
The Use of Translational Modelling and Simulation to Develop Immunomodulatory Therapy as an Adjunct to Antibiotic Treatment in the Context of Pneumonia

 ,
,  , , ,
, , ,  , , and
, , and 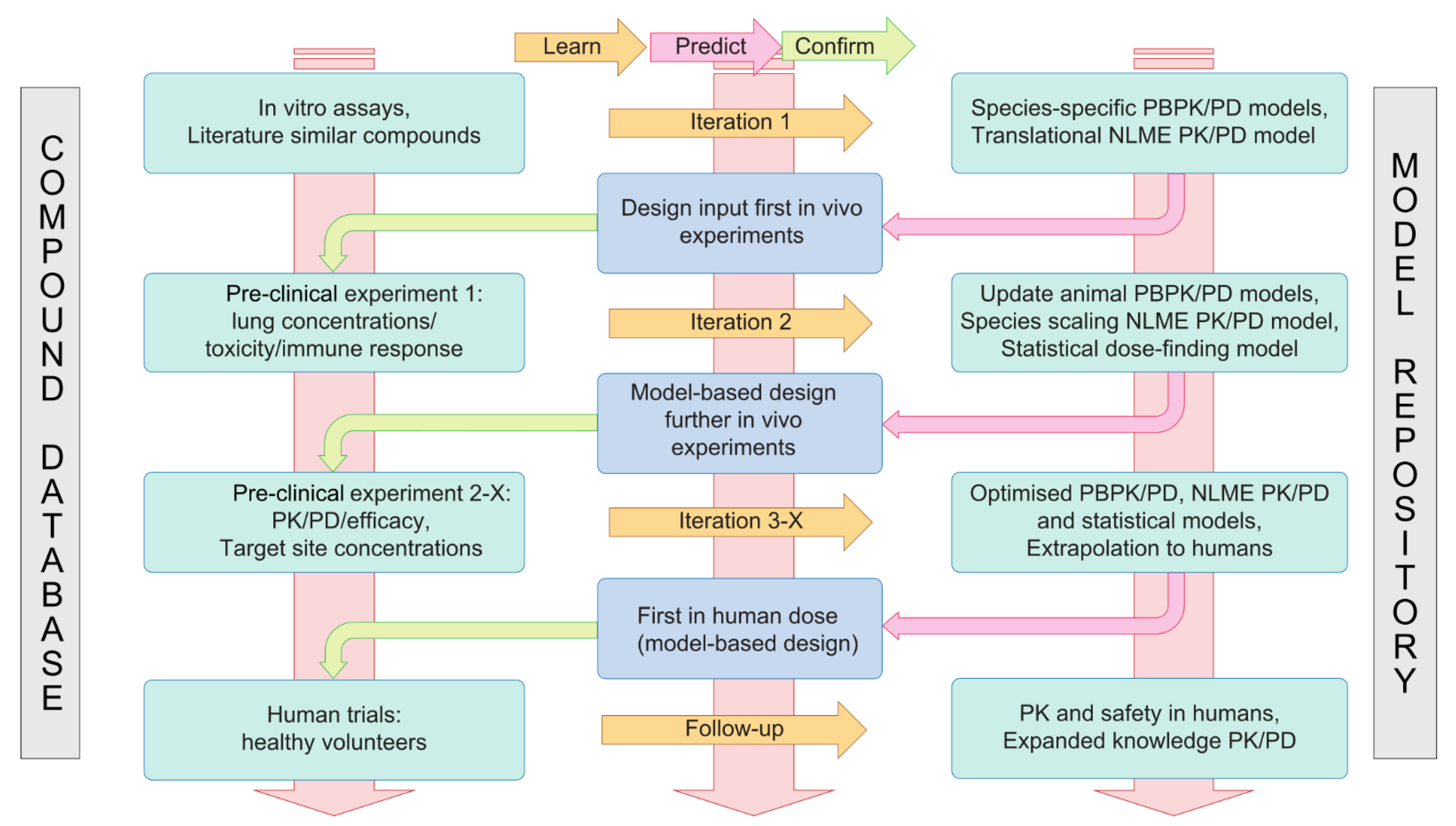{kind=link}
Abstract
:1. Introduction
2. The ABIMMUNE Project
3. The FAIR Project
4. Discussion
4.1. Lessons Learned from ABIMMUNE and FAIR
