
Lipophilicity, Pharmacokinetic Properties, and Molecular Docking Study on SARS-CoV-2 Target for Betulin Triazole Derivatives with Attached 1,4-Quinone

 ,
,  , , and
, , and
Abstract
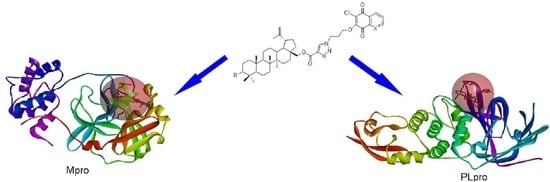
1. Introduction
2. Materials and Methods
2.1. Data Set
2.2. Experimental Lipophilicity
2.3. Theoretical Lipophilicity
2.4. Structure Optimization
2.5. Molecular Docking Study
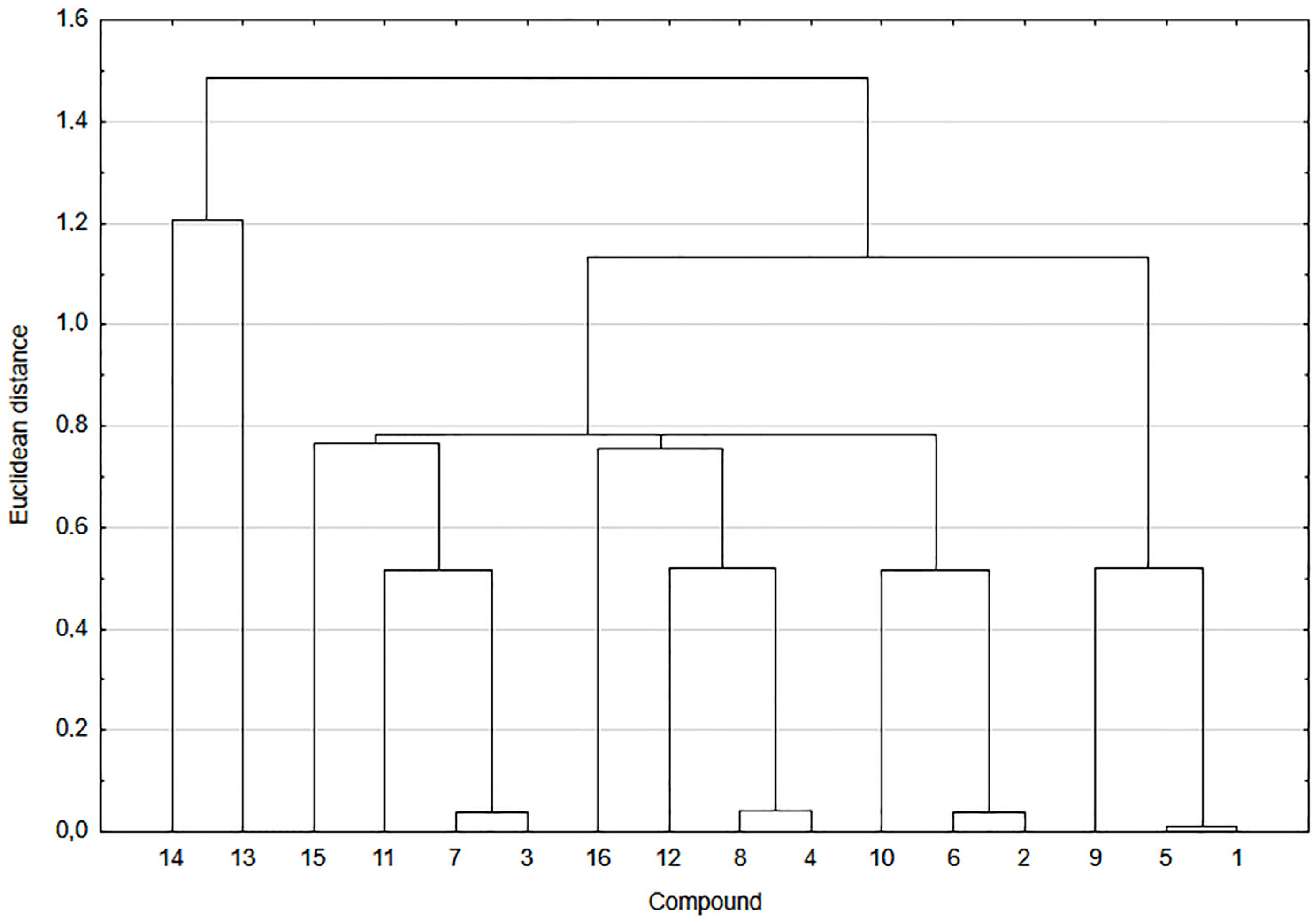
2.6. Correlation and Cluster Analysis
3. Results and Discussion
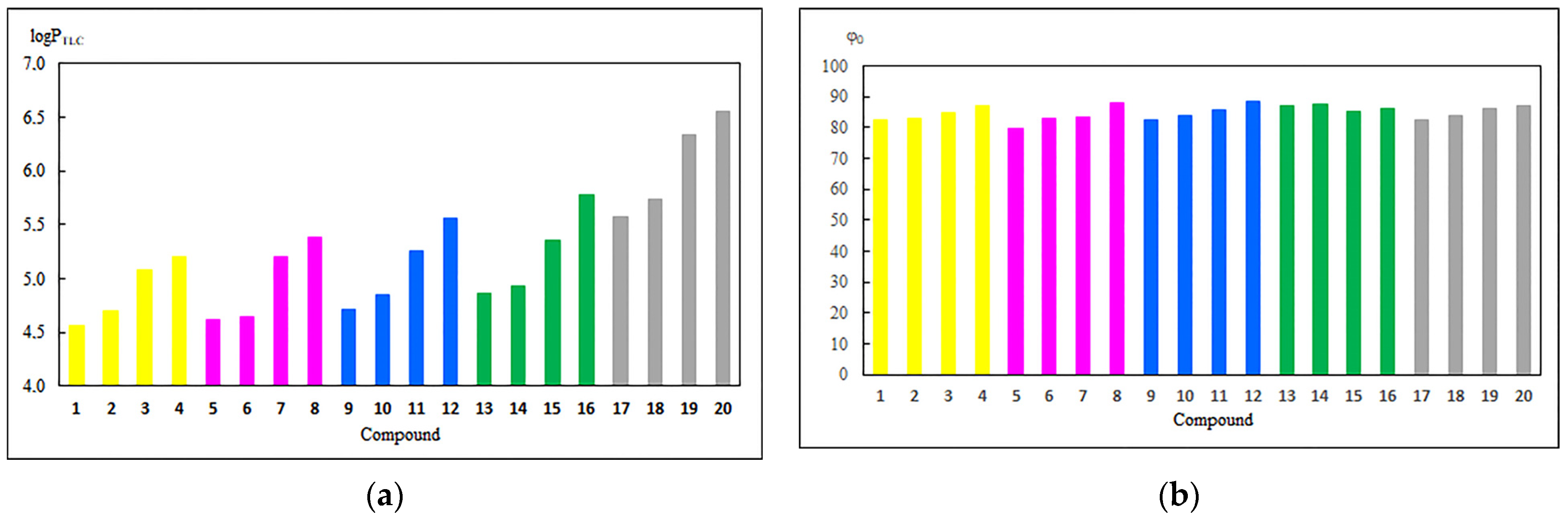
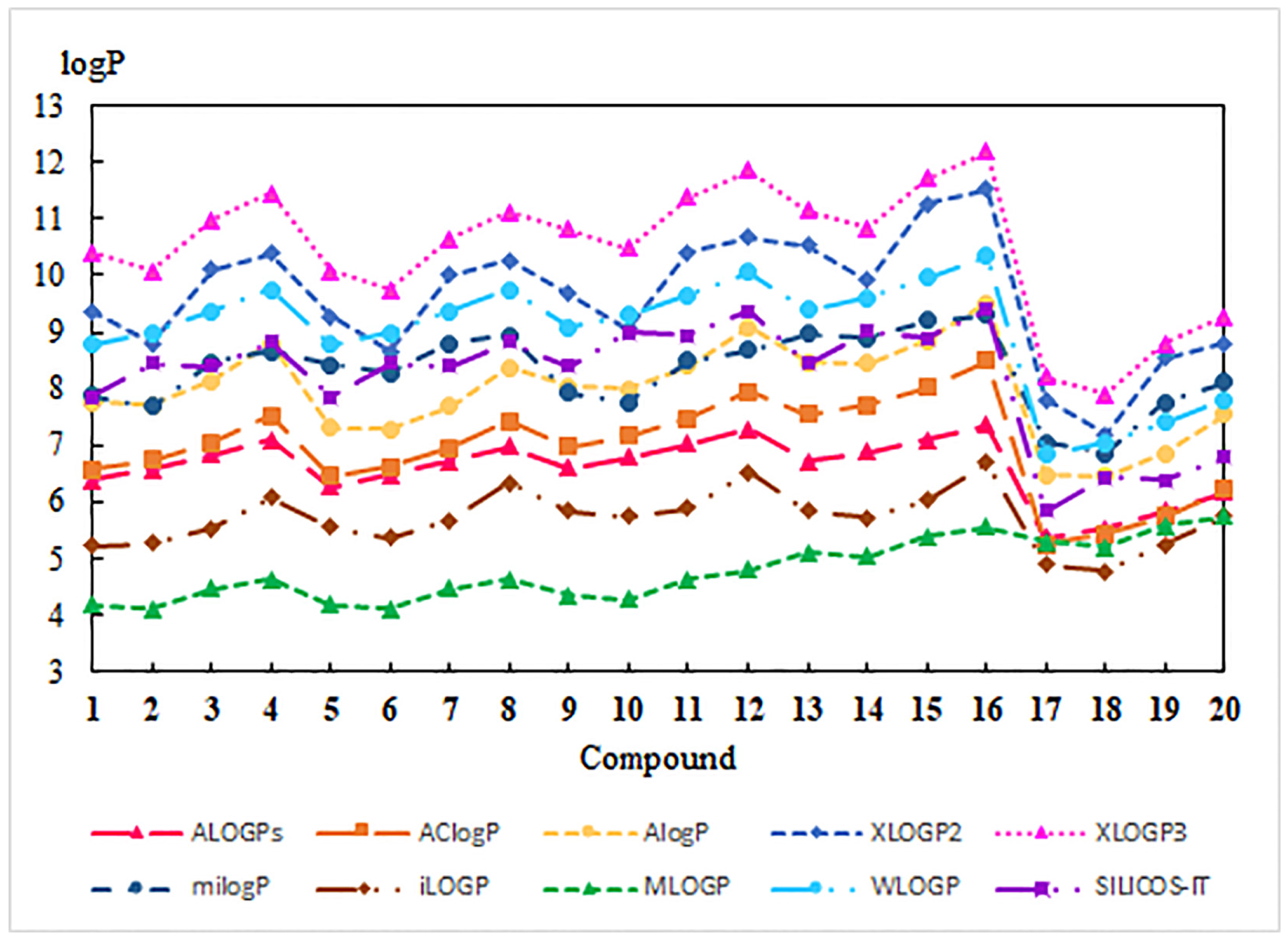
3.1. Experimental and Theoretical Lipophilicity
3.2. Physicochemical and Pharmacokinetic Properties
(r = 0.985, r2 = 0.969, SD = 1.11, VIF = 3.12, F = 126.7)
3.3. Molecular Properties
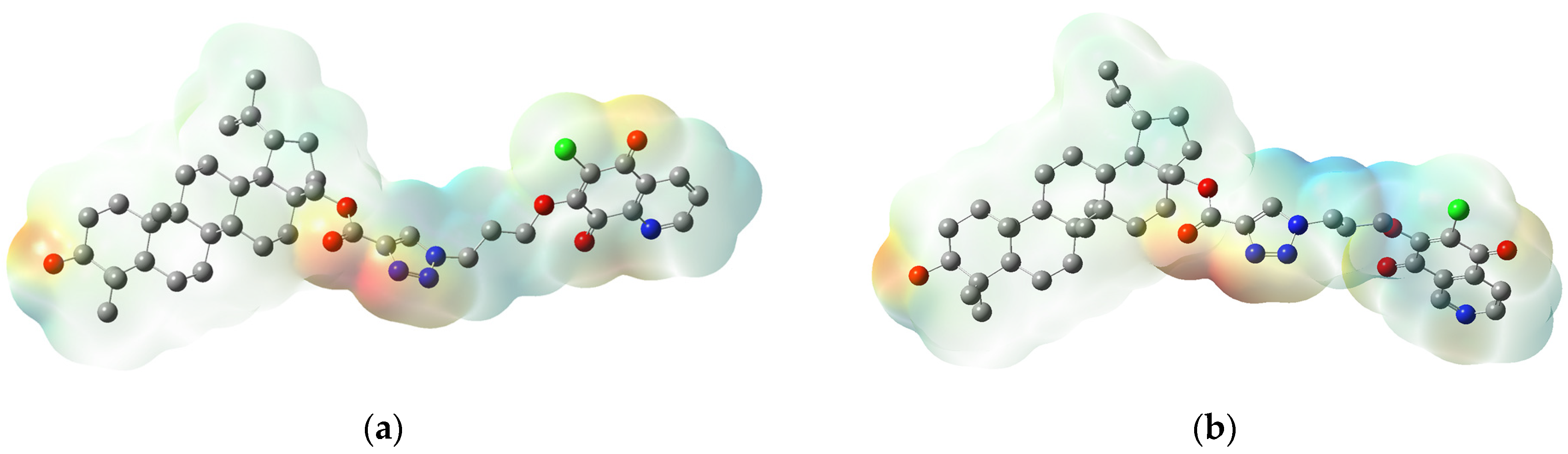
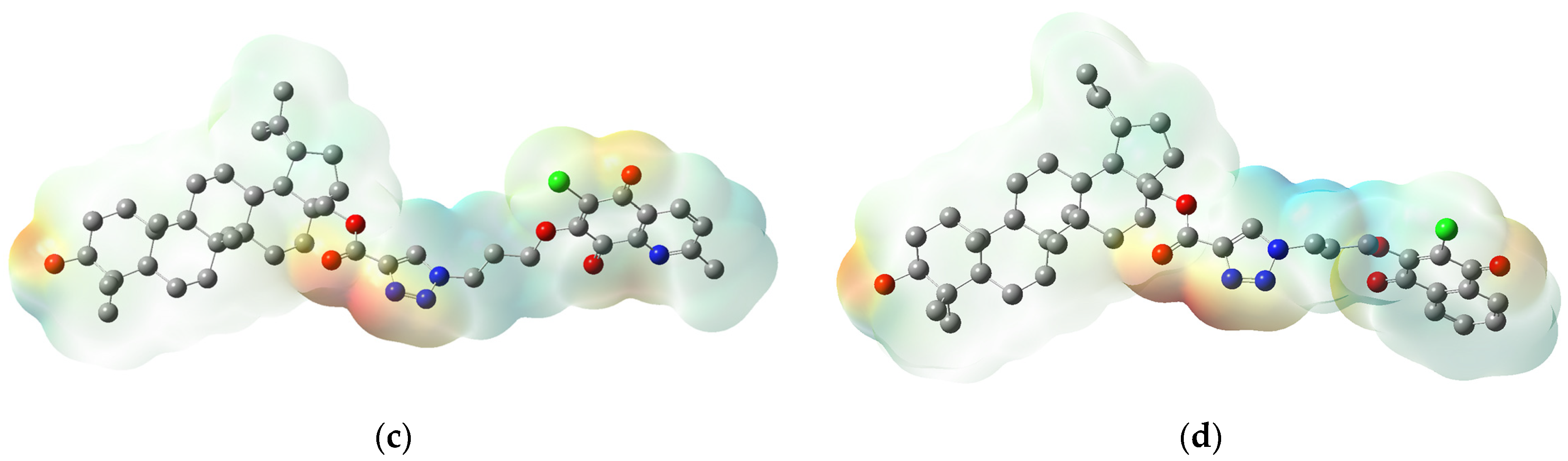
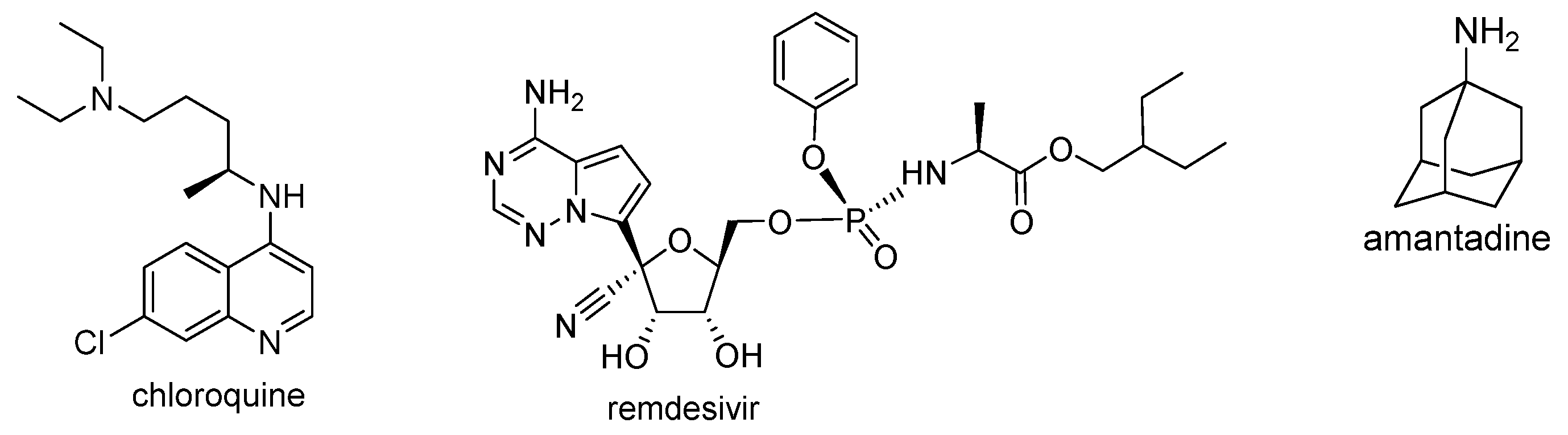
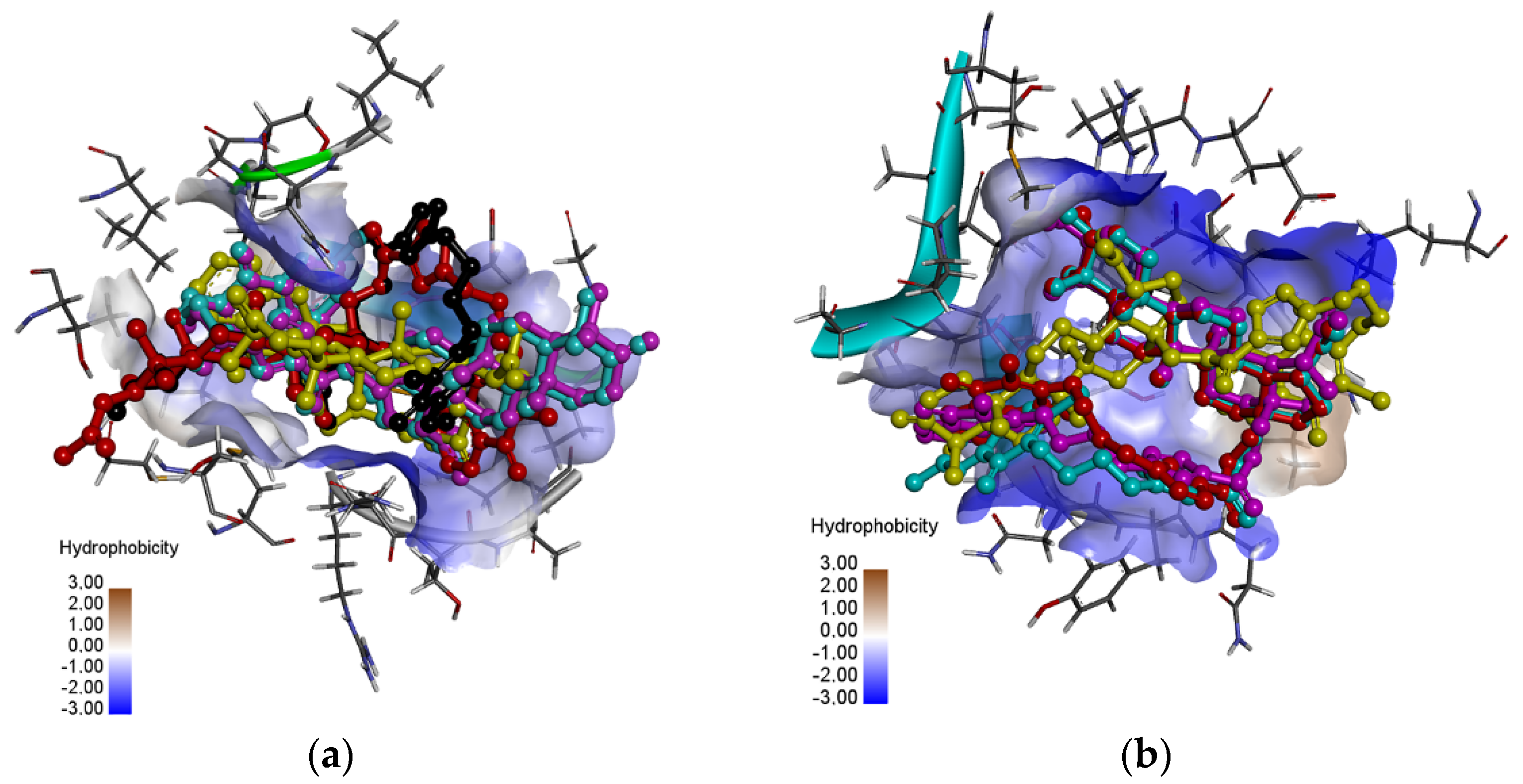
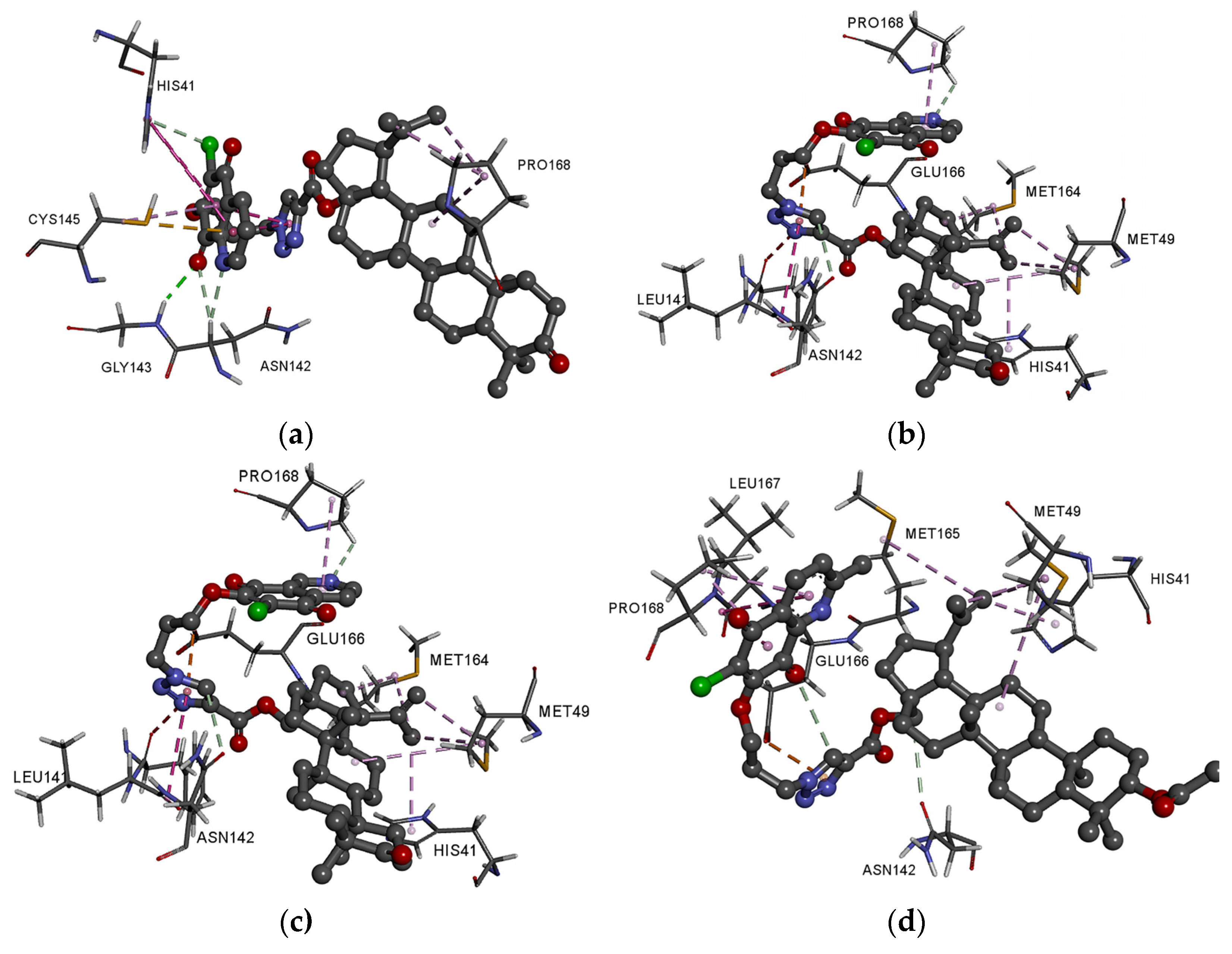
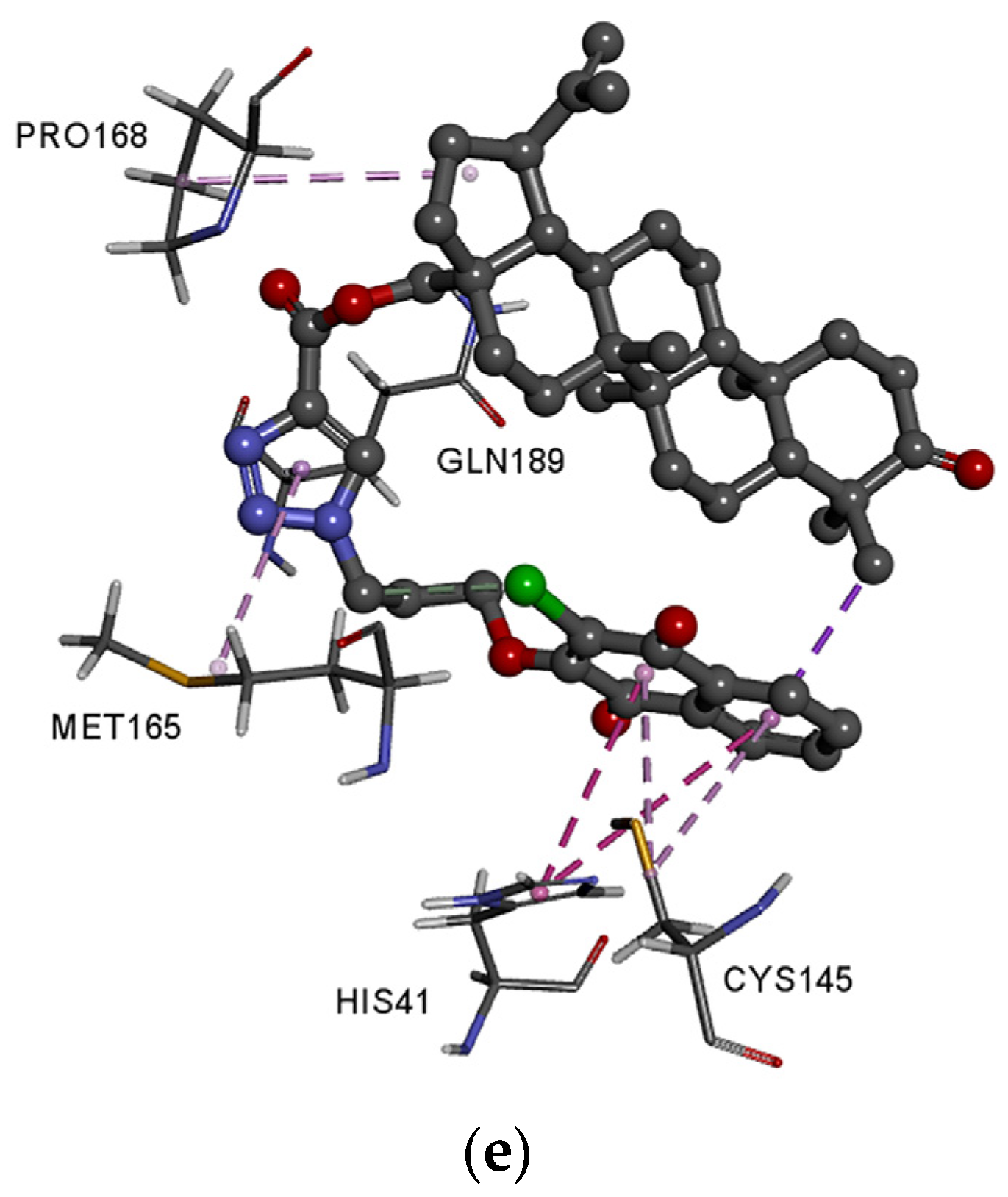
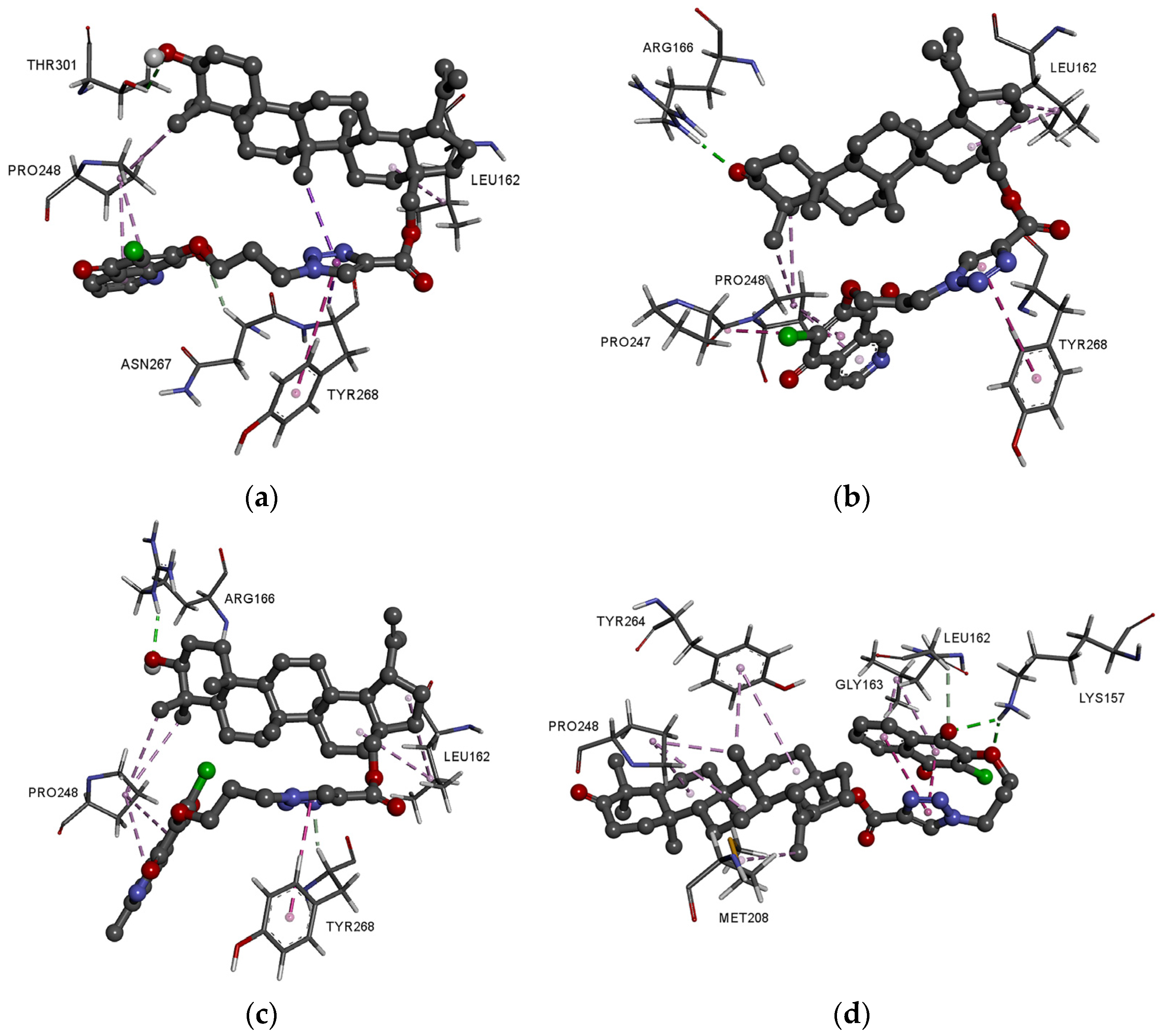
3.4. Molecular Docking Study
(r = 0.788, r2 = 0.621, SD = 0.281, F = 6.559, p = 0.007)
(r = 0.844, r2 = 0.712, SD = 0.135, F = 9.894, p = 0.001)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arnott, J.; Planey, S. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discov. 2021, 7, 863–875. [Google Scholar] [CrossRef]
- Hammoudi, N.-E.-H.; Sobhi, W.; Attoui, A.; Lemaoui, T.; Erto, A.; Benguerba, Y. In silico drug discovery of Acetylcholinesterase and Butyrylcholinesterase enzymes inhibitors based on Quantitative Structure-Activity Relationship (QSAR) and drug-likeness evaluation. J. Mol. Struct. 2021, 1229, 129845. [Google Scholar] [CrossRef]
- Mougin, J.; Bourgaux, C.; Couvreur, P. Elongated self-assembled nanocarriers: From molecular organization to therapeutic applications. Adv. Drug Deliv. Rev. 2021, 172, 127–147. [Google Scholar] [CrossRef] [PubMed]
- Duggal, D. Role of nanotechnology in new drug delivery systems. Int. J. Drug Dev. Res. 2011, 4, 4–8. [Google Scholar]
- Kovačević, S.; Banjac, M.; Milošević, N.; Ćurčić, J.; Marjanović, D.; Todorović, N.; Krmar, J.; Podunavac-Kuzmanović, S.; Banjac, N.; Ušćumlić, G. Comparative chemometric and quantitative structure-retention relationship analysis of aniso-tropic lipophilicity of 1-arylsuccinimide derivatives determined in high-performance thin-layer chromatography system with aprotic solvents. J. Chromatogr. A 2020, 1628, 461439. [Google Scholar] [CrossRef] [PubMed]
