
Development of Physiologically Based Pharmacokinetic Model for Orally Administered Fexuprazan in Humans

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
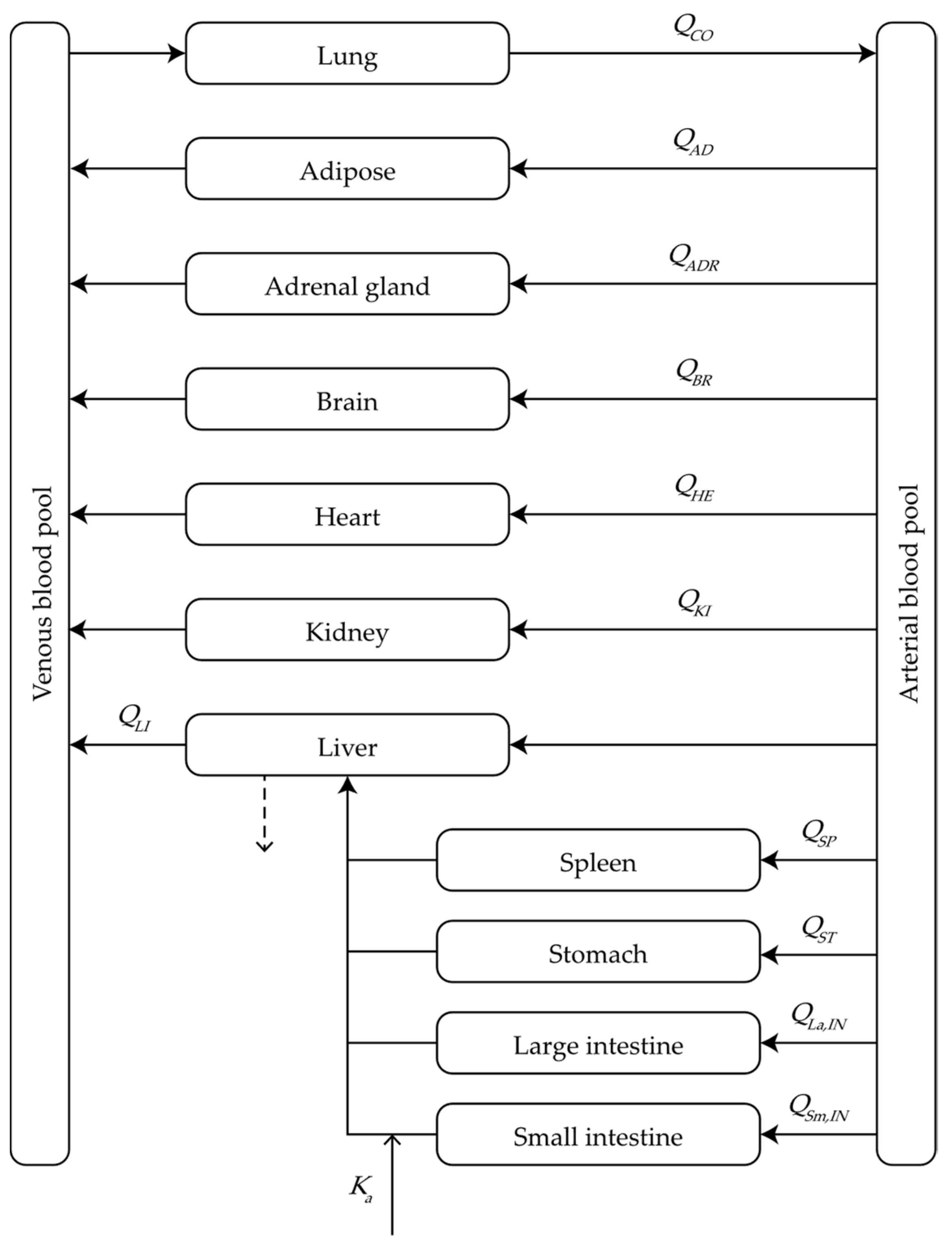
2.1. Model Structure
2.2. Absorption Kinetics
2.3. Distribution Kinetics
2.4. Elimination Kinetics
2.5. PBPK Calculations
2.6. Modelling Strategies
2.7. Statistical Analysis
3. Results
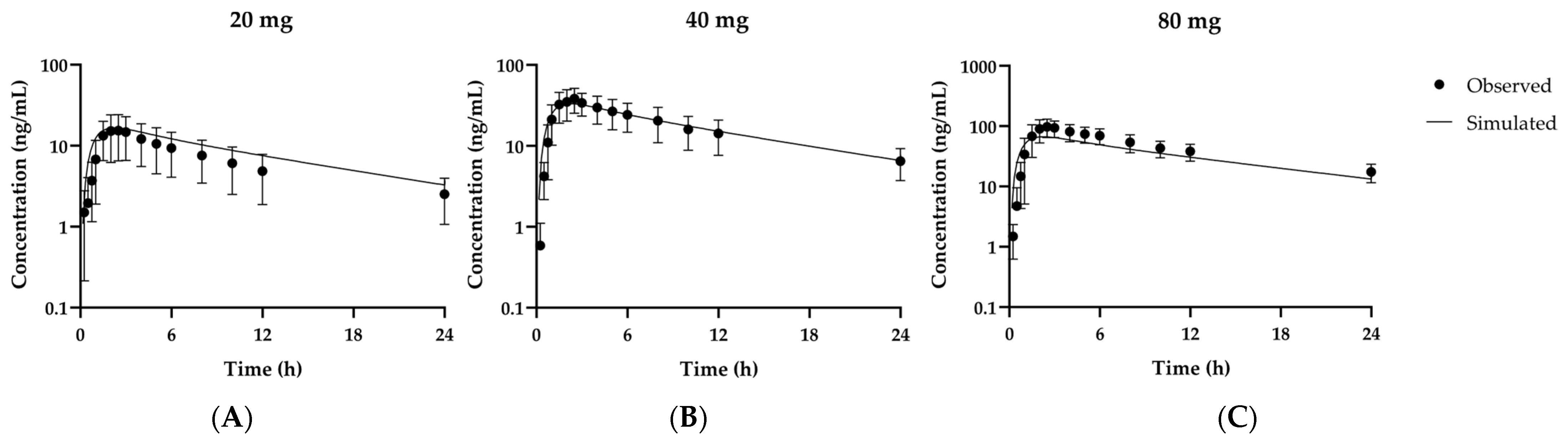
3.1. Establishment and Optimization of the PBPK Model for Fexuprazan in Humans
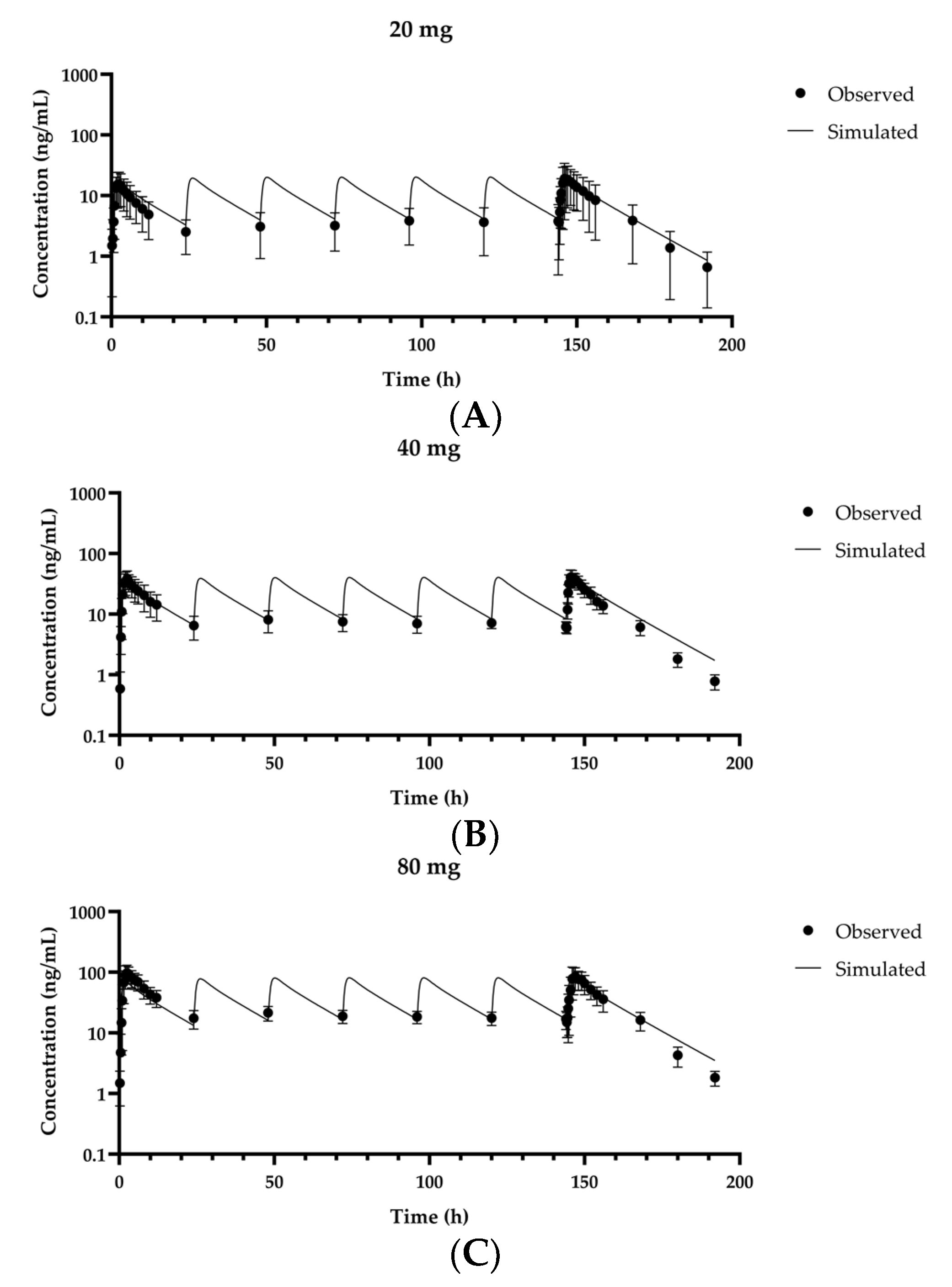
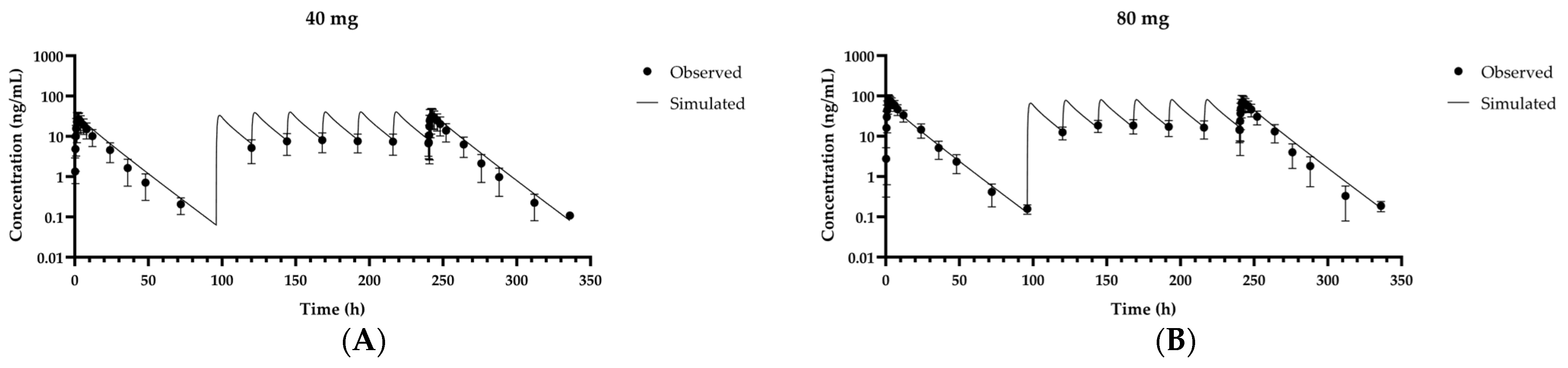
3.2. Validation of the Fexuprazan PBPK Model for Humans
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hwang, J.G.; Jeon, I.; Park, S.A.; Lee, A.; Yu, K.-S.; Jang, I.-J.; Lee, S. Pharmacodynamics and Pharmacokinetics of DWP14012 (Fexuprazan) in Healthy Subjects with Different Ethnicities. Aliment. Pharmacol. Ther. 2020, 52, 1648–1657. [Google Scholar] [CrossRef]
- Chey, W.D.; Leontiadis, G.I.; Howden, C.W.; Moss, S.F. ACG Clinical Guideline: Treatment of Helicobacter pylori Infection. Am. J. Gastroenterol. 2017, 112, 212–239. [Google Scholar] [CrossRef]