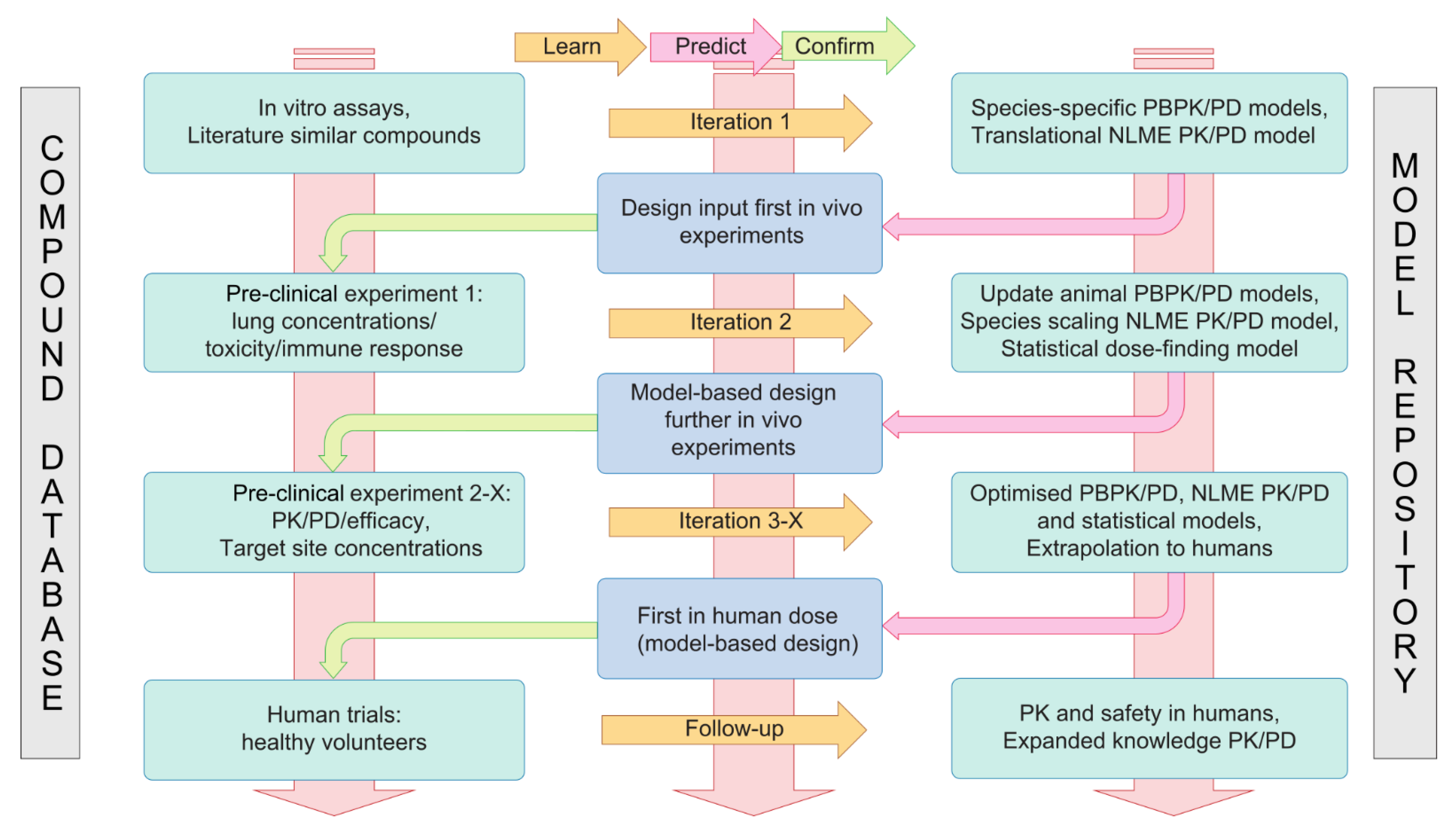
4.2. A Translational Modelling and Simulation Framework for Development of Immunomodulatory Drugs
4.3. Application in the Current Drug Discovery and Development Landscape
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Pneumonia Fact Sheet. Available online: https://www.who.int/news-room/fact-sheets/detail/pneumonia (accessed on 26 February 2021).
