER2-expressing tumors demonstrates that nanobodies are promising vector molecules in combination with the short-lived 213Bi [5].
6.3. Peptides
Peptides have been used extensively in nuclear medicine for peptide receptor radionuclide therapy (PRRT). In the randomized NETTER-1 clinical trial, treatment with 177Lu-DOTATATE resulted in markedly longer progression-free survival and a significantly higher response rate than high-dose octreotide LAR among patients with advanced midgut neuroendocrine tumors [103]. Peptides are designed by rational methods to bind with high specificity and selectivity to their target cells. Due to their ease of synthesis using chemical or molecular biological techniques, peptide sequences can also be easily modified [104]. Oncogenic protein sequences, structures, and pattern interactions are all readily accessible, allowing peptides to be engineered specifically for TAT. Peptides have several important advantages over proteins or antibodies as vector molecule for TRNT: they are small, mostly stable, easy to synthesize, they can be modified to further increase the metabolic stability, and it is straightforward to site-specifically attach a bifunctional chelator, allowing radiolabeling. Furthermore, they are typically less immunogenic than recombinant antibodies or proteins [105].
An in vitro comparison of 213Bi- and 177Lu-based PRRT has been performed. Absorbed doses up to 7 Gy were obtained by 5.2 MBq 213Bi-DOTATATE, and the majority of the dose was caused by α-particle radiation. The cell survival of BON or CA20948 cells after exposure to 213Bi-DTPA and 213Bi-DOTATATE showed a linear-exponential relationship with the absorbed dose, confirming the strong LET character of 213Bi. CA20948 demonstrated the standard curvature of the linear-quadratic model after exposure to 177Lu-DOTATATE and the reference 137Cs. With 213Bi-DOTATATE, 10% CA20948 cell survival was achieved at 3 Gy, which is six times less than the 18 Gy needed for 177Lu-DOTATATE and less than the 5 Gy required after 137Cs external exposure [56].
Important progress has been made in the production and application of 213Bi-optimized vehicles so far. Despite the promising preliminary results, there is still much room for improvement, especially in the development of new coupling chemistries and the elucidation and optimization of in vivo biodistribution.
7. Clinical TAT Studies with 213Bi-labeled Radiopharmaceuticals
It is suggested that TAT is ideally suited for hematologic malignancies due to the easy and fast accessibility of malignant cells in blood, bone marrow, lymph nodes, and spleen, as well as their typical high radiosensitivity [32,70]. The majority of clinical trials for hematologic malignancies using α-particle therapy have focused on acute myeloid leukemia (AML) [71,72]. Nonetheless, preclinical studies have shown activity against other cancer types, including non-Hodgkin’s lymphoma and multiple myeloma [92,106].
To date, 213Bi-labeled radiopharmaceuticals has been used to treat more than 200 patients for leukemia, lymphoma, melanoma, bladder cancer, glioma, and neuroendocrine tumors [7]. The two main approaches currently used for the administration of this radiopharmaceutical are locoregional and systemic administration.
7.1. Locoregional Administration
7.1.1. Intravesical TRNT
A locoregional pilot study to evaluate the feasibility, tolerability, and efficacy of 213Bi-anti-EGFR mAb treatment in patients with bacillus Calmette-Guerin (BCG) refractory carcinoma in situ (CIS) was performed [78]. A single intravesical instillation (or two instillations) of 366–821 MBq of 213Bi-anti-EGFR immunoconjugate (with molar activities in the range of 0.37–0.82 GBq/mg) was tolerated without any adverse effects. Four patients showed a complete response, i.e., no visible CIS, eight weeks after the first or second treatment. Three of the four patients were still tumor-free at the time of the publication, i.e., three months after the second treatment and 30 and 44 months after the first treatment. However, since this was a pilot study, the authors concluded that additional studies are required to confirm this therapeutic outcome [78].
7.1.2. Intracerebral Substance-P PRRT
Interest in PRRT has steadily grown because of the advantages of targeting cellular receptors in vivo with highly tumor-specific targeting and high tumoral activity concentration. Unparalleled attempts to develop radiolabeled receptor-binding somatostatin analogues for the treatment of neuroendocrine tumors have occurred in recent decades, and these efforts have facilitated the evolution of PRRT and paved the way for the development of other receptor-targeting peptides [103,107,108,109]. The highest number of patients treated with locoregional administration of 213Bi was gathered for the treatment of grade II to IV glioma with radiolabeled DOTA-Substance P analog (substance-P binds to GPCR neurokinin type 1 receptor). However, its short plasma half-life presented a challenge to researchers. In a clinical study, 213Bi-DOTA-Substance P was administered locoregionally into the brain tumor or tumor cavity via an implanted catheter. Patients were treated with up to 14.1 GBq 213Bi-DOTA-Substance P, administered in up to eight treatment cycles at two-month intervals. No severe side effects were recorded. The median survival time from the start of 213Bi-DOTA-Substance P was 7.5 months. Several patients showed complete remissions, and no recurrence was observed up to 23.8 months after the end of therapy. A subgroup analysis indicated prolonged survival times for grade IV patients in comparison with standard treatments [75]. These data should encourage future randomized controlled trials.
7.1.3. Intralesional Melanoma TRNT
Injection of a high concentration of a drug directly into skin lesions without significant systemic absorption is also becoming increasingly popular. The rationale for this technique is the establishment of a subepidermal depot that bypasses the superficial barrier zone.
Intralesional melanoma TRNT with 213Bi conjugated to the benign tumor targeting antibody vector 9.2.27 has been studied. Sixteen melanoma patients positive to the monoclonal antibody 9.2.27 were recruited. 213Bi-immunoconjugate activities from 5.5 to 50 MBq injected into lesions of different sizes resulted in massive cell death, as observed by the presence of tumor debris. The 213Bi-immunoconjugate was very efficient in providing a high radiation dose to the tumor lesions while sparing the surrounding tissues. Blood proteins and electrolytes did not display any major changes after intralesional injection [110].
7.2. Systemic Administration
7.2.1. Acute Myeloid Leukemia TRNT
mAb-TAT uses α-emitting radionuclide labeled antibodies directed against a tumor-associated antigen to deliver a lethal radiation dose selectively to tumor cells. Table 4 summarizes the current state of clinical investigation of 213Bi-mAb-TAT radiopharmaceuticals.
Table 4.
Overview of Clinical Studies with 213Bi-labeled Ligands.
An initial phase I study conducted in 18 patients with relapsed or refractory acute myeloid leukemia (AML) demonstrated the safety and antitumor effects of 213Bi-lintuzumab. Subsequently, 213Bi-lintuzumab produced remissions in AML patients after partial cytoreduction with cytarabine in a phase I/II trial [72]. The short half-life of 213Bi and the need for an onsite generator presented a major obstacle to the wide-spread use of mAb-TAT with 213Bi. Therefore, a second-generation construct was developed using 225Ac that was directly conjugated to the antibody. A phase I trial demonstrated that a single infusion of 225Ac-lintuzumab could be given safely at doses up to 111 kBq/kg, with anti-leukemic activity across all studied activity levels. In a second phase I study, 28% of older patients with untreated AML and unfit for intensive chemotherapy had objective responses after receiving fractionated 225Ac-lintuzumab and low-dose cytarabine [111,112].
