
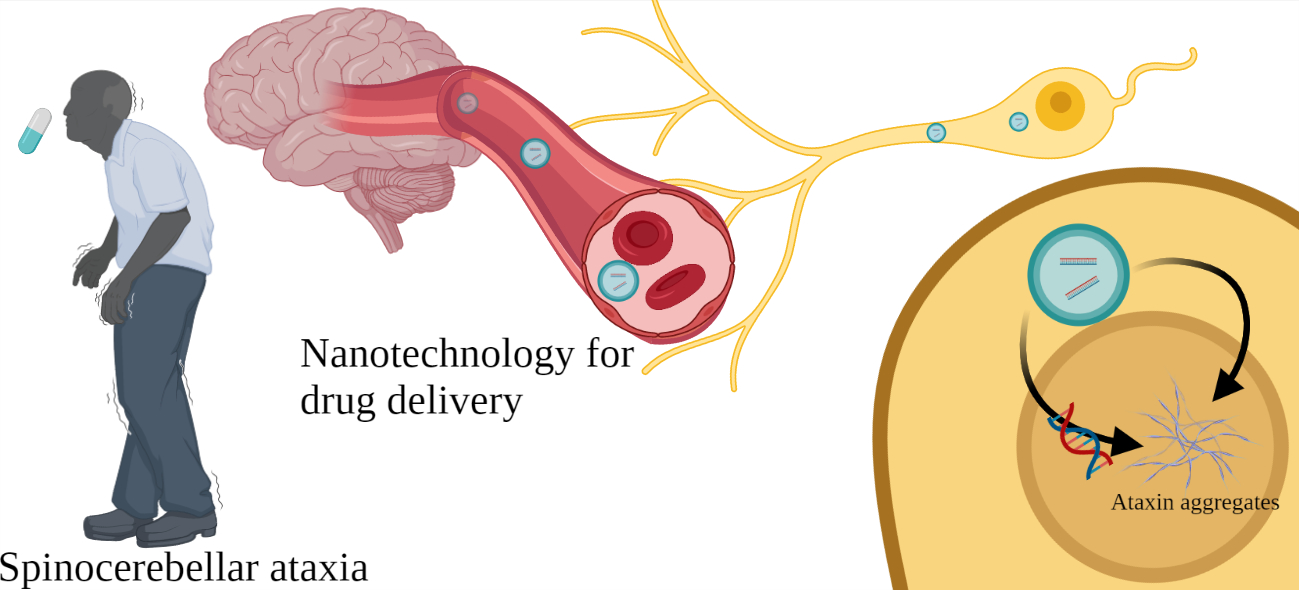
New Perspectives of Gene Therapy on Polyglutamine Spinocerebellar Ataxias: From Molecular Targets to Novel Nanovectors