- World Health Organization (WHO). Pneumococcal Disease. Available online: https://www.who.int/ith/diseases/pneumococcal/en/ (accessed on 26 February 2021).
- The Top 10 Causes of Death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 4 February 2021).
- Ferreira-Coimbra, J.; Sarda, C.; Rello, J. Burden of Community-Acquired Pneumonia and Unmet Clinical Needs. Adv. Ther. 2020, 37, 1302–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, A.; Niederman, M.S.; Chastre, J.; Ewig, S.; Fernandez-Vandellos, P.; Hanberger, H.; Kollef, M.; Li Bassi, G.; Luna, C.M.; Martin-Loeches, I.; et al. International ERS/ESICM/ESCMID/ALAT Guidelines for the Management of Hospital-Acquired Pneumonia and Ventilator-Associated Pneumonia. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [PubMed]
- Keller, S.C.; Cosgrove, S.E. Reducing Antibiotic Resistance through Antibiotic Stewardship in the Ambulatory Setting. Lancet Infect. Dis. 2019, 3994. [Google Scholar] [CrossRef]
- Gullberg, E.; Cao, S.; Berg, O.G.; Ilbäck, C.; Sandegren, L.; Hughes, D.; Andersson, D.I. Selection of Resistant Bacteria at Very Low Antibiotic Concentrations. PLoS Pathog. 2011, 7, e1002158. [Google Scholar] [CrossRef] [Green Version]
- Spellberg, B.; Guidos, R.; Gilbert, D.; Bradley, J.; Boucher, H.W.; Scheld, W.M.; Bartlett, J.G.; Edwards, J. The Epidemic of Antibiotic-Resistant Infections: A Call to Action for the Medical Community from the Infectious Diseases Society of America. Clin. Infect. Dis. 2008, 46, 155–164. [Google Scholar] [CrossRef]
- Laxminarayan, R.; Duse, A.; Wattal, C.; Zaidi, A.K.M.; Wertheim, H.F.L.; Sumpradit, N.; Vlieghe, E.; Hara, G.L.; Gould, I.M.; Goossens, H.; et al. Antibiotic Resistance-the Need for Global Solutions. Lancet Infect. Dis. 2013, 13, 1057–1098. [Google Scholar] [CrossRef] [Green Version]
- Levy, S.B.; Bonnie, M. Antibacterial Resistance Worldwide: Causes, Challenges and Responses. Nat. Med. 2004, 10, S122–S129. [Google Scholar] [CrossRef]
- Ventola, C.L. The Antibiotic Resistance Crisis: Causes and Threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- World Health Organization (WHO). Antibiotic Resistance Fact Sheet. Available online: https://www.who.int/news-room/fact-sheets/detail/antibiotic-resistance (accessed on 4 February 2021).