- Kahrilas, P.J. Gastroesophageal Reflux Disease. N. Engl. J. Med. 2008, 359, 1700–1707. [Google Scholar] [CrossRef]
- Kim, J.-H.; Kim, B.J.; Kim, S.W.; Kim, S.E.; Kim, Y.S.; Sung, H.Y.; Oh, T.-H.; Jeong, I.D.; Park, M.I. Current Issues on Gastroesophageal Reflux Disease. Korean J. Gastroenterol. 2014, 64, 127–132. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Ofosu, A.; Gaduputi, V. Potassium-Competitive Acid Blockers—Are They the Next Generation of Proton Pump Inhibitors? World J. Gastrointest. Pharmacol. Ther. 2018, 9, 63–68. [Google Scholar] [CrossRef]
- Hunt, R.H.; Scarpignato, C. Potent Acid Suppression with PPIs and P-CABs: What’s New? Curr. Treat. Options Gastroenterol. 2018, 16, 570–590. [Google Scholar] [CrossRef] [PubMed]
- Sugano, K. Vonoprazan Fumarate, a Novel Potassium-Competitive Acid Blocker, in the Management of Gastroesophageal Reflux Disease: Safety and Clinical Evidence to Date. Ther. Adv. Gastroenterol. 2018, 11, 1756283X17745776. [Google Scholar] [CrossRef]
- Takahashi, N.; Take, Y. Tegoprazan, a Novel Potassium-Competitive Acid Blocker to Control Gastric Acid Secretion and Motility. J. Pharmacol. Exp. Ther. 2018, 364, 275. [Google Scholar] [CrossRef]