7.2.2. SSTR PRRT
The only 213Bi PRRT study so far was performed by Kratochwil et al., in 2014. Eight patients with multi-resistant neuroendocrine tumors refractory to therapy with beta emitter labeled 90Y-/177Lu-DOTATOC were treated with 213Bi-DOTATOC. Seven of them underwent intra-arterial injection into the hepatic artery, which results in an enriched exposure of the liver metastases but also allows systemic targeting. Even though the patients presented were in an advanced disease setting and had developed resistance against therapy with beta emitting 90Y-/177Lu-DOTATOC, 213Bi-DOTATOC therapy resulted in a considerable number of long-lasting antitumor responses, including one complete remission [19].
7.2.3. 213Bi-PSMA
PSMA has received great attention in nuclear medicine in the last decade, as it is a promising molecular target for small molecules, antibodies, and antibody fragments [113]. A first-in-human treatment with 213Bi-PSMA-617 was performed in a patient with mCRPC (metastatic castration-resistant prostate cancer) that was progressive under conventional therapy. The patient was treated with two cycles of 213Bi-PSMA-617 with a cumulative activity of 592 MBq. Restaging with 68Ga-PSMA PET/CT after 11 months showed an outstanding molecular imaging response. This patient also exhibited a biochemical response (decrease in prostate-specific antigen levels from 237 μg/L to 43 μg/L) [80].
8. Future Perspectives of 213Bi-TAT
The recent data on the remarkable therapeutic efficacy of 213Bi-bioconjugates for cancer therapy have significantly ignited interest in the clinical application of TAT. The implementation of 213Bi-bioconjugates does not only offer an auspicious therapeutic option for cancer treatment but also strongly underlines an important potential of the concept of TAT. Data from clinical studies with 213Bi-DOTATOC and 213Bi-PSMA-617 suggest that patients do not develop resistance to therapy with α-emitters, whereas this is the case with conventional drugs and with β–-emitters therapy. TAT could provide a useful additional treatment option to patients who were progressive under conventional treatments. A major advantage of 213Bi-TAT compared with 225Ac-TAT is that there is no problem of uncontrolled redistribution of recoiled daughter nuclides, which might cause considerable toxic effects to heathy tissues [117]. To successfully translate 213Bi-bioconjugate probes from bench-to-bedside, one needs to consider the half-life of 213Bi (46 min), which should match the tumor and plasma kinetics of the vector molecule. Evidently, a therapeutic agent that dispels most of its energy before reaching its target will cause more harm than good. Low molecular weight peptide ligands and antibody fragments labeled with 213Bi are for this reason promising due to their favorable fast pharmacokinetics.
Additionally, treatment of cancer-associated fibroblasts (CAF), which are highly expressed in the stroma of most tumor entities, is an emerging tumor targeting area. CAFs, in contrast with normal fibroblasts, overexpress fibroblast activation protein α (FAP). Novel radioprobes, both for diagnostic and therapeutic applications, were designed and are based on FAP inhibitors (FAPI) as vector molecule (e.g., 68Ga-DOTA-FAPI-46). FAPI PET images are characterized by rapid kinetics, very low background activity (no uptake in brain, muscle, brown fat, bowel, etc.) and high tumor-to-background contrast. The fast pharmacokinetics of FAPI vector molecules might be an ideal match with the short physical half-life of 213Bi [118,119,120,121]. Hence, this will be an interesting area to explore for future 213Bi radiopharmaceutical development.
For 213Bi chelation chemistry, closer inspection reveals that “gold standard” ligands such as DOTA might not be ideal for clinical application. The chelator should clearly match with the chemical properties of 213Bi, and the thermodynamic stability should be balanced with the formation kinetics of the Bi-complexes. This is particularly important for the practical preparation of 213Bi-radiopharmaceuticals. NETA and DEPA derivatives have so far demonstrated to be an ideal match for 213Bi. However, the potential of NETA and DEPA for use in 213Bi radiopharmaceuticals requires further investigation before it can be translated to a clinical setting.
Another area to consider is in vivo kidney radiotoxicity, due to kidney uptake and retention of 213Bi-radioprobes. The mechanisms responsible for unwanted kidney uptake and retention are well explored. Radioconjugates with a molecular weight of less than 60 kDa (Figure 6) are filtered through the glomerulus and are often reabsorbed and transported to the lysosomal compartment. The long residence time of radionuclides in the kidney causes a high radiation burden for the kidneys. This can lead to tubular necrosis and limits the total therapeutic dose, especially when using the short-lived radionuclide 213Bi. Several strategies have been explored to mitigate the nephrotoxic aspect of 213Bi-labeled radioprobes during cancer treatment [5,57,122,123], and some of these approaches have been approved for clinical applications. The co-infusion of lysine or the plasma expander Gelofusine® with 213Bi-bioconjugates during TAT has been reported to have a 3-fold reduction in kidney uptake and retention [57,124]. Another promising approach could be that 213Bi-radioprobes will be developed with a cleavable linker—positioned between the Bi-chelator complex and vector molecule—that is recognized specifically by renal brush border membrane enzymes, so that the radionuclides are excreted more efficiently toward the bladder instead of being retained in the kidneys.
A study has demonstrated that derivatives of [68Ga]Ga-DOTA-AmBz-MVK-OH could be cleaved specifically by neutral endopeptidase (NEP), and therefore the amino acids linker MVK could reduce kidney uptake of radiolabeled DOTA-conjugated peptides and peptidomimetics [125]. These findings would allow development of radiometal-based radiopharmaceuticals with clinically relevant lower renal radioactivity levels [126].
To conclude, TAT using 213Bi has demonstrated interesting results preclinically and clinically. The limitation is that 213Bi radionuclide has a relatively short half-life (46 min). However, it can be made available to hospitals from a 225Ac/213Bi generator that can be used over a more than 10-day period. 213Bi is an ideal match for vector molecules with fast plasma clearance, fast tumor targeting, but limited tumoral retention.
Author Contributions
Writing—original draft preparation, S.A. and F.C.; writing—review and editing, S.A., I.C., C.M.D., T.C., A.R.B., G.B., M.O., and F.C. All authors have read and agreed to the published version of the manuscript.