 ,
,  and
and
Abstract
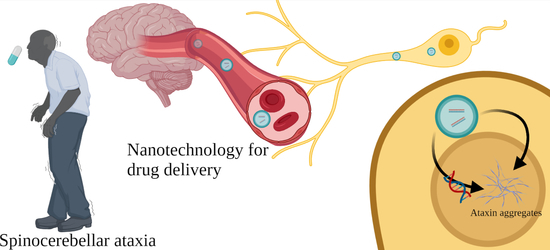
:
1. Introduction
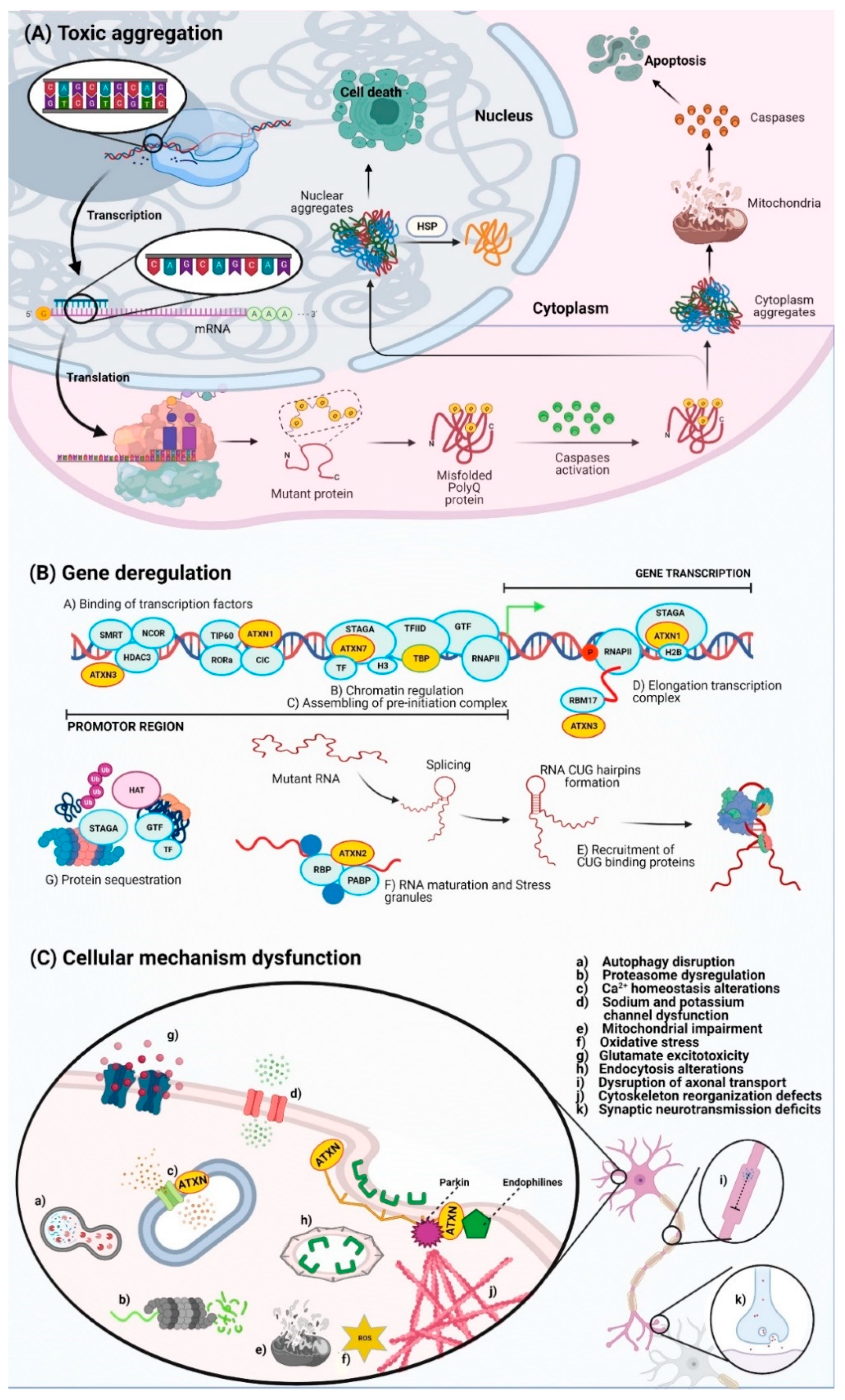