- Sunwoo, J.; Ji, S.C.; Oh, J.; Ban, M.S.; Nam, J.Y.; Kim, B.; Song, G.S.; Yu, K.-S.; Jang, I.-J.; Lee, S. Pharmacodynamics of Tegoprazan and Revaprazan after Single and Multiple Oral Doses in Healthy Subjects. Aliment. Pharmacol. Ther. 2020, 52, 1640–1647. [Google Scholar] [CrossRef] [PubMed]
- Sunwoo, J.; Oh, J.; Moon, S.J.; Ji, S.C.; Lee, S.H.; Yu, K.S.; Kim, H.S.; Lee, A.; Jang, I.J. Safety, Tolerability, Pharmacodynamics and Pharmacokinetics of DWP14012, a Novel Potassium-Competitive Acid Blocker, in Healthy Male Subjects. Aliment. Pharmacol. Ther. 2018, 48, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Galetin, A.; Burt, H.; Gibbons, L.; Houston, J.B. Prediction of Time-Dependent CYP3A4 Drug-Drug Interactions: Impact of Enzyme Degradation, Parallel Elimination Pathways, and Intestinal Inhibition. Drug Metab. Dispos. 2006, 34, 166. [Google Scholar] [CrossRef]
- Pal, D.; Mitra, A.K. MDR- and CYP3A4-Mediated Drug–Drug Interactions. J. Neuroimmune Pharmacol. 2006, 1, 323–339. [Google Scholar] [CrossRef]
