1. Introduction
Acetylsalicylic acid (aspirin) is a common anti-inflammatory drug and is widely prescribed as an anti-platelet to prevent cardiovascular events [
1,
2,
3]. Although aspirin’s main administration route is oral, this route has poor bioavailability (40–50%) [
3,
4] and its prolonged use is associated with gastrointestinal mucosa ulcers and gastrointestinal hemorrhaging in severe cases [
3,
5]. In the literature, there is evidence that aspirin parenteral administration (non-oral) can reduce gastrointestinal side effects [
6]. An alternative to oral administration is developing aspirin local delivery systems by loading the aspirin into polymeric implants for cardiovascular applications, such as stents [
7,
8,
9,
10], scaffolds [
11,
12,
13], and gels [
6].
Several strategies can be used to develop these polymeric drug delivery systems [
14,
15,
16]. Soaking is a conventional method that consists of immersing the polymer in a drug-concentrated solution, allowing the drug to diffuse into its amorphous regions and be retained due to interactions with the polymeric chains [
14,
16,
17]. In the final step, the solvent is removed, usually by evaporation [
18,
19]. The choice of solvent must be accurate: it must solubilize the drug and swell the polymer. Soaking is a simple method and can be used to load a variety of drugs according to the right combination of {solvent + polymer + drug}. In the literature, it has been used mainly to load drugs into sutures [
18,
19], scaffolds [
20], and contact lenses [
21,
22,
23,
24].
The supercritical carbon dioxide (scCO
2) impregnation is based on a similar principle as the soaking method, but in this case, scCO
2 is the solvent. In its supercritical state, above the critical point of 31 °C and 7.4 MPa, CO
2 has a high density and high diffusivity [
25]. The high density provides scCO
2 with a solvating power similar to liquids, solubilizing various nonpolar drugs with low molecular weight [
26,
27]. Moreover, its high diffusivity, similar to a gas, allows it to be absorbed into the amorphous regions of various amorphous and semicrystalline polymers [
28,
29]. Therefore, scCO
2 can solubilize the drug and carry it into the polymeric matrix. Since CO
2 is a gas at atmospheric conditions, the polymeric drug delivery system is obtained without solvent through a simple depressurization step, whereas the drug is trapped between the polymeric chains. Sutures [
30,
31], lenses [
32,
33], and textiles [
34,
35] have been loaded with drugs or other active pharmaceutical substances using this method.
Previous works have compared the soaking method and scCO
2 impregnation method of drug delivery system development based on hydrogels and amorphous matrices [
21,
36,
37]. To the best of our knowledge, such studies have not been performed on thermoplastic matrices, which are extensively used to develop biomedical implants. Moreover, the increased interest in developing drug-eluting implants and scaffolds of thermoplastics polymers using fused deposition modeling (FDM) 3D printing motivates the comparison of the impregnation methods, to decide which should be used to load the drugs into the filaments or printed device, depending on the desired release profile [
20,
38,
39,
40,
41]. In order to select the most appropriate drug loading processes, it is necessary to understand its impact on drug loading, the polymer microstructure, the drug crystalline state, the in vitro polymer behavior, and the drug release profile.
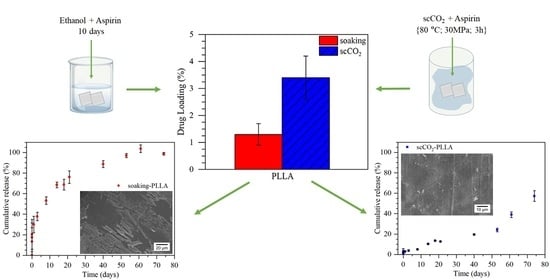
In this paper, we compared the two methods of post-manufacture aspirin loading into polymeric matrices, to highlight their differences and verify if scCO
2 impregnation is a suitable alternative to soaking. Poly(
l-acid lactic) (PLLA) and linear low-density polyethylene (LLDPE) were chosen as model polymeric matrices due to their different physical-chemical properties and the fact that they can interact differently with aspirin and with the solvents. These two polymers are also widely used to produce implants [
42,
43,
44,
45]. The two impregnation methods were compared in terms of drug loading and the characteristics of each process (solvent absorption, residual solvent, impregnation time) for PLLA and LLDPE. Considering that PLLA resulted in higher drug loadings, it was selected for further characterizations. The impact of the impregnation method was evaluated on thermal properties, on the microstructure, in vitro degradation, and in vitro release.