- Tshepelevitsh, S.; Kadam, S.A.; Darnell, A.; Bobacka, J.; Rüütel, A.; Haljasorg, T.; Leito, I. LogP determination for highly lipophilic hydrogen-bonding anion receptor molecules. Anal. Chim. Acta 2020, 1132, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Kulig, K.; Malawska, B. RP-TLC determination of the lipophilicity of 1-substituted pyrrolidin-2-one derivatives. Correlation of lipophilicity with affinity for α-adrenoceptors. JPC J. Planar Chromatogr. Mod. TLC 2009, 22, 141–144. [Google Scholar] [CrossRef]
- Bolzán, A.D.; Bianchi, M.S. Genotoxicity of streptonigrin: A review. Mutat. Res. Rev. Mutat. Res. 2001, 488, 25–37. [Google Scholar] [CrossRef]
- Boger, D.L.; Yasuda, M.; Mitscher, L.A.; Drake, S.D.; Kitos, P.A.; Thompson, S.C. Streptonigrin and lavendamycin partial structures. Probes for the minimum, potent pharmacophore of streptonigrin, lavendamycin, and synthetic quinoline-5,8-diones. J. Med. Chem. 1987, 30, 1918–1928. [Google Scholar] [CrossRef]
- Kadela-Tomanek, M.; Bębenek, E.; Chrobak, E.; Boryczka, S. 5,8-Quinolinedione Scaffold as a Promising Moiety of Bioactive Agents. Molecules 2019, 24, 4115. [Google Scholar] [CrossRef] [PubMed]
- Kadela-Tomanek, M.; Jastrzębska, M.; Chrobak, E.; Bębenek, E.; Latocha, M.; Kusz, J.; Boryczka, S. Structural and spectral characterisation of 2-amino-2H-[1,2,3]triazolo[4,5-g]quinoline-4,9-dione polymorphs. Cytotoxic activity and molecu-lardocking study with NQO1 enzyme. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2020, 230, 118038–118050. [Google Scholar] [CrossRef] [PubMed]
- Kadela-Tomanek, M.; Bębenek, E.; Chrobak, E.; Latocha, M.; Boryczka, S. Alkoxy and enediyne eerivatives containing 1,4-benzoquinone subunits-synthesis and antitumor activity. Molecules 2017, 22, 447. [Google Scholar] [CrossRef] [PubMed]
- Kadela-Tomanek, M.; Jastrzębska, M.; Bębenek, E.; Chrobak, E.; Latocha, M.; Kusz, J.; Tarnawska, D.; Boryczka, S. New Acetylenic Amine Derivatives of 5,8-Quinolinediones: Synthesis, Crystal Structure and Antiproliferative Activity. Crystals 2017, 7, 15. [Google Scholar] [CrossRef]
- Kadela-Tomanek, M.; Bębenek, E.; Chrobak, E.; Marciniec, K.; Latocha, M.; Kuśmierz, D.; Jastrzębska, M.; Boryczka, S. Betulin-1,4-quinone hybrids: Synthesis, anticancer activity and molecular docking study with NQO1 enzyme. Eur. J. Med. Chem. 2019, 177, 302–315. [Google Scholar] [CrossRef]
- Bayrak, N. Novel azanaphtoquinone compounds with aromatic amino moiety: Synthesis, structural characterization, and antimicrobial features. J. Mol. Struct. 2019, 1195, 411–416. [Google Scholar] [CrossRef]
- Gholampour, M.; Sakhteman, A.; Pirhadi, S.; Seradj, H. In silico design of novel diamino-quinoline-5,8-dione derivatives as putative inhibitors of NAD(P)H:Quinone oxidoreductase 1 based on docking studies and molecular dynamics simulations. J. Mol. Struct. 2021, 1230, 129906. [Google Scholar] [CrossRef]