2. Molecular Basis of PolyQ SCAs
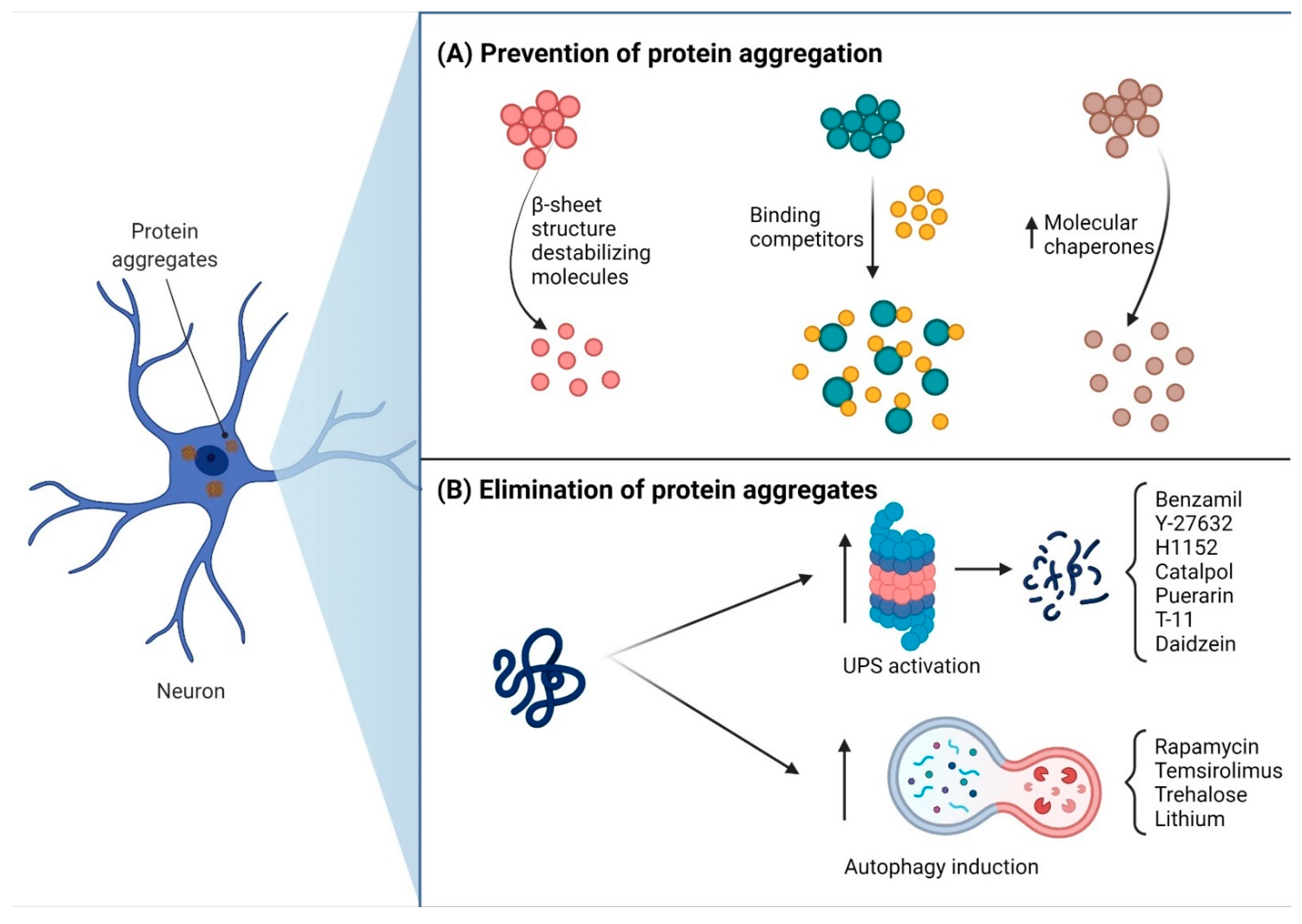
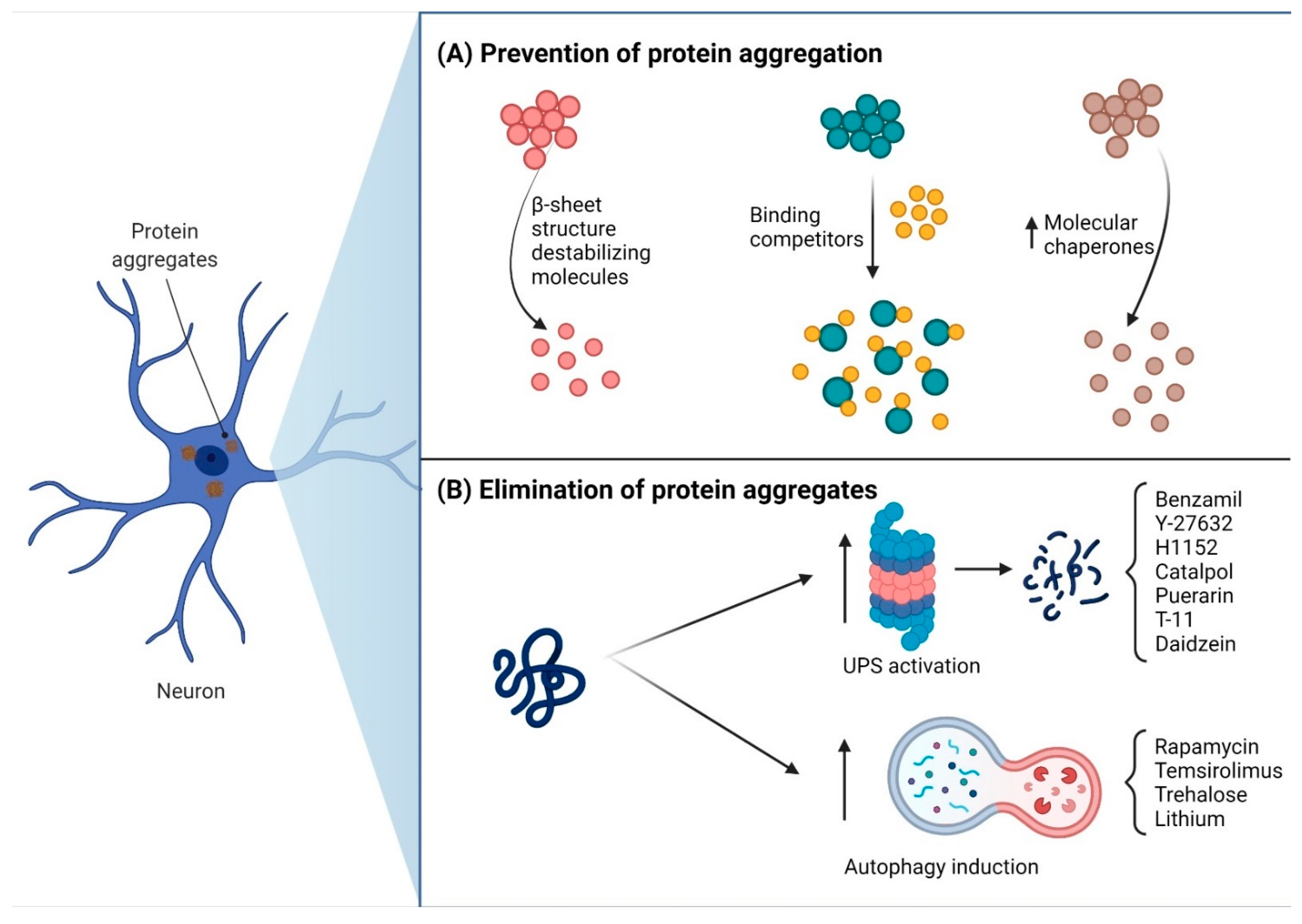
3. Pharmacological Therapy Treatment for PolyQ SCAs