- Graham, D.Y.; Dore, M.P. Update on the Use of Vonoprazan: A Competitive Acid Blocker. Gastroenterology 2018, 154, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Yim, C.-S.; Jeong, Y.-S.; Lee, S.-Y.; Pyeon, W.; Ryu, H.-M.; Lee, J.-H.; Lee, K.-R.; Maeng, H.-J.; Chung, S.-J. Specific Inhibition of the Distribution of Lobeglitazone to the Liver by Atorvastatin in Rats: Evidence for a Rat Organic Anion Transporting Polypeptide 1B2–Mediated Interaction in Hepatic Transport. Drug Metab. Dispos. 2017, 45, 246–259. [Google Scholar] [CrossRef]
- Jamei, M.; Marciniak, S.; Feng, K.; Barnett, A.; Tucker, G.; Rostami-Hodjegan, A. The Simcyp® Population-Based ADME Simulator. Expert Opin. Drug Metab. Toxicol. 2009, 5, 211–223. [Google Scholar] [CrossRef]
- Investigator’s Brochure DWP14012, DWP14012–IB-06 ed.; Daewoong Pharmaceutical Co., Ltd.: Seoul, Korea, 2018.
- Zheng, Y.; Benet, L.Z.; Okochi, H.; Chen, X. pH Dependent but not P-gp Dependent Bidirectional Transport Study of S-propranolol: The Importance of Passive Diffusion. Pharm. Res. 2015, 32, 2516–2526. [Google Scholar] [CrossRef] [PubMed]