- Ling, Y.; Yang, Q.-X.; Teng, Y.-N.; Chen, S.; Gao, W.-J.; Guo, J.; Hsu, P.-L.; Liu, Y.; Morris-Natschke, S.L.; Hung, C.-C.; et al. Development of novel amino-quinoline-5,8-dione derivatives as NAD(P)H:quinone oxidoreductase 1 (NQO1) inhibitors with potent antiproliferative activities. Eur. J. Med. Chem. 2018, 154, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Kadela-Tomanek, M.; Marciniec, K.; Jastrzębska, M.; Bębenek, E.; Chrobak, E.; Boryczka, S. Spectroscopic investigations, computational analysis and molecular docking to SARS-Co-2 targets studies of 5,8-quinolinedione attached to betulin de-rivatives. Crystals 2021, 11, 76. [Google Scholar] [CrossRef]
- Kadela-Tomanek, M.; Pawełczak, B.; Jastrzebska, M.; Bębenek, E.; Chrobak, E.; Latocha, M.; Kusz, J.; Książek, M.; Boryczka, S. Structural, vibrational and quantum chemical investigations for 6,7-dichloro-2-methyl-5,8-quinolinedione. Cytotoxic and molecular docking studies. J. Mol. Struct. 2018, 1168, 73–83. [Google Scholar] [CrossRef]
- Kadela-Tomanek, M.; Bober, K.; Bębenek, E.; Chrobak, E.; Boryczka, S. Application of thin-layer chromatography to evaluate the lipophilicity of 5,8-quinolinedione compounds. J. Planar Chromatogr. Mod. TLC 2017, 30, 219–224. [Google Scholar] [CrossRef]
- Kadela-Tomanek, M.; Jastrzębska, M.; Chrobak, E.; Bębenek, E.; Boryczka, S. Chromatographic and Computational Screening of Lipophilicity and Pharmacokinetics of Newly Synthesized Betulin-1,4-quinone Hybrids. Processes 2021, 9, 376. [Google Scholar] [CrossRef]
- Ezeokonkwo, M.A.; Ibeanu, F.; Eze, C.; Ibezim, A.; Ezeokoye, C.; Ezenwa, O.; Ezeoka, T.; Vincent, O. Synthesis, antimi-crobial activity and molecular docking studies of 7-bromoquinoline-5,8-dione containing aryl sulphonamides. Int. J. Appl. Chem. 2019, 15, 99–112. [Google Scholar]
- Musioł, R.; Jampiledek, J.; Podeszwa, B.; Finster, J.; Tabak, D.; Dohnal, J.; Polański, J. RP-HPLC determination of lipo-philicity in series of quinoline derivatives. Cent. Eur. J. Chem. 2009, 7, 586–597. [Google Scholar]
- Atia, A.; Alrawaiq, N.; Abdullah, A. A review of NAD(P)H: Quinone oxidoreductase 1 (NQO1); a multifunctional anti-oxidant enzyme. J. Appl. Pharm. Sci. 2014, 4, 118–122. [Google Scholar]
- Valdés, K.; Morales, J.; Rodríguez, L.; Günther, G. Potential use of nanocarriers with pentacyclic triterpenes in cancer treatments. Nanomedicine 2016, 11, 3139–3156. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, B.; Al Hoque, A.; Dutta, D.; Paul, B.; Mukherjee, A.; Mallick, S. Nanoformulated Drug Delivery of Potential Betulinic Acid Derivatives: A Promising Approach Toward Cancer Therapy. In Nanomedicine for Bioactives; Springer: New York, NY, USA, 2020; pp. 127–153. [Google Scholar]