Funding
SCK CEN Academy support is gratefully acknowledged. Frederik Cleeren is a Postdoctoral Fellow of FWO (12R3119N). Christophe M. Deroose is a Senior Clinical Investigator at the FWO.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable: No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vermeulen, K.; Vandamme, M.; Bormans, G.; Cleeren, F. Design and Challenges of Radiopharmaceuticals. Semin. Nucl. Med. 2019, 49, 339–356. [Google Scholar] [CrossRef] [PubMed]
- Kostelnik, T.I.; Orvig, C. Radioactive Main Group and Rare Earth Metals for Imaging and Therapy. Chem. Rev. 2019, 2, 902–956. [Google Scholar] [CrossRef] [PubMed]
- Price, E.W.; Orvig, C. Matching chelators to radiometals for radiopharmaceuticals. Chem. Soc. Rev. 2014, 43, 260–290. [Google Scholar] [CrossRef]
- Dekempeneer, Y.; Keyaerts, M.; Krasniqi, A.; Puttemans, J.; Muyldermans, S.; Lahoutte, T.; D’huyvetter, M.; Devoogdt, N. Targeted alpha therapy using short-lived alpha-particles and the promise of nanobodies as targeting vehicle. Expert Opin. Biol. Ther. 2016, 16, 1035–1047. [Google Scholar] [CrossRef]
- Dekempeneer, Y.; Caveliers, V.; Ooms, M.; Maertens, D.; Gysemans, M.; Lahoutte, T.; Xavier, C.; Lecocq, Q.; Maes, K.; Covens, P.; et al. The therapeutic efficacy of 213Bi-labeled sdAbs in a preclinical model of ovarian cancer. Mol. Pharm. 2020, 17, 3553–3566. [Google Scholar] [CrossRef]
- Heskamp, S.; Hernandez, R.; Molkenboer-Kuenen, J.D.M.; Essler, M.; Bruchertseifer, F.; Morgenstern, A.; Steenbergen, E.J.; Cai, W.; Seidl, C.; McBride, W.J.; et al. α-Versus β-Emitting radionuclides for pretargeted radioimmunotherapy of carcinoembryonic antigen-expressing human colon cancer xenografts. J. Nucl. Med. 2017, 58, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Bruchertseifer, F.; Kellerbauer, A.; Malmbeck, R.; Morgenstern, A. Targeted alpha therapy with bismuth-213 and actinium-225: Meeting future demand. J. Label. Compd. Rad. 2019, 62, 794–802. [Google Scholar] [CrossRef]
- Beyls, C.; Haustermans, K.; Deroose, C.M.; Pans, S.; Vanbeckevoort, D.; Verslype, C.; Dekervel, J. Could Autoimmune Disease Contribute to the Abscopal Effect in Metastatic Hepatocellular Carcinoma? Hepatology 2020, 72, 1152–1154. [Google Scholar] [CrossRef] [PubMed]
- Gill, M.R.; Vallis, K.A. Transition metal compounds as cancer radiosensitizers. Chem. Soc. Rev. 2019, 48, 540–557. [Google Scholar] [CrossRef]
- Lacoeuille, F.; Arlicot, N.; Faivre-Chauvet, A. Targeted alpha and beta radiotherapy: An overview of radiopharmaceutical and clinical aspects. Med. Nucl. 2018, 42, 32–44. [Google Scholar] [CrossRef]
- Turner, J.H. Recent advances in theranostics and challenges for the future. Br. J. Radiol. 2018, 91, 20170969. [Google Scholar] [CrossRef]
- Qaim, S.M. Theranostic radionuclides: Recent advances in production methodologies. J. Radioanal. Nucl. Chem. 2019, 322, 1257–1266. [Google Scholar] [CrossRef]
- Morgenstern, A.; Apostolidis, C.; Kratochwil, C.; Sathekge, M.; Krolicki, L.; Bruchertseifer, F. An Overview of Targeted Alpha Therapy with 225Actinium and 213Bismuth. Curr. Radiopharm. 2018, 11, 200–208. [Google Scholar] [CrossRef]
- Sgouros, G.; Roeske, J.C.; McDevitt, M.R.; Palm, S.; Allen, B.J.; Fisher, D.R.; Brill, A.B.; Song, H.; Howell, R.W.; Akabani, G.; et al. MIRD pamphlet No. 22 (Abridged): Radiobiology and dosimetry of α-particle emitters for targeted radionuclide therapy. J. Nucl. Med. 2010, 51, 311–328. [Google Scholar] [CrossRef]
- Jiao, R.; Allen, K.J.H.; Malo, M.E.; Helal, M.; Jiang, Z.; Smart, K.; Buhl, S.V.; Rickles, D.; Bryan, R.A.; Dadachova, E. Evaluation of novel highly specific antibodies to cancer testis antigen Centrin-1 for radioimmunoimaging and radioimmunotherapy of pancreatic cancer. Cancer Med. 2019, 8, 5289–5300. [Google Scholar] [CrossRef]
- Feuerecker, B.; Tauber, R.; Knorr, K.; Heck, M.; Beheshti, A.; Seidl, C.; Bruchertseifer, F.; Pickhard, A.; Gafita, A.; Kratochwil, C.; et al. Activity and Adverse Events of Actinium-225-PSMA-617 in Advanced Metastatic Castration-resistant Prostate Cancer After Failure of Lutetium-177-PSMA. Eur. Urol. Suppl. 2020, 79, 343–350. [Google Scholar] [CrossRef]
- Zacherl, M.J.; Gildehaus, F.J.; Mittlmeier, L.; Boening, G.; Gosewisch, A.; Wenter, V.; Schmidt-Hegemann, N.-S.; Belka, C.; Kretschmer, A.; Casuscelli, J.; et al. First clinical results for PSMA targeted alpha therapy using 225Ac-PSMA-I&T in advanced mCRPC patients. J. Nucl. Med. 2020. [Google Scholar] [CrossRef]
- Kratochwil, C.; Bruchertseifer, F.; Rathke, H.; Bronzel, M.; Apostolidis, C.; Weichert, W.; Haberkorn, U.; Giesel, F.L.; Morgenstern, A. Targeted a-therapy of metastatic castration-resistant prostate cancer with 225Ac-PSMA-617: Dosimetry estimate and empiric dose finding. J. Nucl. Med. 2017, 58, 1624–1631. [Google Scholar] [CrossRef]
- Kratochwil, C.; Giesel, F.L.; Bruchertseifer, F.; Mier, W.; Apostolidis, C.; Boll, R.; Murphy, K.; Haberkorn, U.; Morgenstern, A. 213Bi-DOTATOC receptor-targeted alpha-radionuclide therapy induces remission in neuroendocrine tumours refractory to beta radiation: A first-in-human experience. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 2106–2119. [Google Scholar] [CrossRef]
- Boden, S.; Vints, K.; Cagno, S.; Maertens, D.; Van Hecke, K.; Cardinaels, T. Thorium-229 quantified in historical Thorium-228 capsules. Appl. Radiat. Isot. 2017, 120, 40–44. [Google Scholar] [CrossRef]
- Apostolidis, C.; Molinet, R.; Rasmussen, G.; Morgenstern, A. Production of Ac-225 from Th-229 for targeted α therapy. Anal. Chem. 2005, 77, 6288–6291. [Google Scholar] [CrossRef]