- Haltner-Ukomadu, E.; Gureyeva, S.; Burmaka, O.; Goy, A.; Mueller, L.; Kostyuk, G.; Margitich, V. In Vitro Bioavailability Study of an Antiviral Compound Enisamium Iodide. Sci. Pharm. 2018, 86, 3. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, J.; Wang, X.; Zeng, S. Stereoselective Transport and Uptake of Propranolol across Human Intestinal Caco-2 Cell Monolayers. Chirality 2010, 22, 361–368. [Google Scholar] [CrossRef]
- Rostami-Hodjegan, A.; Tucker, G.T. Simulation and Prediction of in Vivo Drug Metabolism in Human Populations From in Vitro Data. Nat. Rev. Drug Discov. 2007, 6, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.B.; Rostami-Hodjegan, A.; Tucker, G.T.; Yeo, K.R. Prediction of Non-Specific Hepatic Microsomal Binding from Readily Available Physicochemical Properties Available Physicochemical Properties. Drug Metab. Rev. 2006, 38, 162. [Google Scholar]
- ClinicalTrials.gov (NCT02757144): Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of DWP14012 after Oral Administration in Healthy Male Volunteers; Daewoong Pharmaceutical Co., Ltd.: Seoul, Korea, 2017.
- ClinicalTrials.gov (NCT03574415): Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of DWP14012 after Oral Administration in Healthy Japanese, Caucasian and Korean; Daewoong Pharmaceutical Co., Ltd.: Seoul, Korea, 2019.