- Kazakova, O.B.; Smirnova, I.E.; Baltina, L.A.; Boreko, E.I.; Savinova, O.V.; Pokrovskii, A.G. Antiviral Activity of Acyl Derivatives of Betulin and Betulinic and Dihydroquinopimaric Acids. Russ. J. Bioorgan. Chem. 2018, 44, 740–744. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, C.-H.; Morris-Natschke, S.L.; Lee, K.-H. Design, synthesis, and structure activity relationship analysis of new betulinic acid derivatives as potent HIV inhibitors. Eur. J. Med. Chem. 2021, 215, 113287. [Google Scholar] [CrossRef] [PubMed]
- Karagöz, A.Ç.; Leidenberger, M.; Hahn, F.; Hampel, F.; Friedrich, O.; Marschall, M.; Kappes, B.; Tsogoeva, S.B. Synthesis of new betulinic acid/betulin-derived dimers and hybrids with potent antimalarial and antiviral activities. Bioorgan. Med. Chem. 2019, 27, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Bębenek, E.; Jastrzębska, M.; Kadela-Tomanek, M.; Chrobak, E.; Orzechowska, B.; Zwolińska, K.; Latocha, M.; Mertas, A.; Czuba, Z.; Boryczka, S. Novel Triazole Hybrids of Betulin: Synthesis and Biological Activity Profile. Molecules 2017, 22, 1876. [Google Scholar] [CrossRef] [PubMed]
- Amiri, S.; Dastghaib, S.; Ahmadi, M.; Mehrbod, P.; Khadem, F.; Behrouj, H.; Aghanoori, M.; Machaj, F.; Ghamsari, M.; Rosik, J.; et al. Betulin and its de-rivatives as novel compounds with different pharmacological effects. Biotechnol. Adv. 2020, 38, 107409. [Google Scholar] [CrossRef]
- Zhang, D.-H.; Wu, K.-L.; Zhang, X.; Deng, S.-Q.; Peng, B. In silico screening of Chinese herbal medicines with the potential to directly inhibit 2019 novel coronavirus. J. Integr. Med. 2020, 18, 152–158. [Google Scholar] [CrossRef]
- Wen, C.-C.; Kuo, Y.-H.; Jan, J.-T.; Liang, P.-H.; Wang, S.-Y.; Liu, H.-G.; Lee, C.-K.; Chang, S.-T.; Kuo, C.-J.; Lee, S.-S.; et al. Specific Plant Terpenoids and Lignoids Possess Potent Antiviral Activities against Severe Acute Respiratory Syndrome Coronavirus. J. Med. Chem. 2007, 50, 4087–4095. [Google Scholar] [CrossRef] [PubMed]
- Kadioglu, O.; Saeed, M.; Greten, H.J.; Efferth, T. Identification of novel compounds against three targets of SARS CoV-2 coronavirus by combined virtual screening and supervised machine learning. Comput. Biol. Med. 2021, 133, 104359. [Google Scholar] [CrossRef] [PubMed]
- Marciniec, K.; Chrobak, E.; Dąbrowska, A.; Bębenek, E.; Kadela-Tomanek, M.; Pęcak, P.; Boryczka, S. Phosphate deriva-tives of 3-carboxyacylbetulin: Synthesis, in vitro anti-HIV and molecular docking study. Biomoleclues 2020, 10, 1148. [Google Scholar] [CrossRef]