- Apostolidis, C.; Molinet, R.; McGinley, J.; Abbas, K.; Möllenbeck, J.; Morgenstern, A. Cyclotron production of Ac-225 for targeted alpha therapy. Appl. Radiat. Isot. 2005, 62, 383–387. [Google Scholar] [CrossRef]
- Morgenstern, A.; Abbas, K.; Bruchertseifer, F.; Apostolidis, C. Production of Alpha Emitters for Targeted Alpha Therapy. Curr. Radiopharm. 2008, 1, 135–143. [Google Scholar] [CrossRef]
- Nesteruk, K.P.; Ramseyer, L.; Carzaniga, T.S.; Braccini, S. Measurement of the Beam Energy Distribution of a Medical Cyclotron with a Multi-Leaf Faraday Cup. Instruments 2019, 3, 4. [Google Scholar] [CrossRef]
- Engle, J.W.; Mashnik, S.G.; Weidner, J.W.; Wolfsberg, L.E.; Fassbender, M.E.; Jackman, K.; Couture, A.; Bitteker, L.J.; Ullmann, J.L.; Gulley, M.S.; et al. Cross sections from proton irradiation of thorium at 800 MeV. Phys. Rev. C 2013, 88, 014604. [Google Scholar] [CrossRef]
- Weidner, J.W.; Mashnik, S.G.; John, K.D.; Hemez, F.; Ballard, B.; Bach, H.; Birnbaum, E.R.; Bitteker, L.J.; Couture, A.; Dry, D.; et al. Proton-induced cross sections relevant to production of 225Ac and 223Ra in natural thorium targets below 200MeV. Appl. Radiat. Isot. 2012, 70, 2602–2607. [Google Scholar] [CrossRef]
- Melville, G.; Meriarty, H.; Metcalfe, P.; Knittel, T.; Allen, B.J. Production of Ac-225 for cancer therapy by photon-induced transmutation of Ra-226. Appl. Radiat. Isot. 2007, 65, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.K.H.; Ramogida, C.F.; Schaffer, P.; Radchenko, V. Development of 225Ac Radiopharmaceuticals: TRIUMF Perspectives and Experiences. Curr. Radiopharm. 2018, 11, 156–172. [Google Scholar] [CrossRef]
- Morgenstern, A.; Bruchertseifer, F.; Apostolidis, C. Bismuth-213 and Actinium-225—Generator Performance and Evolving Therapeutic Applications of Two Generator-Derived Alpha-Emitting Radioisotopes. Curr. Radiopharm. 2012, 5, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; McDevitt, M.R.; Finn, R.D.; Scheinberg, D.A. Breakthrough of 225Ac and its radionuclide daughters from an 225Ac/213Bi generator: Development of new methods, quantitative characterization, and implications for clinical use. Appl. Radiat. Isot. 2001, 55, 667–678. [Google Scholar] [CrossRef]
- Sinenko, I.L.; Kalmykova, T.P.; Likhosherstova, D.V.; Egorova, B.V.; Zubenko, A.D.; Vasiliev, A.N.; Vasiliev, A.N.; Ermolaev, S.V.; Lapshina, E.V.; Ostapenko, V.S.; et al. 213Bi production and complexation with new picolinate containing ligands. J. Radioanal. Nucl. Chem. 2019, 321, 531–540. [Google Scholar] [CrossRef]
- Morgenstern, A.; Apostolidis, C.; Bruchertseifer, F. Supply and Clinical Application of Actinium-. Semin. Nucl. Med. 2020, 50, 119–123. [Google Scholar] [CrossRef] [PubMed]
- McDevitt, M.R.; Finn, R.D.; Sgouros, G.; Ma, D.; Scheinberg, D.A. An 225Ac/213Bi generator system for therapeutic clinical applications: Construction and operation. Appl. Radiat. Isot. 1999, 50, 895–904. [Google Scholar] [CrossRef]
- Mehring, M. From molecules to bismuth oxide-based materials: Potential homo-and heterometallic precursors and model compounds. Coord. Chem. Rev. 2007, 251, 974–1006. [Google Scholar] [CrossRef]
- Briand, G.G.; Burford, N. Bismuth Compounds and Preparations with Biological or Medicinal Relevance. Chem. Rev. 1999, 99, 2601–2658. [Google Scholar] [PubMed]
- Ananthakrishnan, S.V. The electronic theory of valency. Curr. Sci. 1946, 15, 33–35. [Google Scholar]
- Pyykkö, P. Relativistic Effects in Structural Chemistry. Chem. Rev. 1988, 88, 563–594. [Google Scholar] [CrossRef]
- Ershov, B.G.; Akinshin, M.A.; Gordeev, A.V.; Sukhov, N.L. A pulse radiolysis study of the chloride complexes of Bi(II) and Bi(IV). Int. J. Radiat. Appl. Instrum. Part C Radiat. Phys. Chem. 1986, 27, 91–92. [Google Scholar] [CrossRef]
- Tooth, B.; Etschmann, B.; Pokrovski, G.S.; Testemale, D.; Hazemann, J.L.; Grundler, P.V.; Brugger, J. Bismuth speciation in hydrothermal fluids: An X-ray absorption spectroscopy and solubility study. Geochim. Cosmochim. Acta 2013, 101, 156–172. [Google Scholar] [CrossRef]
- Näslund, J.; Persson, I.; Sandström, M. Solvation of the bismuth(III) ion by water, dimethyl sulfoxide, N,N’-dimethylpropyleneurea, and N,N-dimethylthioformamide. An EXAFS, large-angle X-ray scattering, and crystallographic structural study. Inorg. Chem. 2000, 39, 4012–4021. [Google Scholar] [CrossRef]
- Sun, H.; Li, H.; Sadler, P.J. The biological and medicinal chemistry of bismuth. Chem. Ber. 1997, 130, 669–681. [Google Scholar] [CrossRef]
- Tooth, B. The Hydrothermal Chemistry of Bismuth and the Liquid Bismuth Collector Model. Ph.D. Thesis, University of Adelaide, Adelaide, Australia, 2013. [Google Scholar]
- Pearson, R.G. Hard and soft acids and bases, HSAB, part I: Fundamental principles. J. Chem. Ed. 1968, 45, 581–587. [Google Scholar] [CrossRef]
- Li, M.X.; Zhang, L.Z.; Yang, M.; Niu, J.Y.; Zhou, J. Synthesis, crystal structures, in vitro biological evaluation of zinc(II) and bismuth(III) complexes of 2-acetylpyrazine N(4)-phenylthiosemicarbazone. Bioorganic Med. Chem. Lett. 2012, 22, 2418–2423. [Google Scholar] [CrossRef]
- Ferraz, K.S.O.; Silva, N.F.; Da Silva, J.G.; De Miranda, L.F.; Romeiro, C.F.D.; Souza-Fagundes, E.M.; Mendes, I.C.; Beraldo, H. Investigation on the pharmacological profile of 2,6-diacetylpyridine bis(benzoylhydrazone) derivatives and their antimony(III) and bismuth(III) complexes. Eur. J. Med. Chem. 2012, 53, 98–106. [Google Scholar] [CrossRef]
- Sadler, P.J.; Li, H.; Sun, H. Coordination chemistry of metals in medicine: Target sites for bismuth. Coord. Chem. Rev. 1999, 185–186, 689–709. [Google Scholar] [CrossRef]
- Hancock, R.D.; Baloyi, J.; Mashishi, J. The Affinity of Bismuth (rii) for Nitrogen-donor Ligands. J. Chem. Soc. Dalt. Trans. 1993, 5, 2895–2899. [Google Scholar] [CrossRef]