- The Lancet Antibiotic Resistance: A Final Warning. Lancet 2013, 382, 1072. [CrossRef]
- Sriram, A.; Kalanxhi, E.; Kapoor, G.; Craig, J.; Balasubramanian, R.; Brar, S.; Criscuolo, N.; Hamilton, A.; Klein, E.; Tseng, K.; et al. State of the World’s Antibiotics 2021: A Global Analysis of Antimicrobial Resistance and Its Drivers; Center for Disease Dynamics, Economics & Policy: Washington, DC, USA, 2021. [Google Scholar]
- Theuretzbacher, U.; Bush, K.; Harbarth, S.; Paul, M.; Rex, J.H.; Tacconelli, E.; Thwaites, G.E. Critical Analysis of Antibacterial Agents in Clinical Development. Nat. Rev. Microbiol. 2020, 18, 286–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawson, T.M.; Moore, L.S.P.; Zhu, N.; Ranganathan, N.; Skolimowska, K.; Gilchrist, M.; Satta, G.; Cooke, G.; Holmes, A. Bacterial and Fungal Coinfection in Individuals with Coronavirus: A Rapid Review to Support COVID-19 Antimicrobial Prescribing. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Rawson, T.M.; Ming, D.; Ahmad, R.; Moore, L.S.P.; Holmes, A.H. Antimicrobial Use, Drug-Resistant Infections and COVID-19. Nat. Rev. Microbiol. 2020, 18. [Google Scholar] [CrossRef]
- Ghosh, C.; Sarkar, P.; Issa, R.; Haldar, J. Alternatives to Conventional Antibiotics in the Era of Antimicrobial Resistance. Trends Microbiol. 2019, 27, 323–338. [Google Scholar] [CrossRef]
- Theuretzbacher, U.; Piddock, L.J.V. Non-Traditional Antibacterial Therapeutic Options and Challenges. Cell Host Microbe 2019, 26, 61–72. [Google Scholar] [CrossRef]
- Czaplewski, L.; Bax, R.; Clokie, M.; Dawson, M.; Fairhead, H.; Fischetti, V.A.; Foster, S.; Gilmore, B.F.; Hancock, R.E.W.; Harper, D.; et al. Alternatives to Antibiotics—A Pipeline Portfolio Review. Lancet Infect. Dis. 2016, 16, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Romero-Calle, D.; Guimarães Benevides, R.; Góes-Neto, A.; Billington, C. Bacteriophages as Alternatives to Antibiotics in Clinical Care. Antibiotics 2019, 8, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The Value of Antimicrobial Peptides in the Age of Resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Chen, C.H.; Lu, T.K. Development and Challenges of Antimicrobial Peptides for Therapeutic Applications. Antibiotics 2020, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Mouton, J.W. Combination Therapy as a Tool to Prevent Emergence of Bacterial Resistance. Infection 1999, 27, S24–S28. [Google Scholar] [CrossRef] [PubMed]
- Cattoir, V.; Felden, B. Future Antibacterial Strategies: From Basic Concepts to Clinical Challenges. J. Infect. Dis. 2019, 220, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W.; Nijnik, A.; Philpott, D.J. Modulating Immunity as a Therapy for Bacterial Infections. Nat. Rev. Microbiol. 2012, 10, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- Rex, J.H.; Fernandez Lynch, H.; Cohen, I.G.; Darrow, J.J.; Outterson, K. Designing Development Programs for Non-Traditional Antibacterial Agents. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Imam, M.T.; Venkateshan, S.P.; Tandon, M.; Saha, N.; Pillai, K.K. Comparative Evaluation of US Food and Drug Administration and Pharmacologically Guided Approaches to Determine the Maximum Recommended Starting Dose for First-in-Human Clinical Trials in Adult Healthy Men. J. Clin. Pharmacol. 2011, 51. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration (FDA). Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers; FDA: Rockville, MD, USA, 2005. [Google Scholar]
- Collin s, J.M.; Grieshaber, C.K.; Chabner, B.A. Pharmacologically Guided Phase I Clinical Trials Based Upon Preclinical Drug Development. J. Natl. Cancer Inst. 1990, 82. [Google Scholar] [CrossRef] [PubMed]