- Kadela-Tomanek, M.; Jastrzębska, M.; Marciniec, K.; Chrobak, E.; Bębenek, E.; Latocha, M.; Kuśmierz, D.; Boryczka, S. Design, synthesis and biological activity of 1,4-quinone moiety attached to betulin derivatives as potent DT-diaphorase substrate. Bioorganic Chem. 2021, 106, 104478. [Google Scholar] [CrossRef] [PubMed]
- Bębenek, E.; Bober-Majnusz, K.; Siudak, S.; Chrobak, E.; Kadela-Tomanek, M.; Wietrzyk, J.; Boryczka, S. Application of TLC to Evaluate the Lipophilicity of Newly Synthesized Betulin Derivatives. J. Chromatogr. Sci. 2020, 58, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Virtual Computational Chemistry Laboratory. Available online: http://www.vcclab.org/lab/alogps/ (accessed on 24 April 2021).
- Molinspiration. Available online: https://www.molinspiration.com/services/logp.html (accessed on 24 April 2021).
- SwissADME. Available online: http://www.swissadme.ch/index.php (accessed on 24 April 2021).
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- pkCSM. Available online: http://biosig.unimelb.edu.au/pkcsm/prediction# (accessed on 24 April 2021).
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16, Revision A. 03. Wallingford 2016, 1, 3. [Google Scholar]
- Politzer, P.; Laurence, P.; Jayasuriya, K. Molecular electrostatic potentials: An effective tool for the elucidation of bio-chemical phenomena. Environ. Health Perspect. 1985, 61, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.; Keith, T.; Millam, J. GaussView Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- PerkinElmer ChemOffice Suite. Available online: https//www.perkinelmer.com (accessed on 24 April 2021).
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [PubMed]
- Dessault Systemes BIOVIA. Available online: https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/ (accessed on 24 April 2021).
- Mannhold, R.; Cruciani, G.; Dross, K.; Rekker, R. Multivariate analysis of experimental and computational descriptors of molecular lipophilicity. J. Comput. Mol. Des. 1998, 12, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Bodor, N.; Gabanyi, Z.; Wong, C.K. A new method for the estimation of partition coefficient. J. Am. Chem. Soc. 1989, 11, 3783–3786. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Šegan, S.; Penjišević, J.; Šukalović, V.; Andrić, D.; Milojković-Opsenica, D.; Kostić-Rajačić, S. Investigation of lipophilicity and pharmacokinetic properties of 2-(methoxy)phenylpiperazine dopamine D2 ligands. J. Chromatogr. B 2019, 1124, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.W.; Dress, K.R.; Edwards, M. Using the Golden Triangle to optimize clearance and oral absorption. Bioorganic Med. Chem. Lett. 2009, 19, 5560–5564. [Google Scholar] [CrossRef]