- Guo, Z.; Sadler, P.J. Metals in Medicine: Metal-based drugs. Angew. Chem. Int. Ed. 1999, 38, 1512–1531. [Google Scholar] [CrossRef]
- Dorso, L.; Bigot-Corbel, E.; Abadie, J.; Diab, M.; Gouard, S.; Bruchertseifer, F.; Morgenstern, A.; Maurel, C.; Chérel, M.; Davodeau, F. Long-term toxicity of 213Bi-labelled BSA in mice. PLoS ONE 2016, 11, e0151330. [Google Scholar] [CrossRef]
- Garmestani, K.; Yao, Z.; Zhang, M.; Wong, K.; Park, C.W.; Pastan, I.; Carrasquillo, J.A.; Brechbiel, M.W. Synthesis and evaluation of a macrocyclic bifunctional chelating agent for use with bismuth radionuclides. Nucl. Med. Biol. 2001, 28, 409–418. [Google Scholar] [CrossRef]
- Bomanji, J.B.; Papathanasiou, N.D. 111In-DTPA0-octreotide (Octreoscan), 131I-MIBG and other agents for radionuclide therapy of NETs. Eur. J. Nucl. Med. Mol. Imaging 2012, 39 (Suppl. S1), S112–S125. [Google Scholar] [CrossRef] [PubMed]
- Rizzieri, D. Zevalin® (ibritumomab tiuxetan): After more than a decade of treatment experience, what have we learned? Crit. Rev. Oncol. Hematol. 2016, 105, 5–17. [Google Scholar] [CrossRef]
- Montavon, G.; Le Du, A.; Champion, J.; Rabung, T.; Morgenstern, A. DTPA complexation of bismuth in human blood serum. Dalt. Trans. 2012, 41, 8615–8623. [Google Scholar] [CrossRef]
- Brechbiel, M.W.; Gansow, O.A. Synthesis of C-functionalized trans-cyclohexyldiethylenetriaminepenta-acetic acids for labelling of monoclonal antibodies with the bismuth-212 α-particle emitter. J. Chem. Soc. Perkin Trans. 1 1992, 1173–1178. [Google Scholar] [CrossRef]
- Milenic, D.E.; Roselli, M.; Mirzadeh, S.; Pippin, C.G.; Gansow, O.A.; Colcher, D.; Brechbiel, M.W.; Schlom, J. In Vivo evaluation of bismuth-labeled monoclonal antibody comparing DTPA-derived bifunctional chelates. Cancer Biother. Radio. 2001, 16, 133–146. [Google Scholar] [CrossRef]
- Chan, H.S.; de Blois, E.; Morgenstern, A.; Bruchertseifer, F.; de Jong, M.; Breeman, W.; Konijnenberg, M. In Vitro comparison of 213Bi- and 177Lu-radiation for peptide receptor radionuclide therapy. PLoS ONE 2017, 12, e0181473. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.S.; Konijnenberg, M.W.; Daniels, T.; Nysus, M.; Makvandi, M.; de Blois, E.; Breeman, W.A.; Atcher, R.W.; de Jong, M.; Norenberg, J.P. Improved safety and efficacy of 213Bi-DOTATATE-targeted alpha therapy of somatostatin receptor-expressing neuroendocrine tumors in mice pre-treated with l-lysine. EJNMMI Res. 2016, 6, 83. [Google Scholar] [CrossRef] [PubMed]
- Csajbók, É.; Baranyai, Z.; Bányai, I.; Brücher, E.; Király, R.; Müller-Fahrnow, A.; Platzek, J.; Radüchel, B.; Schäfer, M. Equilibrium, 1 H and 13 C NMR Spectroscopy, and X-ray Diffraction Studies on the Complexes Bi(DOTA)-and Bi(DO3A-Bu). Inorg. Chem. 2003, 7, 2342–2349. [Google Scholar] [CrossRef] [PubMed]
- Šimeček, J.; Hermann, P.; Seidl, C.; Bruchertseifer, F.; Morgenstern, A.; Wester, H.J.; Notni, J. Efficient formation of inert Bi-213 chelates by tetraphosphorus acid analogues of DOTA: Towards improved alpha-therapeutics. EJNMMI Res. 2018, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.M.P.; Beyler, M.; Delgado, R.; Platas-Iglesias, C.; Tripier, R. Investigating the Complexation of the Pb2+/Bi3+ Pair with Dipicolinate Cyclen Ligands. Inorg. Chem. 2015, 54, 7045–7057. [Google Scholar] [CrossRef]
- Lima, L.M.P.; Beyler, M.; Oukhatar, F.; Le Saec, P.; Faivre-Chauvet, A.; Platas-Iglesias, C.; Delgado, R.; Tripier, R. H2Me-do2pa: An attractive chelator with fast, stable and inert natBi3+ and 213Bi3+ complexation for potential α-radioimmunotherapy applications. Chem. Comm. 2014, 50, 12371–12374. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.S.; Song, H.A.; Birch, N.; Le, T.; Lim, S.; Ma, X. Efficient synthesis and evaluation of bimodal ligand NETA. Bioorg. Med. Chem. Lett. 2008, 18, 3436–3439. [Google Scholar] [CrossRef]
- Song, H.A.; Kang, C.S.; Baidoo, K.E.; Milenic, D.E.; Chen, Y.; Dai, A.; Brechbiel, M.W.; Chong, H.-S. Efficient Bifunctional Decadentate Ligand 3p-C-DEPA for Targeted α-Radioimmunotherapy Applications. Bioconjug. Chem. 2011, 22, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.S.; Song, H.A.; Ma, X.; Milenic, D.E.; Brady, E.D.; Lim, S.; Lee, H.; Baidoo, K.; Cheng, D.; Brechbiel, M.W. Novel bimodal bifunctional ligands for radioimmunotherapy and targeted MRI. Bioconjug. Chem. 2008, 19, 1439–1447. [Google Scholar] [CrossRef][Green Version]
- Kang, C.S.; Song, H.A.; Milenic, D.E.; Baidoo, K.E.; Brechbiel, M.W.; Chong, H.S. Preclinical evaluation of NETA-based bifunctional ligand for radioimmunotherapy applications using 212Bi and 213Bi: Radiolabeling, serum stability, and biodistribution and tumor uptake studies. Nucl. Med. Biol. 2013, 40, 600–605. [Google Scholar] [CrossRef]
- Dadwal, M.; Kang, C.S.; Song, H.A.; Sun, X.; Dai, A.; Baidoo, K.E.; Brechbiel, M.W.; Chong, H.-S. Synthesis and evaluation of a bifunctional chelate for development of Bi(III)-labeled radioimmunoconjugates. Bioorg. Med. Chem. Lett. 2011, 21, 7513–7515. [Google Scholar] [CrossRef]
- Chong, H.S.; Lim, S.; Baidoo, K.E.; Milenic, D.E.; Ma, X.; Jia, F.; Song, H.A.; Brechbiel, M.W.; Lewis, M.R. Synthesis and biological evaluation of a novel decadentate ligand DEPA. Bioorg. Med. Chem. Lett. 2008, 18, 5792–5795. [Google Scholar] [CrossRef] [PubMed]
- Spreckelmeyer, S.; Ramogida, C.F.; Rousseau, J.; Arane, K.; Bratanovic, I.; Colpo, N.; Jermilova, U.; Dias, G.M.; Dude, I.; Jaraquemada-Peláez, M.G.; et al. p-NO2-Bn-H4neunpa and H4neunpa-Trastuzumab: Bifunctional Chelator for Radiometalpharmaceuticals and 111In Immuno-Single Photon Emission Computed Tomography Imaging. Bioconjug. Chem. 2017, 28, 2145–2159. [Google Scholar] [CrossRef]