2. Materials and Methods
2.1. Materials
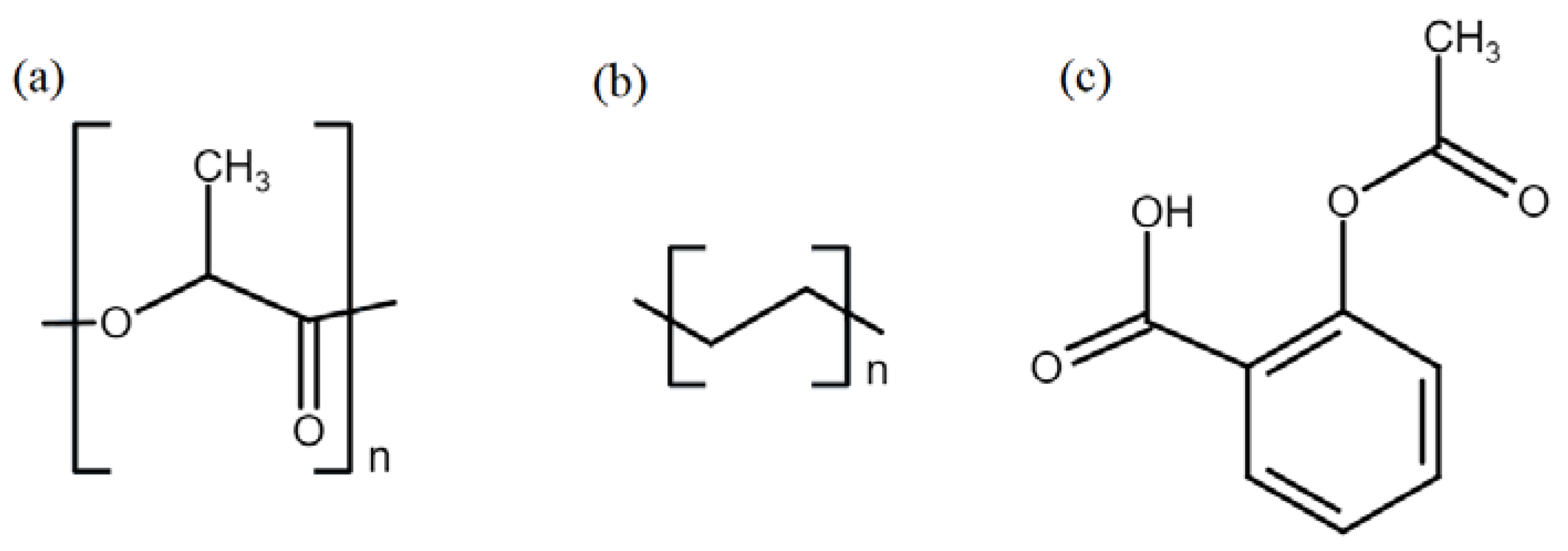
PLLA filament was purchased from Cliever (Belo Horizonte, MG, Brazil). LLDPE pellets were supplied by Braskem (São Paulo, SP, Brazil). Carbon dioxide (purity 99.985%) was purchased from Oxilumen (São Paulo, SP, Brazil). Acetylsalicylic Acid (aspirin) (purity 99.0%) was purchased from Sigma-Aldrich (Barueri, SP, Brazil) and was macerated before use to reduce particle size and facilitate its solubilization. The structures of the polymers and aspirin are presented in
Figure 1. Silicone oil (Synth, 350 cps) was used for thermostated baths. Ethanol (purity 99%) and Isopropanol (purity 99%) were obtained from Synth (São Paulo, SP, Brazil) and used as received. Sodium chloride (NaCl), potassium chloride (KCl), sodium phosphate dibasic (Na
2HPO
4), monopotassium phosphate (KH
2PO
4), were purchased from Sigma-Aldrich and used as received to prepare the 1 M phosphate-buffered saline (PBS) solution for in vitro degradation and release analysis. Deuterated chloroform (CDCl
3, 99.8%) containing 0.05%
v/
v tetramethylsilane (TMS) as the internal reference was purchased from Cambridge Isotope Laboratories and used in the NMR analysis.