- Kulkarni, A.; Han, Y.; Hopfinger, A. Predicting Caco-2 cell permeation coefficients of organic molecules using mem-brane-interaction QSAR analysis. J. Chem. Inf. Comput. Sci. 2002, 42, 331–342. [Google Scholar] [CrossRef]
- Feher, M.; Schmidt, J.M. Property Distributions: Differences between Drugs, Natural Products, and Molecules from Combinatorial Chemistry. J. Chem. Inf. Comput. Sci. 2002, 43, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Hutter, M.C. Prediction of blood–brain barrier permeation using quantum chemically derived information. J. Comput. Mol. Des. 2003, 17, 415–443. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, M.; Karabacak, M.; Periandy, S.; Tanuja, D. Spectroscopic (FT-IR, FT-Raman, UV and NMR) investigation and NLO, HOMO–LUMO, NBO analysis of organic 2,4,5-trichloroaniline. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 97, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Saini, V.; Maurya, I.K.; Sindhu, J.; Kumari, M.; Kataria, R.; Kumar, V. Design, synthesis, DFT, docking studies and ADME prediction of some new coumarinyl linked pyrazolylthiazoles: Potential standalone or adjuvant antimicrobial agents. PLoS ONE 2018, 13, e0196016. [Google Scholar] [CrossRef] [PubMed]
- Singha, P.; Islama, S.; Ahmada, H.; Prabaharan, A. Spectroscopic investigation (FT-IR, FT-Raman), HOMO-LUMO, NBO, and molecular docking analysis of N-ethyl-N-nitrosourea, a potential anticancer agent. J. Mol. Struct. 2018, 1154, 39–50. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, Y.; Ren, Q.; Yu, D.; Lu, H. Synthesis, crystal structure and DFT study of a novel compound N-(4-(2,4-dimorpholinopyrido[2,3-d]pyrimidin-6-yl)phenyl)pyrrolidine-1-carboxamide. J. Mol. Struct. 2021, 1235, 130261. [Google Scholar] [CrossRef]
- Mary, Y.S.; Mary, Y.S.; Krátký, M.; Vinsova, J.; Baraldi, C.; Gamberin, M.C. DFT, molecular docking and SERS (concen-tration and solvent dependant) investigations of a methylisoxazole derivative with potential antimicrobial activity. J. Mol. Struct. 2021, 1232, 130034. [Google Scholar] [CrossRef]
- Chandrasekaran, K.; Kumar, R.T. Structural, spectral, thermodynamical, NLO, HOMO, LUMO and NBO analysis of fluconazole. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 150, 974–991. [Google Scholar] [CrossRef]
- World Health Organization. WHO Coronavirus Disease (COVID-19) Dashboard. 2020. Available online: https://covid19.who.int/ (accessed on 3 March 2021).