- Milenic, D.E.; Brady, E.D.; Garmestani, K.; Albert, P.S.; Abdulla, A.; Brechbiel, M.W. Improved efficacy of α-particle-targeted radiation therapy: Dual targeting of human epidermal growth factor receptor-2 and tumor-associated glycoprotein 72. Cancer 2010, 116 (Suppl. S4), 1059–1066. [Google Scholar] [CrossRef]
- Jurcic, J. Alpha-Particle Therapy for Acute Myeloid Leukemia. J. Med Imaging Radiat. Sci. 2019, 50, S86–S87. [Google Scholar] [CrossRef]
- Rosenblat, T.L.; McDevitt, M.R.; Mulford, D.A.; Pandit-Taskar, N.; Divgi, C.R.; Panageas, K.S.; Heaney, M.L.; Chanel, S.; Morgenstern, A.; Sgouros, G.; et al. Sequential cytarabine and α-particle immunotherapy with bismuth-213-lintuzumab (HuM195) for acute myeloid leukemia. Clin. Cancer Res. 2010, 16, 5303–5311. [Google Scholar] [CrossRef]
- Jurcic, J.G.; Larson, S.M.; Sgouros, G.; McDevitt, M.R.; Finn, R.D.; Divgi, C.R.; Ballangrud, Å.M.; Hamacher, K.A.; Ma, D.; Humm, J.L.; et al. Targeted α particle immunotherapy for myeloid leukemia. Blood 2002, 100, 1233–1239. [Google Scholar] [CrossRef]
- Bethge, W.A.; Wilbur, D.S.; Storb, R.; Hamlin, D.K.; Santos, E.B.; Brechbiel, M.W.; Sandmaier, B.M. Radioimmunotherapy with bismuth-213 as conditioning for nonmyeloablative allogeneic hematopoietic cell transplantation in dogs: A dose deescalation study. Transplantation 2004, 78, 352–359. [Google Scholar] [CrossRef]
- Kneifel, S.; Cordier, D.; Good, S.; Ionescu, M.C.S.; Ghaffari, A.; Hofer, S.; Kretzschmar, M.; Tolnay, M.; Apostolidis, C.; Waser, B.; et al. Local targeting of malignant gliomas by the diffusible peptidic vector 1.,4,7,10-tetraazacyclododecane-1-glutaric acid-4,7,10-triacetic acid-substance P. Clin. Cancer Res. 2006, 12, 3843–3850. [Google Scholar] [CrossRef]
- Królicki, L.; Bruchertseifer, F.; Kunikowska, J.; Koziara, H.; Królicki, B.; Jakuciński, M.; Pawlak, D.; Apostolidis, C.; Mirzadeh, S.; Rola, R.; et al. Safety and efficacy of targeted alpha therapy with 213 Bi-DOTA-substance P in recurrent glioblastoma. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Cordier, D.; Forrer, F.; Bruchertseifer, F.; Morgenstern, A.; Apostolidis, C.; Good, S.; Müller-Brand, J.; Mäcke, H.; Reubi, J.C.; Merlo, A. Targeted alpha-radionuclide therapy of functionally critically located gliomas with 213Bi-DOTA-[Thi8,Met(O2)11]- substance P: A pilot trial. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, C.; Schmidt, K.; Afshar-Oromieh, A.; Bruchertseifer, F.; Rathke, H.; Morgenstern, A.; Haberkorn, U.; Giesel, F. Targeted alpha therapy of mCRPC: Dosimetry estimate of 213Bismuth-PSMA-617. Eur J. Nucl. Med. Mol. Imaging. 2018, 45, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Autenrieth, M.E.; Seidl, C.; Bruchertseifer, F.; Horn, T.; Kurtz, F.; Feuerecker, B.; D’Alessandria, C.; Pfob, C.; Nekolla, S.; Apostolidis, C.; et al. Treatment of carcinoma in situ of the urinary bladder with an alpha-emitter immunoconjugate targeting the epidermal growth factor receptor: A pilot study. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1364–1371. [Google Scholar] [CrossRef]
- Hong, H.; Sun, J.; Cai, W. Radionuclide-Based Cancer Imaging Targeting the Carcinoembryonic Antigen. Biomark. Insights 2008, 3, 435–451. [Google Scholar] [CrossRef]
- Sathekge, M.; Knoesen, O.; Meckel, M.; Modiselle, M.; Vorster, M.; Marx, S. 213Bi-PSMA-617 targeted alpha-radionuclide therapy in metastatic castration-resistant prostate cancer. Eur. J. Nucl. Med. 2017, 44, 1099–1100. [Google Scholar] [CrossRef]
- Fazel, J.; Rötzer, S.; Seidl, C.; Feuerecker, B.; Autenrieth, M.; Weirich, G.; Bruchertseifer, F.; Morgenstern, A.; Senekowitsch-Schmidtke, R. Fractionated intravesical radioimmunotherapy with 213Bi-anti-EGFR-MAb is effective without toxic side-effects in a nude mouse model of advanced human bladder carcinoma. Cancer Biol. Ther. 2015, 16, 1526–1534. [Google Scholar] [CrossRef]
- Jiao, R.; Allen, K.J.H.; Malo, M.E.; Rickles, D.; Dadachova, E. Evaluating the combination of radioimmunotherapy and immunotherapy in a melanoma mouse model. Int. J. Mol. Sci. 2020, 21, 773. [Google Scholar] [CrossRef] [PubMed]
- Nosanchuk, J.D.; Jeyakumar, A.; Ray, A.; Revskaya, E.; Jiang, Z.; Bryan, R.A.; Allen, K.J.H.; Jiao, R.; Malo, M.E.; Gómez, B.L.; et al. Structure-function analysis and therapeutic efficacy of antibodies to fungal melanin for melanoma radioimmunotherapy. Nat. Sci. Rep. 2018, 8, 5466. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson-Lutz, A.; Bäck, T.; Aneheim, E.; Hultborn, R.; Palm, S.; Jacobsson, L.; Morgenstern, A.; Bruchertseifer, F.; Albertsson, P.; Lindegren, S. Therapeutic efficacy of α-radioimmunotherapy with different activity levels of the 213Bi-labeled monoclonal antibody MX35 in an ovarian cancer model. EJNMMI Res. 2017, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Revskaya, E.; Jiang, Z.; Morgenstern, A.; Bruchertseifer, F.; Sesay, M.; Walker, S.; Fuller, S.; Lebowitz, M.S.; Gravekamp, C.; Ghanbari, H.A.; et al. A Radiolabeled Fully Human Antibody to Human Aspartyl (Asparaginyl) β-Hydroxylase Is a Promising Agent for Imaging and Therapy of Metastatic Breast Cancer. Cancer Biother. Radiopharm. 2017, 32, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Derrien, A.; Gouard, S.; Maurel, C.; Gaugler, M.H.; Bruchertseifer, F.; Morgenstern, A.; Faivre-Chauvet, A.; Classe, J.M.; Chérel, M. Therapeutic efficacy of alpha-RIT using a 213Bi-anti-hCD138 antibody in a mouse model of ovarian peritoneal carcinomatosis. Front. Med. 2015, 2, 88. [Google Scholar] [CrossRef] [PubMed]