2.2. Preparation of Polymeric Films
PLLA filaments cut in pieces of approximately 1 cm and LLDPE pellets as received were processed into films through hot pressing on a hydraulic press with heating (model SL-11, Solab, Piracicaba, Brazil). A pressure gradient of up to 6 tons was applied for 10 min at 215 °C to PLLA and at 165 °C to LLDPE, using Teflon sheets for the contact between the press and the samples. The films of 0.4 mm thickness were cut into squares of approximately 1 cm2, which corresponded to approximately 40 mg per sample.
2.3. Solvent Absorption and Residual Solvent
Ethanol and isopropanol were used as solvents for the soaking impregnation method, and CO
2 was used as the solvent for the scCO
2 impregnation method. The solvent absorption and residual solvent content in the polymeric matrices were investigated in the absence of aspirin and evaluated gravimetrically using a precision balance (10
−4 g, model AY-220, Marte, Shimadzu, São Paulo, Brazil). The measurements were performed in triplicate. The residual solvent content of ethanol was also determined by proton nuclear magnetic resonance (1H NMR) analysis (
Supplementary Materials Section S1.1).
In order to confirm the presence or absence of solvent, Fourier-transform infrared spectroscopy (FTIR) analyses were performed using a spectrophotometer (Perkin Elmer, Model Spectrum Two, São Paulo, Brazil) in attenuated total reflectance (ATR) mode (diamond doped with zinc selenide crystal). The absorbance spectra were obtained with 16 scans and a resolution of 2 cm−1, between 700 and 4000 cm−1, at room temperature.
2.3.1. Ethanol and Isopropanol
Neat PLLA and LLDPE films were weighted (m