- Oscanoa, T.; Romero-Ortuno, R.; Carvajal, A.; Savarino, A. A pharmacological perspective of chloroquine in SARS-CoV-2 infection: An old drug for the fight against a new coronavirus? Int. J. Antimicrob. Agents 2020, 56, 106078. [Google Scholar] [CrossRef]
- Ko, W.; Rolain, J.; Lee, N.; Chen, P.; Huang, C.; Lee, P.; Hsueh, P. Arguments in favour of remdesivir for treating SARS-CoV-2 infections. Int. J. Antimicrob. Agents 2020, 55, 105933. [Google Scholar] [CrossRef]
- Borra, A. Does amantadine have a protective effect against COVID-19? Neurol. Neurochir. Pol. 2020, 54, 284–285. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Arya, R.; Das, A.; Prashar, V.; Kumar, M. Potential inhibitors against papain-like protease of novel coronavirus (SARS-CoV-2) from FDA approved drugs. ChemRxiv 2020, 1–10. [Google Scholar] [CrossRef]
- Oashi, T.; Ringer, A.L.; Raman, E.P.; MacKerell, A.D. Automated Selection of Compounds with Physicochemical Properties to Maximize Bioavailability and Druglikeness. J. Chem. Inf. Model. 2010, 51, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Douguet, D.; Miteva, M.A.; Villoutreix, B.O. Computational analysis of calculated physicochemical and ADMET properties of protein-protein interaction inhibitors. Sci. Rep. 2017, 7, srep46277. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Chemical Structure | Compound | Chemical Structure |
|---|---|---|---|
| 1 |  | 2 |  |
| 3 |  | 4 |  |
| 5 |  | 6 |  |
| 7 |  | 8 |  |
| 9 |  | 10 |  |
| 11 |  | 12 |  |
| 13 |  | 14 |  |
| 15 |  | 16 |  |
| 17 |  | 18 |  |
| 19 |  | 20 |  |
| Compound | RM0 | b | r | SD |
|---|---|---|---|---|
| 1 | 3.51 | −0.04 | 0.995 | 0.051 |
| 2 | 3.64 | −0.04 | 0.992 | 0.067 |
| 3 | 3.97 | −0.05 | 0.996 | 0.047 |
| 4 | 4.08 | −0.05 | 0.992 | 0.072 |
| 5 | 3.56 | −0.04 | 0.983 | 0.100 |
| 6 | 3.59 | −0.04 | 0.997 | 0.041 |
| 7 | 4.07 | −0.05 | 0.985 | 0.101 |
| 8 | 4.24 | −0.05 | 0.993 | 0.065 |
| 9 | 3.65 | −0.04 | 0.996 | 0.047 |
| 10 | 3.77 | −0.04 | 0.996 | 0.048 |
| 11 | 4.12 | −0.05 | 0.995 | 0.054 |
| 12 | 4.39 | −0.05 | 0.981 | 0.115 |
| 13 | 3.78 | −0.04 | 0.998 | 0.028 |
| 14 | 3.84 | −0.04 | 0.992 | 0.067 |
| 15 | 4.21 | −0.05 | 0.979 | 0.120 |
| 16 | 4.58 | −0.05 | 0.960 | 0.183 |
| 17 | 4.40 | −0.05 | 0.995 | 0.065 |
| 18 | 4.54 | −0.05 | 0.989 | 0.098 |
| 19 | 5.07 | −0.06 | 0.995 | 0.073 |
| 20 | 5.26 | −0.06 | 0.976 | 0.161 |
| Compound | logPlit | RM0 | b | r | SD | logPTLC |
|---|---|---|---|---|---|---|
| Acetanilide | 1.21 | 0.55 | −0.01 | 0.954 | 0.051 | 1.19 |
| Prednisone | 1.63 | 0.73 | −0.02 | 0.932 | 0.077 | 1.39 |
| 4-Bromoacetophenone | 2.43 | 1.89 | −0.03 | 0.993 | 0.037 | 2.72 |
| Benzophenone | 3.18 | 2.37 | −0.03 | 0.993 | 0.046 | 3.26 |
| Anthracene | 4.45 | 3.48 | −0.04 | 0.994 | 0.053 | 4.52 |
| Dibenzyl | 4.79 | 3.65 | −0.04 | 0.997 | 0.043 | 4.72 |
| DDT | 6.01 | 4.87 | −0.06 | 0.985 | 0.041 | 6.17 |
| 9-Phenylanthracene | 6.38 | 4.93 | −0.06 | 0.993 | 0.080 | 6.11 |
| Program | Correlation Equation | r | SD |
|---|---|---|---|
| Compounds 1–16 | |||
| ALOGPs | logPTLC = 1.094 logPCALC − 2.416 | 0.932 | 0.137 |
| AClogP | logPTLC = 0.540 logPCALC + 1.104 | 0.854 | 0.196 |
| AlogP | logPTLC = 0.492 logPCALC + 0.987 | 0.843 | 0.203 |
| XLOGP2 | logPTLC = 0.378 logPCALC + 1.268 | 0.854 | 0.196 |
| XLOGP3 | logPTLC = 0.472 logPCALC − 0.121 | 0.887 | 0.174 |
| milogP | logPTLC = 0.527 logPCALC + 0.549 | 0.729 | 0.258 |
| iLOGP | logPTLC = 0.758 logPCALC + 0.620 | 0.887 | 0.174 |
| WLOGP | logPTLC = 0.755 logPCALC − 2.091 | 0.953 | 0.114 |
| MLOGP | logPTLC = 0.582 logPCALC + 2.353 | 0.723 | 0.260 |
| SILICOS-IT | logPTLC = 0.634 logPCALC − 0.443 | 0.798 | 0.227 |
| Compounds 17–20 | |||
| ALOGPs | logPTLC = 1.313 logPCALC − 1.485 | 0.980 | 0.114 |
| AClogP | logPTLC = 1.078 logPCALC − 0.058 | 0.962 | 0.157 |
| AlogP | logPTLC = 0.847 logPCALC + 0.251 | 0.962 | 0.232 |
| XLOGP2 | logPTLC = 0.567 logPCALC + 1.462 | 0.880 | 0.273 |
| XLOGP3 | logPTLC = 0.718 logPCALC − 0.097 | 0.929 | 0.213 |
| milogP | logPTLC = 0.766 logPCALC + 0.345 | 0.960 | 0.160 |
| iLOGP | logPTLC = 0.994 logPCALC + 0.916 | 0.924 | 0.220 |
| WLOGP | logPTLC = 1.083 logPCALC − 1.828 | 0.979 | 0.116 |
| MLOGP | logPTLC = 1.754 logPCALC − 3.543 | 0.955 | 0.170 |
| SILICOS-IT | logPTLC = 0.965 logPCALC − 0.092 | 0.815 | 0.332 |
| Compound | MW (g/mol) | HA | HD | RB | TPSA (Å) |
|---|---|---|---|---|---|
| 1 | 787.42 | 10 | 1 | 9 | 133.50 |
| 2 | 785.41 | 10 | 0 | 9 | 130.34 |
| 3 | 829.46 | 11 | 0 | 10 | 139.57 |
| 4 | 843.49 | 11 | 0 | 11 | 139.57 |
| 5 | 787.42 | 10 | 1 | 9 | 133.50 |
| 6 | 785.41 | 10 | 0 | 9 | 130.45 |
| 7 | 829.46 | 11 | 0 | 10 | 139.57 |
| 8 | 843.39 | 11 | 0 | 11 | 139.57 |
| 9 | 801.45 | 10 | 1 | 9 | 133.50 |
| 10 | 799.43 | 10 | 0 | 9 | 130.34 |
| 11 | 843.49 | 11 | 0 | 10 | 139.57 |
| 12 | 857.51 | 11 | 0 | 11 | 139.57 |
| 13 | 786.44 | 9 | 1 | 9 | 120.61 |