- Fichou, N.; Gouard, S.; Maurel, C.; Barbet, J.; Ferrer, L.; Morgenstern, A.; Bruchertseifer, F.; Faivre-Chauvet, A.; Bigot-Corbel, E.; Davodeau, F.; et al. Single-dose anti-CD138 radioimmunotherapy: Bismuth-213 is more efficient than lutetium-177 for treatment of multiple myeloma in a preclinical model. Front. Med. 2015, 2, 76. [Google Scholar] [CrossRef]
- Feuerecker, B.; Michalik, M.; Hundshammer, C.; Schwaiger, M.; Bruchertseifer, F.; Morgenstern, A.; Seidl, C. Assessment of 213Bi-anti-EGFR MAb treatment efficacy in malignant cancer cells with [1−13C]pyruvate and [18F]FDG. Nat. Sci. Rep. 2019, 9, 8294. [Google Scholar] [CrossRef] [PubMed]
- Ménager, J.; Gorin, J.B.; Maurel, C.; Drujont, L.; Gouard, S.; Louvet, C.; Chérel, M.; Faivre-Chauvet, A.; Morgenstern, A.; Bruchertseifer, F.; et al. Combining α-radioimmunotherapy and adoptive T cell therapy to potentiate tumor destruction. PLoS ONE 2015, 10, e0130249. [Google Scholar] [CrossRef]
- Teiluf, K.; Seidl, C.; Blechert, B.; Gaertner, F.C.; Gilbertz, K.P.; Fernandez, V.; Bassermann, F.; Endell, J.; Boxhammer, R.; Leclair, S.; et al. α-radioimmunotherapy with 213Bi-anti-CD38 immunoconjugates is effective in a mouse model of human multiple myeloma. Oncotarget 2015, 6, 4692–4703. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Hedayati, M.; Hobbs, R.F.; Shao, C.; Bruchertseifer, F.; Morgenstern, A.; DeWeese, T.L.; Sgouros, G. Targeting aberrant DNA double-strand break repair in triple-negative breast cancer with alpha-particle emitter radiolabeled anti-EGFR antibody. Mol. Cancer Ther. 2013, 12, 2043–2054. [Google Scholar] [CrossRef]
- Roscher, M.; Hormann, I.; Leib, O.; Marx, S.; Moreno, J.; Miltner, E.; Friesen, C. Targeted alpha-therapy using [Bi-213]anti-CD20 as novel treatment option for radio-and chemoresistant non-Hodgkin lymphoma cells. Oncotarget 2013, 4, 218–230. [Google Scholar] [CrossRef]
- Pagel, J.M.; Kenoyer, A.L.; Bäck, T.; Hamlin, D.K.; Wilbur, D.S.; Fisher, D.R.; Park, S.I.; Frayo, S.; Axtman, A.; Orgun, N.; et al. Anti-CD45 pretargeted radioimmunotherapy using bismuth-213: High rates of complete remission and long-term survival in a mouse myeloid leukemia xenograft model. Blood 2011, 118, 703–711. [Google Scholar] [CrossRef]
- Allen, B.J.; Abbas Rizvi, S.M.; Qu, C.F.; Smith, R.C. Targeted alpha therapy approach to the management of pancreatic cancer. Cancers 2011, 3, 1821–1843. [Google Scholar] [CrossRef] [PubMed]
- Park, S.I.; Shenoi, J.; Page, J.M.; Hamlin, D.K.; Wilbur, D.S.; Orgun, N.; Kenoyer, A.L.; Frayo, S.; Axtman, A.; Bäck, T.; et al. Conventional and pretargeted radioimmunotherapy using bismuth-213 to target and treat non-Hodgkin lymphomas expressing CD20: A preclinical model toward optimal consolidation therapy to eradicate minimal residual disease. Blood 2010, 116, 4231–4239. [Google Scholar] [CrossRef] [PubMed]
- Lingappa, M.; Song, H.; Thompson, S.; Bruchertseifer, F.; Morgenstern, A.; Sgouros, G. Immunoliposomal delivery of 213Bi for α-emitter targeting of metastatic breast cancer. Cancer Res. 2010, 70, 6815–6823. [Google Scholar] [CrossRef] [PubMed]
- Drecoll, E.; Gaertner, F.C.; Miederer, M.; Blechert, B.; Vallon, M.; Müller, J.M.; Alke, A.; Seidl, C.; Bruchertseifer, F.; Morgenstern, A.; et al. Treatment of peritoneal carcinomatosis by targeted delivery of the radio-labeled tumor homing peptide 213Bi-DTPA-[F3]2 into the nucleus of tumor cells. PLoS ONE 2009, 4, e5715. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Rizvi, S.M.A.; Ranson, M.; Allen, B.J. 213Bi-PAI2 conjugate selectively induces apoptosis in PC3 metastatic prostate cancer cell line and shows anti-cancer activity in a xenograft animal model. Br. J. Cancer 2002, 86, 1197–1203. [Google Scholar] [CrossRef]
- Verhoeven, M.; Seimbille, Y.; Dalm, S.U. Therapeutic applications of pretargeting. Pharmaceutics 2019, 11, 434. [Google Scholar] [CrossRef]
- Listek, M.; Hönow, A.; Gossen, M.; Hanack, K. A novel selection strategy for antibody producing hybridoma cells based on a new transgenic fusion cell line. Nat. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef]
- Adams, G.P.; Shaller, C.C.; Chappell, L.L.; Wu, C.; Horak, E.M.; Simmons, H.H.; Litwin, S.; Marks, J.D.; Weiner, L.M.; Brechbiel, M.W. Delivery of the α-emitting radioisotope bismuth-213 to solid tumors via single-chain Fv and diabody molecules. Nucl. Med. Biol. 2000, 27, 339–346. [Google Scholar] [CrossRef]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen EM, B.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; Bushnell, D.; et al. Phase 3 Trial of 177Lu-Dotatate for Midgut Neuroendocrine Tumors. N. Engl. J. Med. 2017, 2, 125–135. [Google Scholar] [CrossRef]
- Bidwell, G.L.; Raucher, D. Therapeutic peptides for cancer therapy. Part I—Peptide inhibitors of signal transduction cascades. Expert Opin. Drug Deliv. 2009, 6, 1033–1047. [Google Scholar] [CrossRef] [PubMed]
- McGregor, D.P. Discovering and improving novel peptide therapeutics. Curr. Opin. Pharmacol. 2008, 8, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Fjelde, A.; Id, M.; Saidi, A.; Torgue, J.; Heyerdahl, H.; Stallons, T.A.R.; Kolstad, A.; Dahle, J. Targeted alpha therapy for chronic lymphocytic leukaemia and non-Hodgkin’s lymphoma with the anti-CD37 radioimmunoconjugate 212Pb-NNV003. PLoS ONE 2020, 15, e023. [Google Scholar]
- Ebbers, S.C.; Braat, A.J.A.T.; Moelker, A.; Stokkel, M.P.M.; Lam, M.G.E.H.; Barentsz, M.W. Intra-arterial versus standard intravenous administration of lutetium-177-DOTA-octreotate in patients with NET liver metastases: Study protocol for a multicenter, randomized controlled trial (LUTIA trial). Trials 2020, 21, 141. [Google Scholar] [CrossRef]