- Brown, R.P.; Delp, M.D.; Lindstedt, S.L.; Rhomberg, L.R.; Beliles, R.P. Physiological Parameter Values for Physiologically Based Pharmacokinetic Models. Toxicol. Ind. Health 1997, 13, 407–484. [Google Scholar] [CrossRef]
- Gertz, M.; Harrison, A.; Houston, J.B.; Galetin, A. Prediction of Human Intestinal First-Pass Metabolism of 25 CYP3A Substrates from In Vitro Clearance and Permeability Data. Drug Metab. Dispos. 2010, 38, 1147. [Google Scholar] [CrossRef]
- Øie, S.; Tozer, T.N. Effect of Altered Plasma Protein Binding on Apparent Volume of Distribution. J. Pharm. Sci. 1979, 68, 1203–1205. [Google Scholar] [CrossRef]
- Billett, H.H. Clinical Methods: The History, Physical, and Laboratory Examinations, 3rd ed.; Walker, H.K., Hall, W.D., Hurst, J.W., Eds.; Butterworths: Boston, MA, USA, 1990. [Google Scholar]
- Berezhkovskiy, L.M. A Valid Equation for the Well-Stirred Perfusion Limited Physiologically Based Pharmacokinetic Model that Consistently Accounts for the Blood–Tissue Drug Distribution in the Organ and the Corresponding Valid Equation for the Steady State Volume of Distribution. J. Pharm. Sci. 2010, 99, 475–485. [Google Scholar] [CrossRef]
- Kiriyama, A.; Honbo, A.; Iga, K. Analysis of Hepatic Metabolism Affecting Pharmacokinetics of Propranolol in Humans. Int. J. Pharm. 2008, 349, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Tsamandouras, N.; Rostami-Hodjegan, A.; Aarons, L. Combining the ‘Bottom up’ and ‘Top down’ Approaches in Pharmacokinetic Modelling: Fitting PBPK Models to Observed Clinical Data. Br. J. Clin. Pharmacol. 2015, 79, 48–55. [Google Scholar] [CrossRef]
- Kogame, A.; Takeuchi, T.; Nonaka, M.; Yamasaki, H.; Kawaguchi, N.; Bernards, A.; Tagawa, Y.; Morohashi, A.; Kondo, T.; Moriwaki, T.; et al. Disposition and Metabolism of TAK-438 (Vonoprazan Fumarate), a Novel Potassium-Competitive Acid Blocker, in Rats and Dogs. Xenobiotica 2017, 47, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Gandhi, Y.A.; Duignan, D.B.; Morris, M.E. Prediction of Biliary Excretion in Rats and Humans Using Molecular Weight and Quantitative Structure-Pharmacokinetic Relationships. AAPS J. 2009, 11, 511–525. [Google Scholar] [CrossRef]
- Hatton, G.B.; Yadav, V.; Basit, A.W.; Merchant, H.A. Animal Farm: Considerations in Animal Gastrointestinal Physiology and Relevance to Drug Delivery in Humans. J. Pharm. Sci. 2015, 104, 2747–2776. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Lee, S.L.; Yu, L.X. Mechanistic Approaches to Predicting Oral Drug Absorption. AAPS J. 2009, 11, 217–224. [Google Scholar] [CrossRef]
- HK inno.N Corp. K-CAB. Package Insert. 2020. Available online: https://nedrug.mfds.go.kr/pbp/CCBBB01/getItemDetail?itemSeq=201802815 (accessed on 28 May 2021).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | Volume (mL) | Blood Flow (mL/min) |
|---|---|---|
| Adipose | 15,000 | 270 |
| Adrenal gland | 14 | 15.6 |
| Brain | 1400 | 593 |
| Heart | 329 | 208 |
| Kidney | 308 | 910 |
| Large Intestine | 371 | 208 |
| Liver | 1800 | 1326 |
| Lung | 532 | 5200 |
| Small Intestine | 520 | 520 |
| Spleen | 182 | 104 |
| Stomach | 147 | 52 |
| Venous blood | 3470 | |
| Arterial blood | 1730 |