- Reigner, B.G.; Blesch, K. Estimating the Starting Dose for Entry into Humans: Principles and Practice. Eur. J. Clin. Pharmacol. 2002, 57. [Google Scholar] [CrossRef]
- Penel, N.; Kramar, A. What Does a Modified-Fibonacci Dose-Escalation Actually Correspond To? BMC Med. Res. Methodol. 2012, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Quigley, J.; Pepe, M.; Fisher, L. Continual Reassessment Method: A Practical Design for Phase 1 Clinical Trials in Cancer. Biometrics 1990, 46, 33. [Google Scholar] [CrossRef] [PubMed]
- Neuenschwander, B.; Branson, M.; Gsponer, T. Critical Aspects of the Bayesian Approach to Phase I Cancer Trials. Stat. Med. 2008, 27. [Google Scholar] [CrossRef]
- Comets, E.; Zohar, S. A Survey of the Way Pharmacokinetics Are Reported in Published Phase I Clinical Trials, with an Emphasis on Oncology. Clin. Pharmacokinet. 2009, 48. [Google Scholar] [CrossRef] [PubMed]
- Ursino, M.; Zohar, S.; Lentz, F.; Alberti, C.; Friede, T.; Stallard, N.; Comets, E. Dose-Finding Methods for Phase I Clinical Trials Using Pharmacokinetics in Small Populations. Biom. J. 2017, 59. [Google Scholar] [CrossRef]
- Günhan, B.K.; Weber, S.; Friede, T. A Bayesian Time-to-event Pharmacokinetic Model for Phase I Dose-escalation Trials with Multiple Schedules. Stat. Med. 2020, 39. [Google Scholar] [CrossRef]
- Gerard, E.; Zohar, S.; Thai, H.; Lorenzato, C.; Riviere, M.; Ursino, M. Bayesian Dose-regimen Assessment in Early Phase Oncology Incorporating Pharmacokinetics and Pharmacodynamics. Biometrics 2021. [Google Scholar] [CrossRef]
- Mould, D.R.; Upton, R.N. Basic Concepts in Population Modeling, Simulation, and Model-Based Drug Development. CPT Pharmacomet. Syst. Pharmacol. 2012, 1, e6. [Google Scholar] [CrossRef] [PubMed]
- Mould, D.R.; Upton, R.N. Basic Concepts in Population Modeling, Simulation, and Model-Based Drug Development-Part 2: Introduction to Pharmacokinetic Modeling Methods. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, e38. [Google Scholar] [CrossRef] [PubMed]
- Visser, S.A.G.; Manolis, E.; Danhof, M.; Kerbusch, T. Modeling and Simulation at the Interface of Nonclinical and Early Clinical Drug Development. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Staab, A.; Rook, E.; Maliepaard, M.; Aarons, L.; Benson, C. Modeling and Simulation in Clinical Pharmacology and Dose Finding. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, 2–4. [Google Scholar] [CrossRef]
- Harnisch, L.; Shepard, T.; Pons, G.; della Pasqua, O. Modeling and Simulation as a Tool to Bridge Efficacy and Safety Data in Special Populations. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.S.; Fossler, M.J.; Cadieu, K.D.; Gastonguay, M.R. Pharmacometrics: A Multidisciplinary Field to Facilitate Critical Thinking in Drug Development and Translational Research Settings. J. Clin. Pharmacol. 2008, 48, 632–649. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, R.; Bergström, E.L.; Janzén, D.L.I.; Fridén, M.; Eriksson, U.; Grime, K.; Ferguson, D. Translational Model to Predict Pulmonary Pharmacokinetics and Efficacy in Man for Inhaled Bronchodilators. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 147–157. [Google Scholar] [CrossRef]
- Borghardt, J.M.; Weber, B.; Staab, A.; Kloft, C. Pharmacometric Models for Characterizing the Pharmacokinetics of Orally Inhaled Drugs. AAPS J. 2015, 17, 853–870. [Google Scholar] [CrossRef] [Green Version]