- Graf, J.; Pape, U.F.; Jann, H.; Denecke, T.; Arsenic, R.; Brenner, W.; Pavel, M.; Prasad, V. Prognostic Significance of Somatostatin Receptor Heterogeneity in Progressive Neuroendocrine Tumor Treated with Lu-177 DOTATOC or Lu-177 DOTATATE. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 881–894. [Google Scholar] [CrossRef]
- Delpassand, E.S.; Samarghandi, A.; Zamanian, S.; Wolin, E.M.; Hamiditabar, M.; Espenan, G.D.; Erion, J.L.; O’Dorisio, T.M.; Kvols, L.K.; Simon, J.; et al. Peptide receptor radionuclide therapy with 177Lu-DOTATATE for patients with somatostatin receptor-expressing neuroendocrine tumors: The first US phase 2 experience. Pancreas 2014, 43, 518–525. [Google Scholar] [CrossRef]
- Allen, B.J.; Raja, C.; Rizvi, S.; Li, Y.; Tsui, W.; Graham, P.; Thompson, J.F.; Reisfeld, R.A.; Kearsley, J.; Morgenstern, A.; et al. Intralesional targeted alpha therapy for metastatic melanoma. Cancer Biol. Ther. 2005, 4, 1318–1324. [Google Scholar] [CrossRef]
- Finn, L.E.; Levy, M.; Orozco, J.J.; Park, J.H.; Atallah, E.; Craig, M.; Perl, A.E.; Scheinberg, D.A.; Cicic, D.; Bergonio, G.R.; et al. A Phase 2 Study of Actinium-225 (225Ac)-Lintuzumab in Older Patients with Previously Untreated Acute Myeloid Leukemia (AML) Unfit for Intensive Chemotherapy. Blood 2017, 130 (Suppl. S1), 2638. [Google Scholar]
- Jurcic, J.G.; Ravandi, F.; Pagel, J.M.; Park, J.H.; Smith, B.D.; Douer, D.; Levy, M.Y.; Estey, E.; Kantarjian, H.M.; Earle, D.; et al. Phase I trial of α-particle therapy with actinium-225 (225Ac)-lintuzumab (anti-CD33) and low-dose cytarabine (LDAC) in older patients with untreated acute myeloid leukemia (AML. J. Clin. Oncol. 2015, 33 (Suppl. S15), 7050. [Google Scholar] [CrossRef]
- Juzeniene, A.; Stenberg, V.Y.; Bruland, Ø.S.; Larsen, R.H. Preclinical and Clinical Status of PSMA-Targeted Alpha Therapy for Metastatic Castration-Resistant Prostate Cancer. Cancers 2021, 13, 779. [Google Scholar] [CrossRef]
- Allen, B.J.; Singla, A.A.; Rizvi, S.M.A.; Graham, P.; Bruchertseifer, F.; Apostolidis, C.; Morgenstern, A. Analysis of patient survival in a Phase i trial of systemic targeted α?-therapy for metastatic melanoma. Immunotherapy 2011, 3, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Raja, C.; Graham, P.; Abbas Rizvi, S.M.; Song, E.; Goldsmith, H.; Thompson, J.; Bosserhoff, A.; Morgenstern, A.; Apostolidis, C.; Kearsley, J.; et al. Interim analysis of toxicity and response in phase 1 trial of systemic targeted alpha therapy for metastatic melanoma. Cancer Biol. Ther. 2007, 6, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Krolicki, L.; Bruchertseifer, F.; Kunikowska, J.; Koziara, H.; Królicki, B.; Jakuciński, M.; Pawlak, D.; Apostolidis, C.; Mirzadeh, S.; Rola, R.; et al. Prolonged survival in secondary glioblastoma following local injection of targeted alpha therapy with 213Bi-substance P analogue. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1636–1644. [Google Scholar] [CrossRef]
- De Kruijff, R.; Wolterbeek, H.; Denkova, A. A Critical Review of Alpha Radionuclide Therapy—How to Deal with Recoiling Daughters? Pharmaceuticals 2015, 8, 321–336. [Google Scholar] [PubMed]
- Jansen, K.; Heirbaut, L.; Cheng, J.D.; Joossens, J.; Ryabtsova, O.; Cos, P.; Maes, L.; Lambeir, A.M.; De Meester, I.; Augustyns, K.; et al. Selective inhibitors of fibroblast activation protein (FAP) with a (4-quinolinoyl)-glycyl-2-cyanopyrrolidine scaffold. ACS Med. Chem. Lett. 2013, 4, 491–496. [Google Scholar] [CrossRef]
- Giesel, F.L.; Kratochwil, C.; Lindner, T.; Marschalek, M.M.; Loktev, A.; Lehnert, W.; Debus, J.; Jäger, D.; Flechsig, P.; Altmann, A.; et al. 68Ga-FAPI PET/CT: Biodistribution and preliminary dosimetry estimate of 2 DOTA-containing FAP-targeting agents in patients with various cancers. J. Nucl. Med. 2019, 60, 386–392. [Google Scholar] [CrossRef]
- Lindner, T.; Loktev, A.; Altmann, A.; Giesel, F.; Kratochwil, C.; Debus, J.; Jäger, D.; Mier, W.; Haberkorn, U. Development of quinoline-based theranostic ligands for the targeting of fibroblast activation protein. J. Nucl. Med. 2018, 59, 1415–1422. [Google Scholar] [CrossRef]
- Loktev, A.; Lindner, T.; Mier, W.; Debus, J.; Altmann, A.; Jäger, D.; Giesel, F.; Kratochwil, C.; Barthe, P.; Roumestand, C.; et al. A tumor-imaging method targeting cancer-associated fibroblasts. J. Nucl. Med. 2018, 59, 1423–1429. [Google Scholar] [CrossRef]
- Jaggi, J.S.; Seshan, S.V.; McDevitt, M.R.; Sgouros, G.; Hyjek, E.; Scheinberg, D.A. Mitigation of radiation nephropathy after internal α-particle irradiation of kidneys. Int. J. Radiat. Oncol. Biol. Phys. 2006, 64, 1503–1512. [Google Scholar] [CrossRef]
- Chatal, J.F.; Davodeau, F.; Cherel, M.; Barbet, J. Different ways to improve the clinical effectiveness of radioimmunotherapy in solid tumors. J. Cancer Res. Ther. 2009, 5 (Suppl. S1), S36–S40. [Google Scholar] [CrossRef] [PubMed]
- Song, E.Y.; Abbas Rizvi, S.M.; Qu, C.F.; Raja, C.; Brechbiel, M.W.; Morgenstern, A.; Apostolidis, C.; Allen, B.J. Pharmacokinetics and toxicity of 213Bi-labeled PAI2 in preclinical targeted alpha therapy for cancer. Cancer Biol. Ther. 2007, 6, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Bendre, S.; Zhang, Z.; Kuo, H.-T.; Rousseau, J.; Zhang, C.; Merkens, H.; Roxin, Á.; Bénard, F.; Lin, K.-S. Evaluation of Met-Val-Lys as a Renal Brush Border Enzyme-Cleavable Linker to Reduce Kidney Uptake of 68Ga-Labeled DOTA-Conjugated Peptides and Peptidomimetics. Molecules 2020, 25, 3854. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, C.; Uehara, T.; Kanazawa, N.; Wada, S.; Suzuki, H.; Arano, Y. Preferential Cleavage of a Tripeptide Linkage by Enzymes on Renal Brush Border Membrane To Reduce Renal Radioactivity Levels of Radiolabeled Antibody Fragments. J. Med. Chem. 2018, 61, 5257–5268. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).