| Tissue | Kp,SS |
|---|---|
| Adipose | 11.7 |
| Adrenal gland | 56.1 |
| Brain | 3.55 |
| Heart | 12.4 |
| Kidney | 44.2 |
| Large Intestine | 110 |
| Liver | 417 |
| Lung | 236 |
| Small Intestine | 637 |
| Spleen | 47.9 |
| Stomach | 519 |
| Category | Parameter (unit) | Value | Comments |
|---|---|---|---|
| Physicochemical Properties and Blood Binding | Compound type | Base | |
| pKa | 9.04 | Determined [16] | |
| logP | 2.38 | ||
| fup | 0.0645 | ||
| B/P ratio () | 0.8 | ||
| Absorption | Ka (min−1) | 0.0606 | Predicted (See text) |
| Fa | 0.761 | Optimized (See text) | |
| Distribution (Kp) * | Adrenal gland | 20.8 | Corrected by Kp,scalar (See text) |
| Adipose | 4.32 | ||
| Brain | 1.32 | ||
| Heart | 4.60 | ||
| Kidney | 16.4 | ||
| Liver | 303 | ||
| Lung | 87.6 | ||
| Large Intestine | 40.8 | ||
| Small Intestine | 124 | ||
| Spleen | 17.8 | ||
| Stomach | 193 | ||
| Elimination | 0.904 | Predicted (See text) | |
| (L/min) | 12.9 | Optimized (See text) | |
| M14 Formation by CYP3A4 | (nmol/min) | 248 | Determined [16] |
| (μM) | 0.093 | ||
| M11 Formation by CYP3A4 | (nmol/min) | 800 | Determined [16] |
| (μM) | 15.95 |
| Dose | AUCobs | AUCpred | AUCratio | Cmax,obs | Cmax,pred | Cmax ratio |
|---|---|---|---|---|---|---|
| Training set (1st day MAD)1 | ||||||
| 20 mg | 9020 | 11,900 | 1.32 | 16.3 | 16.6 | 1.02 |
| 40 mg | 23,700 | 23,900 | 1.01 | 40.4 | 33.2 | 0.822 |
| 80 mg | 62,400 | 48,000 | 0.770 | 99.1 | 66.6 | 0.672 |
| Validation set (7th day MAD)1 | ||||||
| 20 mg/day | 16,300 | 14,900 | 0.916 | 20.8 | 20.2 | 0.972 |
| 40 mg/day | 28,300 | 30,000 | 1.06 | 43.2 | 40.4 | 0.935 |
| 80 mg/day | 68,700 | 60,400 | 0.880 | 94.4 | 81.2 | 0.861 |
| Validation set (1st dose)2 | ||||||
| 40 mg | 21,000 | 29,800 | 1.42 | 28.8 | 33.2 | 1.15 |
| 80 mg | 66,300 | 60,000 | 0.905 | 86.4 | 66.6 | 0.770 |
| Validation set (8th dose)2 | ||||||
| 40 mg/day | 28,300 | 37,500 | 1.32 | 35.5 | 40.4 | 1.14 |
| 80 mg/day | 61,800 | 75,700 | 1.23 | 78.9 | 81.2 | 1.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, Y.-S.; Kim, M.-S.; Lee, N.; Lee, A.; Chae, Y.-J.; Chung, S.-J.; Lee, K.-R. Development of Physiologically Based Pharmacokinetic Model for Orally Administered Fexuprazan in Humans. Pharmaceutics 2021, 13, 813. https://doi.org/10.3390/pharmaceutics13060813
Jeong Y-S, Kim M-S, Lee N, Lee A, Chae Y-J, Chung S-J, Lee K-R. Development of Physiologically Based Pharmacokinetic Model for Orally Administered Fexuprazan in Humans. Pharmaceutics. 2021; 13(6):813. https://doi.org/10.3390/pharmaceutics13060813
Chicago/Turabian StyleJeong, Yoo-Seong, Min-Soo Kim, Nora Lee, Areum Lee, Yoon-Jee Chae, Suk-Jae Chung, and Kyeong-Ryoon Lee. 2021. "Development of Physiologically Based Pharmacokinetic Model for Orally Administered Fexuprazan in Humans" Pharmaceutics 13, no. 6: 813. https://doi.org/10.3390/pharmaceutics13060813
APA StyleJeong, Y.-S., Kim, M.-S., Lee, N., Lee, A., Chae, Y.-J., Chung, S.-J., & Lee, K.-R. (2021). Development of Physiologically Based Pharmacokinetic Model for Orally Administered Fexuprazan in Humans. Pharmaceutics, 13(6), 813. https://doi.org/10.3390/pharmaceutics13060813