0) and immersed in ethanol and isopropanol, respectively. The films were removed after 10 days of soaking to ensure maximal solvent absorption (
Supplementary Materials Section S2, Figures S2 and S3). The surface was gently dried with tissue paper, and the films were weighed again (m
s). The percentage of solvent absorption was calculated using Equation (1).
These samples were then dried at 80 °C for 3 h, for the solvent removal, and weighted (m
d). The residual solvent was calculated using Equation (2):
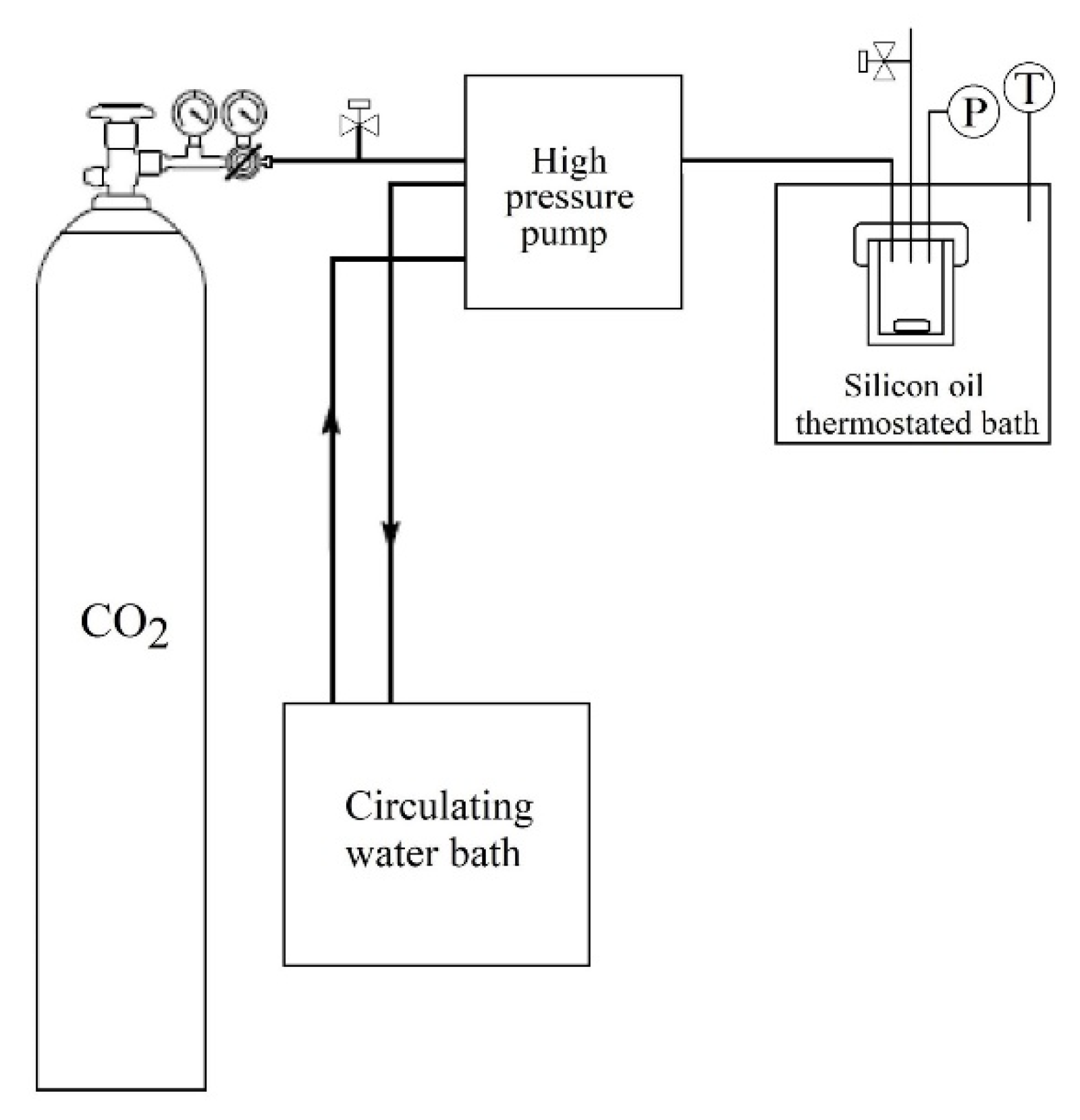
2.3.2. CO2
PLLA and LLDPE films were weighted (m
0) and placed vertically into a 10 mL stainless steel high-pressure cell. The reactor was immersed in a silicone oil thermostated bath at 80 °C, and CO
2 was introduced into the reactor using a high-pressure pump (model SFC P-50A, Thar Technologies, Pittsburgh, PA, USA) until a pressure of 30 MPa was achieved (
Figure 2). The temperature and pressure were kept constant for 3 h. The depressurization was performed by dipping the reactor in dry ice to freeze the CO
2 to prevent its desorption from the polymeric matrices [
46]. The samples were immediately weighed after being removed from the high-pressure cell (m
s) and continuously weighted until mass stabilization (m
d). The amount of CO
2 (m
co2) sorbed into the films during the scCO
2 impregnation process was not desorbed during the depressurization process. Similar to the soaking process, the scCO
2 absorption was calculated using Equation (1).
Preliminary measurements showed that the mass of the films was stabilized 5 days after their impregnation (md). For this reason, all posterior analyses were performed after this period. Similar to the soaking method, the residual solvent was then obtained using Equation (2).