- Himstedt, A.; Braun, C.; Wicha, S.G.; Borghardt, J.M. Towards a Quantitative Mechanistic Understanding of Localized Pulmonary Tissue Retention—A Combined In Vivo/In Silico Approach Based on Four Model Drugs. Pharmaceutics 2020, 12, 408. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, M.O.; Anehall, T.; Friberg, L.E.; Henningsson, A.; Kloft, C.; Sandström, M. Pharmacokinetic / Pharmacodynamic Modelling in Oncological Drug Development. Basic Clin. Pharmacol. Toxicol. 2005, 23, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Haddish-Berhane, N.; Shah, D.K.; Ma, D.; Leal, M.; Gerber, H.P.; Sapra, P.; Barton, H.A.; Betts, A.M. On Translation of Antibody Drug Conjugates Efficacy from Mouse Experimental Tumors to the Clinic: A PK/PD Approach. J. Pharmacokinet. Pharmacodyn. 2013, 40, 557–571. [Google Scholar] [CrossRef] [PubMed]
- Bellanti, F.; della Pasqua, O. Modelling and Simulation as Research Tools in Paediatric Drug Development. Eur. J. Clin. Pharmacol. 2011, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, C.; Hartung, N.; de Wiljes, J.; Kloft, C.; Huisinga, W. Bayesian Data Assimilation to Support Informed Decision Making in Individualized Chemotherapy. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 153–164. [Google Scholar] [CrossRef]
- Zheng, H.; Hampson, L.V.; Wandel, S. A Robust Bayesian Meta-Analytic Approach to Incorporate Animal Data into Phase I Oncology Trials. Stat. Methods Med. Res. 2020, 29. [Google Scholar] [CrossRef] [Green Version]
- Wakefield, J. The Bayesian Analysis of Population Pharmacokinetic Models. J. Am. Stat. Assoc. 1996, 91, 62. [Google Scholar] [CrossRef]
- Kerioui, M.; Mercier, F.; Bertrand, J.; Tardivon, C.; Bruno, R.; Guedj, J.; Desmée, S. Bayesian Inference Using Hamiltonian Monte-Carlo Algorithm for Nonlinear Joint Modeling in the Context of Cancer Immunotherapy. Stat. Med. 2020, 39. [Google Scholar] [CrossRef]
- la Gamba, F.; Jacobs, T.; Geys, H.; Jaki, T.; Serroyen, J.; Ursino, M.; Russu, A.; Faes, C. Bayesian Sequential Integration within a Preclinical Pharmacokinetic and Pharmacodynamic Modeling Framework: Lessons Learned. Pharm. Stat. 2019. [Google Scholar] [CrossRef] [Green Version]
- Gueorguieva, I.; Aarons, L.; Rowland, M. Diazepam Pharamacokinetics from Preclinical to Phase I Using a Bayesian Population Physiologically Based Pharmacokinetic Model with Informative Prior Distributions in Winbugs. J. Pharmacokinet. Pharmacodyn. 2006, 33. [Google Scholar] [CrossRef] [PubMed]
- Petit, C.; Samson, A.; Morita, S.; Ursino, M.; Guedj, J.; Jullien, V.; Comets, E.; Zohar, S. Unified Approach for Extrapolation and Bridging of Adult Information in Early-Phase Dose-Finding Paediatric Studies. Stat. Methods Med. Res. 2018, 27. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G.; Sher, A. Cooperation of Toll-like Receptor Signals in Innate Immune Defence. Nat. Rev. Immunol. 2007, 7, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Casilag, F.; Franck, S.; Matarazzo, L.; Figeac, M.; Michelet, R.; Kloft, C.; Carnoy, C.; Sirard, J.C. Boosting Toll-like Receptor 4 Signaling Enhances the Therapeutic Outcome of Antibiotic Therapy in Pneumococcal Pneumonia. bioRxiv 2020. [Google Scholar] [CrossRef]
- Franck, S.; Fuhrmann-Selter, T.; Joseph, J.F.; Michelet, R.; Casilag, F.; Sirard, J.-C.; Wicha, S.G.; Kloft, C. A Rapid, Simple and Sensitive Liquid Chromatography Tandem Mass Spectrometry Assay to Determine Amoxicillin Concentrations in Biological Matrix of Little Volume. Talanta 2019, 201. [Google Scholar] [CrossRef] [PubMed]
- Franck, S.; Michelet, R.; Casilag, F.; Sirard, J.-C.; Wicha, S.G.; Kloft, C. A Model-Based Pharmacokinetic/Pharmacodynamic Analysis of the Combination of Amoxicillin and Monophosphoryl Lipid a against S. pneumoniae in Mice. Pharmaceutics 2021, 13, 469. [Google Scholar] [CrossRef]
- Anderson, B.J.; Holford, N.H.G. Mechanistic Basis of Using Body Size and Maturation to Predict Clearance in Humans. Drug Metab. Pharmacokinet. 2009, 24. [Google Scholar] [CrossRef]