| 14 | 784.42 | 9 | 0 | 9 | 117.45 |
| 15 | 828.47 | 10 | 0 | 10 | 126.68 |
| 16 | 842.50 | 10 | 0 | 11 | 126.68 |
| Compound | logBB | logPS | logKp | logPapp | HIA |
|---|---|---|---|---|---|
| 1 | −1.459 | −2.481 | −2.734 | 0.641 | 97.226 |
| 2 | −1.581 | −2.513 | −2.735 | 0.639 | 98.193 |
| 3 | −1.804 | −2.442 | −2.735 | 0.195 | 98.510 |
| 4 | −1.831 | −2.434 | −2.735 | 0.215 | 98.358 |
| 5 | −1.457 | −2.466 | −2.735 | 0.672 | 97.226 |
| 6 | −1.579 | −2.499 | −2.735 | 0.671 | 98.193 |
| 7 | −1.802 | −2.428 | −2.735 | 0.158 | 98.510 |
| 8 | −1.830 | −2.420 | −2.735 | 0.178 | 98.358 |
| 9 | −1.447 | −2.443 | −2.734 | 0.598 | 97.235 |
| 10 | −1.569 | −2.475 | −2.735 | 0.597 | 98.090 |
| 11 | −1.792 | −2.404 | −2.735 | 0.181 | 98.358 |
| 12 | −1.820 | −2.396 | −2.735 | 0.200 | 98.231 |
| 13 | −1.219 | −2.215 | −2.734 | 0.747 | 97.410 |
| 14 | −1.341 | −2.248 | −2.735 | 0.745 | 97.838 |
| 15 | −1.564 | −2.177 | −2.735 | 0.733 | 97.885 |
| 16 | −1.592 | −2.169 | −2.735 | 0.728 | 97.879 |
| Hybrid | EHOMO (kcal/mol) | ELUMO (kcal/mol) | ΔE (kcal/mol) | I (kcal/mol) | A (kcal/mol) | η (kcal/mol) | µ (kcal/mol) | χ (kcal/mol) | ω (kcal/mol) | DM (D) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | −6.227 | −3.918 | −2.310 | 6.227 | 3.918 | 1.155 | −5.073 | 5.073 | 11.140 | 5.6507 |
| 2 | −6.510 | −3.989 | −2.521 | 6.510 | 3.989 | 1.261 | −5.250 | 5.250 | 10.930 | 9.563 |
| 3 | −6.423 | −3.980 | −2.443 | 6.423 | 3.980 | 1.222 | −5.202 | 5.202 | 11.076 | 6.3035 |
| 4 | −6.422 | −3.984 | −2.438 | 6.422 | 3.984 | 1.219 | −5.203 | 5.203 | 11.106 | 6.5282 |
| 5 | −6.503 | −3.816 | −2.687 | 6.503 | 3.816 | 1.344 | −5.159 | 5.159 | 9.906 | 5.8305 |
| 6 | −6.228 | −3.869 | −2.360 | 6.228 | 3.869 | 1.180 | −5.049 | 5.049 | 10.801 | 8.9646 |
| 7 | −6.421 | −3.867 | −2.554 | 6.421 | 3.867 | 1.277 | −5.144 | 5.144 | 10.359 | 6.548 |
| 8 | −6.426 | −3.866 | −2.560 | 6.426 | 3.866 | 1.280 | −5.146 | 5.146 | 10.345 | 6.4041 |
| 9 | −6.499 | −4.151 | −2.348 | 6.499 | 4.151 | 1.174 | −5.325 | 5.325 | 12.078 | 6.7295 |
| 10 | −6.248 | −4.183 | −2.065 | 6.248 | 4.183 | 1.032 | −5.216 | 5.216 | 13.176 | 7.2985 |
| 11 | −6.450 | −4.162 | −2.288 | 6.450 | 4.162 | 1.144 | −5.306 | 5.306 | 12.306 | 4.5204 |
| 12 | −6.450 | −4.165 | −2.285 | 6.450 | 4.165 | 1.142 | −5.307 | 5.307 | 12.328 | 4.6037 |
| 13 | −6.485 | −3.786 | −2.699 | 6.485 | 3.786 | 1.349 | −5.135 | 5.135 | 9.772 | 7.0345 |
| 14 | −6.239 | −3.816 | −2.424 | 6.239 | 3.816 | 1.212 | −5.027 | 5.027 | 10.428 | 8.3989 |
| 15 | −6.428 | −3.792 | −2.637 | 6.428 | 3.792 | 1.318 | −5.110 | 5.110 | 9.903 | 5.0970 |
| 16 | −6.442 | −3.793 | −2.649 | 6.442 | 3.793 | 1.325 | −5.117 | 5.117 | 9.885 | 5.6097 |
| Compound | Mpro | PLpro | Compound | Mpro | PLpro | ||||
|---|---|---|---|---|---|---|---|---|---|
| ΔG | pKI | ΔG | pKI | ΔG | pKI | ΔG | pKI | ||
| 1 | −8.8 | 6.46 | −8.3 | 6.09 | 11 | −9.3 | 6.82 | −6.4 | 4.70 |
| 2 | −8.9 | 6.53 | −6.9 | 5.06 | 12 | −9.1 | 6.68 | −7.0 | 5.14 |
| 3 | −8.6 | 6.31 | −6.5 | 4.77 | 13 | −8.6 | 6.31 | −7.6 | 5.58 |
| 4 | −7.8 | 5.72 | −6.5 | 4.77 | 14 | −8.6 | 6.31 | −8.2 | 6.02 |
| 5 | −8.8 | 6.46 | −7.9 | 5.80 | 15 | −8.6 | 6.31 | −6.4 | 4.70 |
| 6 | −8.9 | 6.53 | −8.1 | 5.94 | 16 | −8.4 | 6.16 | −7.0 | 5.14 |
| 7 | −8.0 | 5.87 | −6.9 | 5.06 | Chloroquine | −5.7 | 4.18 | −5.2 | 3.82 |
| 8 | −8.5 | 6.24 | −6.4 | 4.70 | Remdesivir | −7.4 | 5.43 | −5.7 | 4.18 |
| 9 | −9.1 | 6.68 | −8.1 | 5.94 | Amantadine | −4.5 | 3.30 | −4.1 | 3.01 |
| 10 | −9.2 | 6.75 | −6.8 | 4.99 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadela-Tomanek, M.; Jastrzębska, M.; Marciniec, K.; Chrobak, E.; Bębenek, E.; Boryczka, S. Lipophilicity, Pharmacokinetic Properties, and Molecular Docking Study on SARS-CoV-2 Target for Betulin Triazole Derivatives with Attached 1,4-Quinone. Pharmaceutics 2021, 13, 781. https://doi.org/10.3390/pharmaceutics13060781
Kadela-Tomanek M, Jastrzębska M, Marciniec K, Chrobak E, Bębenek E, Boryczka S. Lipophilicity, Pharmacokinetic Properties, and Molecular Docking Study on SARS-CoV-2 Target for Betulin Triazole Derivatives with Attached 1,4-Quinone. Pharmaceutics. 2021; 13(6):781. https://doi.org/10.3390/pharmaceutics13060781
Chicago/Turabian StyleKadela-Tomanek, Monika, Maria Jastrzębska, Krzysztof Marciniec, Elwira Chrobak, Ewa Bębenek, and Stanisław Boryczka. 2021. "Lipophilicity, Pharmacokinetic Properties, and Molecular Docking Study on SARS-CoV-2 Target for Betulin Triazole Derivatives with Attached 1,4-Quinone" Pharmaceutics 13, no. 6: 781. https://doi.org/10.3390/pharmaceutics13060781
APA StyleKadela-Tomanek, M., Jastrzębska, M., Marciniec, K., Chrobak, E., Bębenek, E., & Boryczka, S. (2021). Lipophilicity, Pharmacokinetic Properties, and Molecular Docking Study on SARS-CoV-2 Target for Betulin Triazole Derivatives with Attached 1,4-Quinone. Pharmaceutics, 13(6), 781. https://doi.org/10.3390/pharmaceutics13060781