2.4. Impregnation
2.4.1. Soaking Method
Five PLLA and LLDPE films were immersed in 100 mL of aspirin solution and kept at room temperature without stirring for 10 days. PLLA and LLDPE films were soaked in aspirin solution in ethanol (0.08 g∙mL−1) and isopropanol (0.1 g∙mL−1), respectively. After 10 days of soaking, the films were removed from the solutions, placed in a petri dish, and dried at 80 °C for 3 h in an oven for solvent removal.
2.4.2. scCO2 Impregnation Method
The scCO
2 impregnation was carried out in a batch process with the same protocol as previous work [
47]. Three PLLA films and three LLDPE films were loaded into a 10 mL stainless steel high-pressure cell containing approximately 50 mg of aspirin, to ensure aspirin saturation throughout the experiment (2.8 mg∙mL
−1), and a stirrer bar. The films were placed in the vertical position in a sample holder, ensuring the films did not touch each other and were physically separated from aspirin. As in the swelling solvent absorption measurements, the reactor was immersed in a silicone oil bath at 80 °C and the CO
2 was introduced into the reactor until the pressure of 30 MPa was achieved. The temperature and pressure were kept constant for 3 h, and the magnetic stirring was maintained at 100 rpm. The depressurization was performed by dipping the reactor in dry ice (−78 °C) to freeze the CO
2 and avoid aspirin desorption during venting [
46]. The scCO
2 conditions {80 °C; 30 MPa; 3 h} were chosen because they were above the glass transition temperatures (T
g) of the PLLA (62 °C) and the LLDPE (−110 °C) and because they have been reported to achieve high aspirin loading in PLLA [
46].
2.5. Drug Loading Quantification
The drug loading (DL%) was defined as the mass of aspirin impregnated per mass of polymer and was obtained using Equation (3):
where m
f is the film mass 5 days after impregnation for the scCO
2 impregnation and the film mass after the drying step for the soaking method, and m
0 is the film mass before the impregnation measured with precision balance (10
−4 g, model AY-220, Marte, Shimadzu, São Paulo, Brazil). For each method, the impregnation was performed at least in triplicate, and the average drug loading is presented.
The difference between the drug loading of each polymer and method was statistically evaluated through ANOVA, using the Tukey test with 5% of significance.
2.6. Scanning Electron Microscope (SEM)
SEM analysis (JEOL JSM6010LA, São Paulo, Brazil) was used to evaluate possible aspirin precipitation on the surface of the films and morphological changes in the cross-section of PLLA samples. The fracture of the samples was performed by cryofracture using liquid nitrogen, and the samples were sputtered with a 15 nm layer of gold.
2.7. Thermal Analysis
The impact of the two impregnation processes on PLLA thermal properties and crystallinity was evaluated by differential scanning calorimetry (DSC) analyses (Q200, TA Instruments, Barueri, Brazil). The DSC curve of the neat LLDPE film was also obtained. The films were cut into small pieces, and 7 to 8.5 mg was sealed in aluminum pans. The samples were heated from 25 °C to 200 °C for PLLA and 150 °C for LLDPE with a heating rate of 10 °C∙min
−1 (first heating), then cooled to −80 °C with a rate of 20 °C∙min
−1 and heated up again to 200 °C for PLLA and 150 °C for LLDPE with a rate of 10 °C∙min
−1 (second heating). The polymer crystallinity was obtained using Equation (4):
where ∆H
f (J·g
−1) is the experimental fusion enthalpy, ∆H
c (J·g
−1) is the experimental crystallization enthalpy, W is the polymer mass fraction and ∆H
100f is the fusion enthalpy of 100% crystalline polymer, 93.1 J·g
−1 for PLLA [
48], and 288.7 J·g
−1 for LLDPE [
49].
2.8. X-ray Diffraction (XRD)
XRD analyses were performed to evaluate the crystalline state of impregnated aspirin in PLLA. The changes in PLLA crystallinity after the impregnation were also evaluated. The XRD patterns of the neat films, films only treated with solvent (prepared in
Section 2.3), and impregnated films were obtained with a D8 Focus diffractometer (Bruker, Atibaia, Brazil), using Cu Kα
1 radiation (wavelength = 1.54051 Å) at 40 kV and 40 mA, in the 2θ range of 5°–60° at the rate of 5°∙min
−1. Before the analyses, the impregnated samples were gently cleaned with wet tissue paper to remove any precipitated aspirin from the surface.