- Carlier, M.; Noë, M.; de Waele, J.J.; Stove, V.; Verstraete, A.G.; Lipman, J.; Roberts, J.A. Population Pharmacokinetics and Dosing Simulations of Amoxicillin/Clavulanic Acid in Critically Ill Patients. J. Antimicrob. Chemother. 2013, 68, 2600–2608. [Google Scholar] [CrossRef] [Green Version]
- Chentouh, R.; Fitting, C.; Cavaillon, J.M. Specific Features of Human Monocytes Activation by Monophosphoryl Lipid A. Sci. Rep. 2018, 8, 7096. [Google Scholar] [CrossRef] [Green Version]
- Nempont, C.; Cayet, D.; Rumbo, M.; Bompard, C.; Villeret, V.; Sirard, J.-C. Deletion of Flagellin’s Hypervariable Region Abrogates Antibody-Mediated Neutralization and Systemic Activation of TLR5-Dependent Immunity. J. Immunol. 2008, 181. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, N.; van Maele, L.; Marqués, J.M.; Rial, A.; Sirard, J.-C.; Chabalgoity, J.A. Mucosal Administration of Flagellin Protects Mice from Streptococcus Pneumoniae Lung Infection. Infect. Immun. 2010, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matarazzo, L.; Casilag, F.; Porte, R.; Wallet, F.; Cayet, D.; Faveeuw, C.; Carnoy, C.; Sirard, J.-C. Therapeutic Synergy Between Antibiotics and Pulmonary Toll-Like Receptor 5 Stimulation in Antibiotic-Sensitive or -Resistant Pneumonia. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Porte, R.; Fougeron, D.; Muñoz-Wolf, N.; Tabareau, J.; Georgel, A.-F.; Wallet, F.; Paget, C.; Trottein, F.; Chabalgoity, J.A.; Carnoy, C.; et al. A Toll-Like Receptor 5 Agonist Improves the Efficacy of Antibiotics in Treatment of Primary and Influenza Virus-Associated Pneumococcal Mouse Infections. Antimicrob. Agents Chemother. 2015, 59. [Google Scholar] [CrossRef] [Green Version]
- Home—FAIR Website. Available online: https://fair-flagellin.eu/ (accessed on 19 February 2021).
- Miller, F.; Zohar, S.; Stallard, N.; Madan, J.; Posch, M.; Hee, S.W.; Pearce, M.; Vågerö, M.; Day, S. Approaches to Sample Size Calculation for Clinical Trials in Rare Diseases. Pharm. Stat. 2018, 17. [Google Scholar] [CrossRef] [Green Version]
- Stallard, N.; Miller, F.; Day, S.; Hee, S.W.; Madan, J.; Zohar, S.; Posch, M. Determination of the Optimal Sample Size for a Clinical Trial Accounting for the Population Size. Biom. J. 2017, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheiner, L.B. Learning versus Confirming in Clinical Drug Development. Clin. Pharmacol. Ther. 1997, 61, 275–291. [Google Scholar] [CrossRef]
- Pena-Miller, R.; Laehnemann, D.; Jansen, G.; Fuentes-Hernandez, A.; Rosenstiel, P.; Schulenburg, H.; Beardmore, R. When the Most Potent Combination of Antibiotics Selects for the Greatest Bacterial Load: The Smile-Frown Transition. PLoS Biol. 2013, 11. [Google Scholar] [CrossRef] [Green Version]
- Handel, A.; Margolis, E.; Levin, B.R. Exploring the Role of the Immune Response in Preventing Antibiotic Resistance. J. Theor. Biol. 2009, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Baeder, D.Y.; Regoes, R.R.; Rolff, J. Predicting Drug Resistance Evolution: Insights from Antimicrobial Peptides and Antibiotics. Proc. R. Soc. B: Biol. Sci. 2018, 285. [Google Scholar] [CrossRef] [Green Version]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the Pharmaceutical Industry: New Estimates of R&D Costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [CrossRef] [Green Version]
- Marshall, S.F.; Burghaus, R.; Cosson, V.; Cheung, S.; Chenel, M.; DellaPasqua, O.; Frey, N.; Hamrén, B.; Harnisch, L.; Ivanow, F.; et al. Good Practices in Model-Informed Drug Discovery and Development: Practice, Application, and Documentation. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 93–122. [Google Scholar] [CrossRef] [Green Version]
- Sachs, J.R.; Mayawala, K.; Gadamsetty, S.; Kang, S.P.; de Alwis, D.P. Optimal Dosing for Targeted Therapies in Oncology: Drug Development Cases Leading by Example. Clin. Cancer Res. 2016, 22, 1318–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sou, T.; Hansen, J.; Liepinsh, E.; Backlund, M.; Ercan, O.; Grinberga, S.; Cao, S.; Giachou, P.; Petersson, A.; Tomczak, M.; et al. Model-Informed Drug Development for Antimicrobials: Translational PK and PK/PD Modeling to Predict an Efficacious Human Dose for Apramycin. Clin. Pharmacol. Ther. 2020, 109, 1063–1073. [Google Scholar] [CrossRef]
- Van Wijk, R.C.; Ayoun Alsoud, R.; Lennernäs, H.; Simonsson, U.S.H. Model-Informed Drug Discovery and Development Strategy for the Rapid Development of Anti-Tuberculosis Drug Combinations. Appl. Sci. 2020, 10, 2376. [Google Scholar] [CrossRef] [Green Version]
- Rayner, C.R.; Smith, P.F.; Andes, D.; Andrews, K.; Derendorf, H.; Friberg, L.E.; Hanna, D.; Lepak, A.; Mills, E.; Polasek, T.M.; et al. Model-Informed Drug Development for Anti-Infectives: State of the Art and Future. Clin. Pharmacol. Ther. 2021, 109, 867–891. [Google Scholar] [CrossRef]
- Betts, A.; Keunecke, A.; van Steeg, T.J.; van der Graaf, P.H.; Avery, L.B.; Jones, H.; Berkhout, J. Linear Pharmacokinetic Parameters for Monoclonal Antibodies Are Similar within a Species and across Different Pharmacological Targets: A Comparison between Human, Cynomolgus Monkey and HFcRn Tg32 Transgenic Mouse Using a Population-Modeling Approach. mAbs 2018, 10, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.M.; Zhang, Z.; Jasper, P.; Luo, H.; Avery, L.B.; King, L.E.; Neubert, H.; Barton, H.A.; Betts, A.M.; Webster, R. A Physiologically-Based Pharmacokinetic Model for the Prediction of Monoclonal Antibody Pharmacokinetics From In Vitro Data. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 738–747. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Wiszniewski, L.; Constant, S.; Roggen, E. Potential of in Vitro Reconstituted 3D Human Airway Epithelia (MucilAirTM) to Assess Respiratory Sensitizers. Toxicol. In Vitro 2013, 27, 1151–1156. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michelet, R.; Ursino, M.; Boulet, S.; Franck, S.; Casilag, F.; Baldry, M.; Rolff, J.; van Dyk, M.; Wicha, S.G.; Sirard, J.-C.; et al. The Use of Translational Modelling and Simulation to Develop Immunomodulatory Therapy as an Adjunct to Antibiotic Treatment in the Context of Pneumonia. Pharmaceutics 2021, 13, 601. https://doi.org/10.3390/pharmaceutics13050601
Michelet R, Ursino M, Boulet S, Franck S, Casilag F, Baldry M, Rolff J, van Dyk M, Wicha SG, Sirard J-C, et al. The Use of Translational Modelling and Simulation to Develop Immunomodulatory Therapy as an Adjunct to Antibiotic Treatment in the Context of Pneumonia. Pharmaceutics. 2021; 13(5):601. https://doi.org/10.3390/pharmaceutics13050601
Chicago/Turabian StyleMichelet, Robin, Moreno Ursino, Sandrine Boulet, Sebastian Franck, Fiordiligie Casilag, Mara Baldry, Jens Rolff, Madelé van Dyk, Sebastian G. Wicha, Jean-Claude Sirard, and et al. 2021. "The Use of Translational Modelling and Simulation to Develop Immunomodulatory Therapy as an Adjunct to Antibiotic Treatment in the Context of Pneumonia" Pharmaceutics 13, no. 5: 601. https://doi.org/10.3390/pharmaceutics13050601
APA StyleMichelet, R., Ursino, M., Boulet, S., Franck, S., Casilag, F., Baldry, M., Rolff, J., van Dyk, M., Wicha, S. G., Sirard, J. -C., Comets, E., Zohar, S., & Kloft, C. (2021). The Use of Translational Modelling and Simulation to Develop Immunomodulatory Therapy as an Adjunct to Antibiotic Treatment in the Context of Pneumonia. Pharmaceutics, 13(5), 601. https://doi.org/10.3390/pharmaceutics13050601