2.9. In Vitro PLLA Degradation
The study of in vitro degradation was performed with neat PLLA, PLLA impregnated using the scCO
2 method, and PLLA impregnated using the soaking method. PLLA was chosen because it has higher drug loadings and is known to degrade during its in vivo application, whereas LLDPE is expected to last longer (>years) [
43,
45]. Of each sample, 10 specimens were separately immersed in 30 mL of 1 M PBS, pH 7.4 at 37 ± 1 °C, following ASTM F1635-16 [
50]. The pH of the PBS solutions was periodically monitored throughout the degradation study, and when a pH variation of ±0.2 was detected, the solution was replaced with fresh PBS. The PLLA films were removed at predetermined intervals and dried in a vacuum oven at 30 °C for 24 h. The degradation study was carried out for 112 days.
1H NMR analyses were performed to evaluate the decrease in the polymer’s number-average molecular weight (M
n). Approximately 10 mg of sample was dissolved in 0.5 mL of CDCl
3 and the spectra were recorded in a Varian VNMRS 500 MHz at 27 °C, using 800 transients. The number-average molecular weights for PLLA were estimated from the comparison of the signal integration of the PLLA methine ester end group (δ = 4.36 ppm) and the PLLA methine ester group (δ = 5.20 ppm) [
51]. The PLLA crystallinity and thermal properties evolution were analyzed through DSC using the same protocol described in
Section 2.8.
2.10. In Vitro Aspirin Release
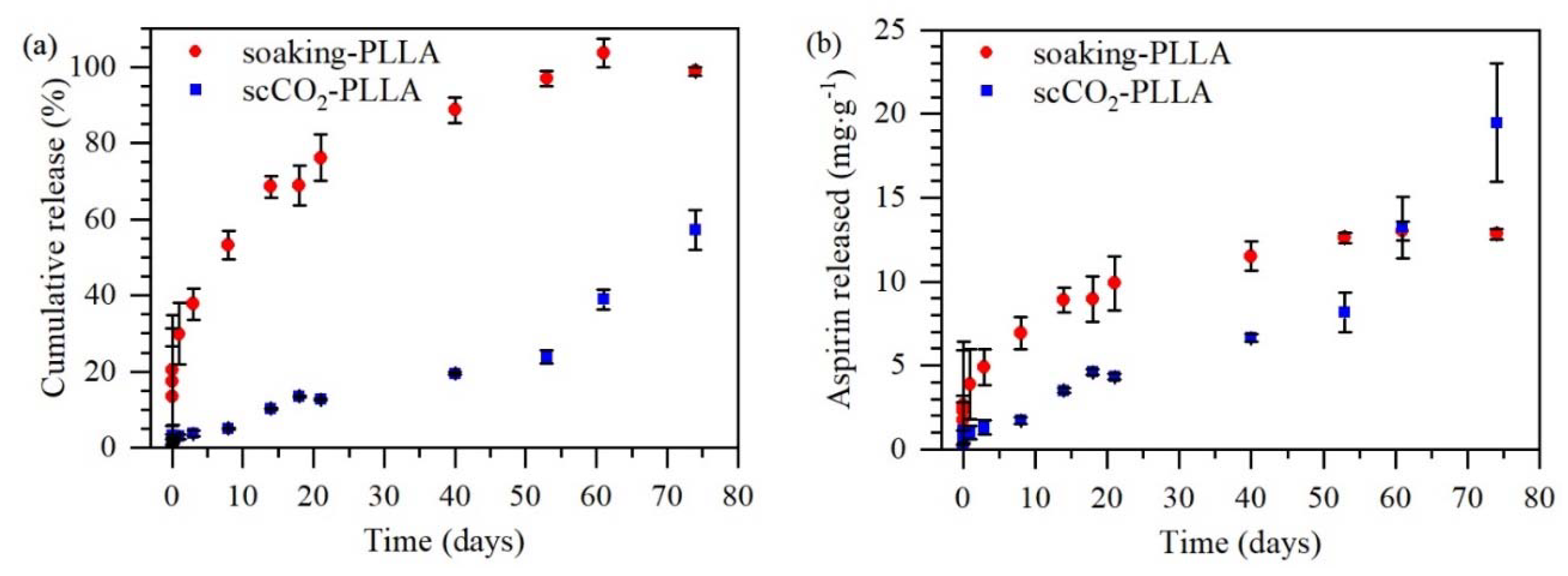
The in vitro aspirin release from the PLLA films impregnated using the soaking method and the scCO
2 impregnation method was carried out by immersing one film in 10 mL of 1 M PBS at 37 ± 1 °C. At predetermined intervals, 2 mL of the PBS solution were withdrawn and replaced with 2 mL of fresh PBS. The collected PBS samples were analyzed in a UV-Vis spectrometer (model UV-3600, Shimadzu, São Paulo, Brazil) at 298 nm for the aspirin quantification. The calibration curve was performed, and the molar absorptivity was found to be 0.3292 m
2mol
−1 at 298 nm. The release experiment was performed in triplicate and the cumulative release was calculated using Equation (5), considering that the 100% release is the release of the total amount of aspirin impregnated in each sample:
where m
t is the total mass of the compound released at the time t and m
impregnated is the total aspirin mass impregnated in the film. The release kinetics and mechanism were evaluated, considering the possibility of a burst effect, using the Korsmeyer–Peppas model that can be applied to data up to 60% of the total release following the Equation (6) [
52,
53]:
where M
t is the released mass at the time t, M is the mass released at infinite time (which corresponds to m
impregnated), k is a kinetic constant, b is the effect of a burst release, and n is the diffusional coefficient, which corresponds to a specific release mechanism as described in
Table 1 [
52].
2.11. Dissolution of the Aspirin Coating
The soaking-PLLA sample was covered with crystals of aspirin. The time necessary to dissolve the aspirin coating tcoating was determined using SEM images of soaking-PLLA films, that were immersed in 1 M PBS at 37 °C for different times. After the immersion in PBS, the samples were dried in a vacuum oven at 30 °C for 24 h and sputtered with a 15 nm layer of gold.
5. Conclusions
This paper investigated aspirin loading in PLLA and LLDPE through soaking and supercritical CO2 (scCO2) impregnation methods to develop local delivery systems as an alternative to oral administration. The scCO2 impregnation method was a faster process, promoted higher drug loadings in PLLA, and resulted in systems free of solvent residues. Higher drug loadings were obtained in PLLA samples due to their higher solvent absorption and affinity with aspirin, in addition, needle-shaped aspirin crystals were observed on the PLLA sample impregnated by soaking. Both methods increased the PLLA crystallinity and resulted in amorphous aspirin in the matrix, evidenced by the absence of aspirin Tm and crystalline peaks of aspirin in DSC and XRD curves, respectively. The in vitro degradation of PLLA samples from both methods was similar, showing a faster degradation rate than neat PLLA as an effect of the aspirin acid groups that catalyze PLLA hydrolysis. Finally, aspirin in vitro release from soaking-PLLA was faster and governed by Fickian diffusion with a burst release of 15% in the first 1.5 h, whereas the release from scCO2-PLLA was slower, with 3% released in the first 24 h, and depended not only on aspirin diffusion but also matrix degradation leading to c.a. 60% of aspirin released after 74 days vs. 100% after 60 days for soaking-PLLA. This prolonged release from scCO2-PLLA could be a substitution for oral long-term aspirin administration, which may cause gastrointestinal adverse events. The scCO2 impregnation method presented a good alternative to the conventional soaking, due to its higher drug loading, similar degradation profile, and more sustained release.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}