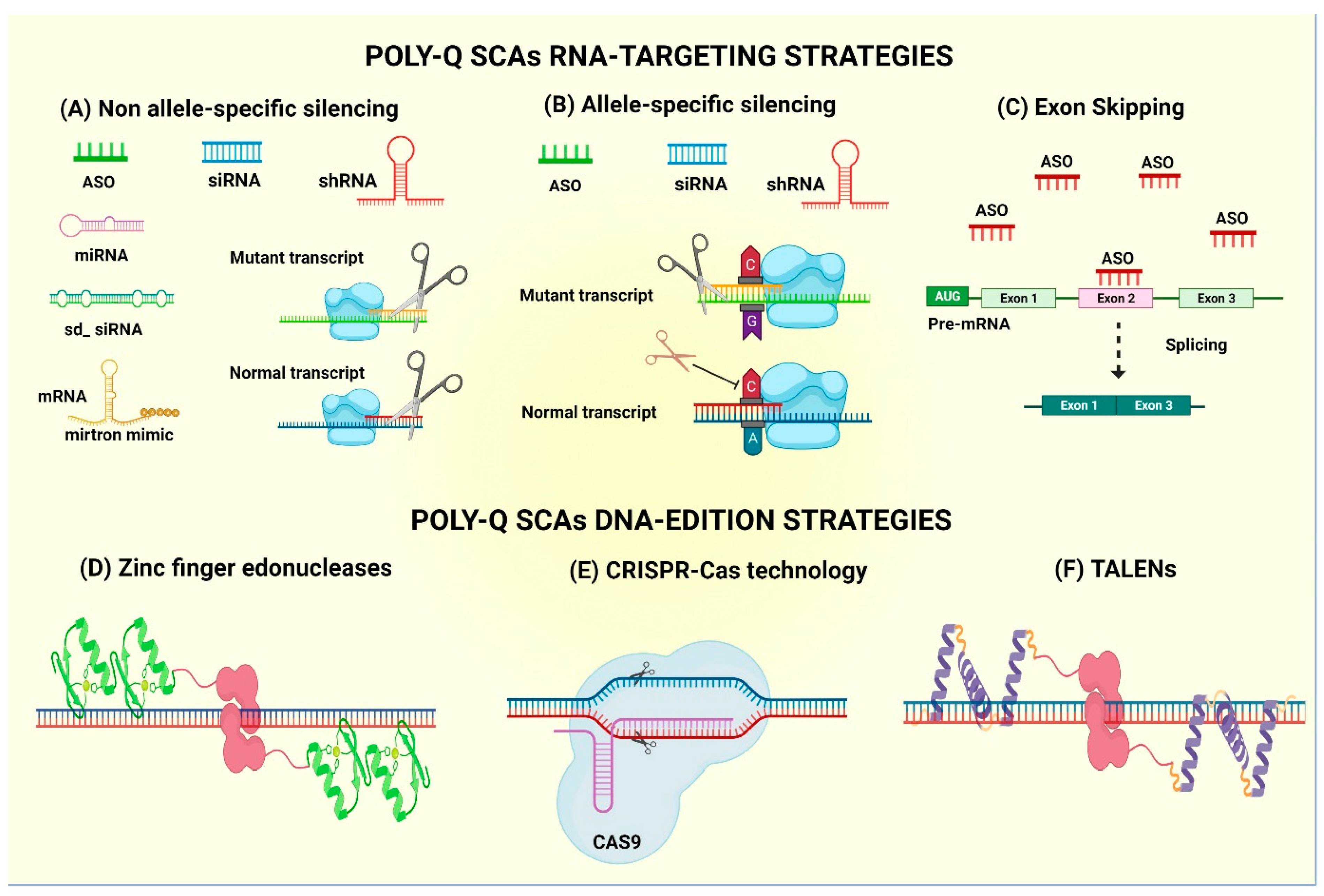
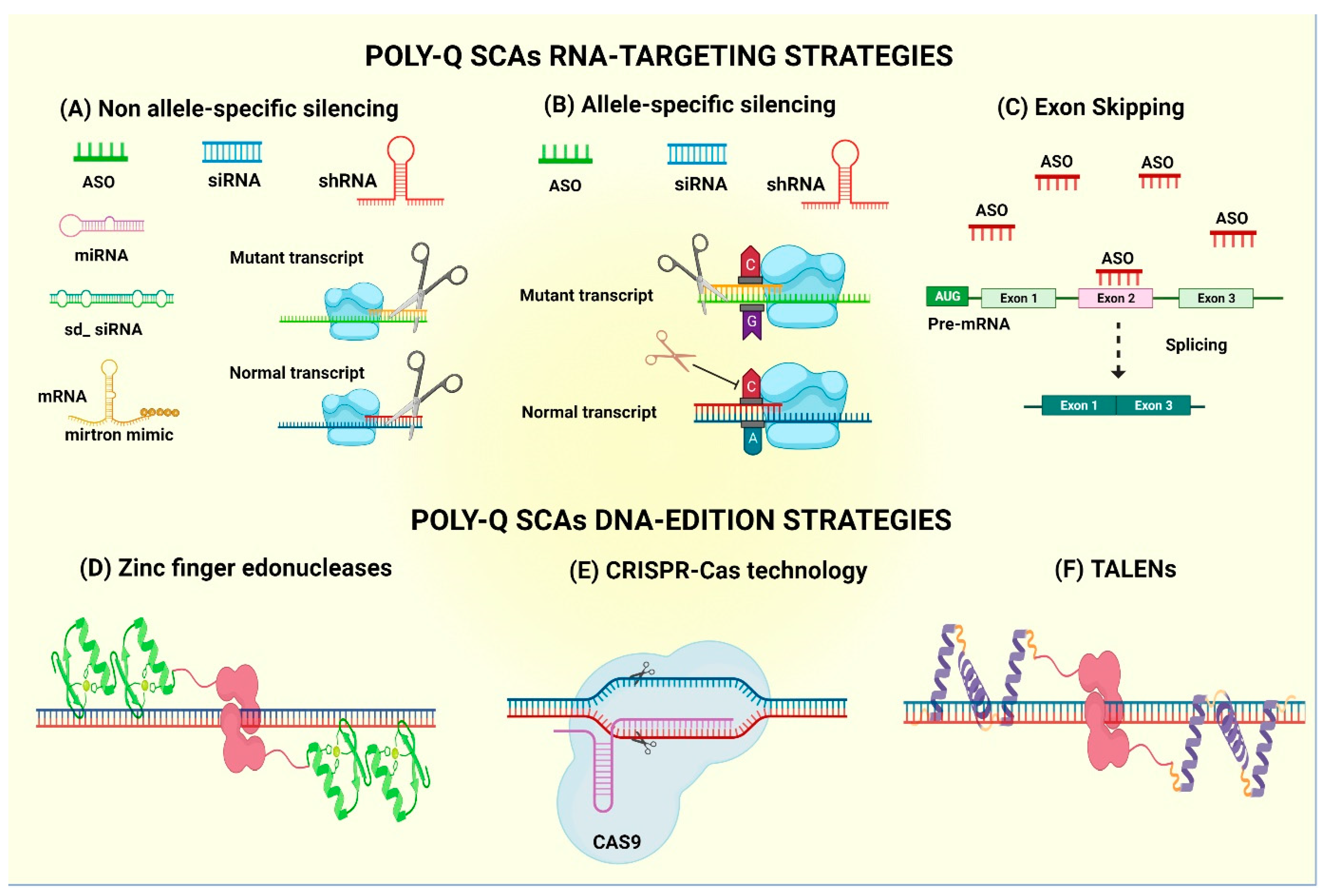
4. Gene Therapy Treatment for PolyQ SCAs
4.1. mRNA-Based Technology—Antisense Oligonucleotides
4.2. mRNA-Based Technology—Allele-Specific Antisense Oligonucleotides
4.3. Exon Skipping by ASOs
4.4. Non-Allele Interferent Gene Silencing
4.5. Allele-Specific siRNA-Mediated Gene Silencing
4.6. Gene Editing
5. Limitations of Gene Therapy
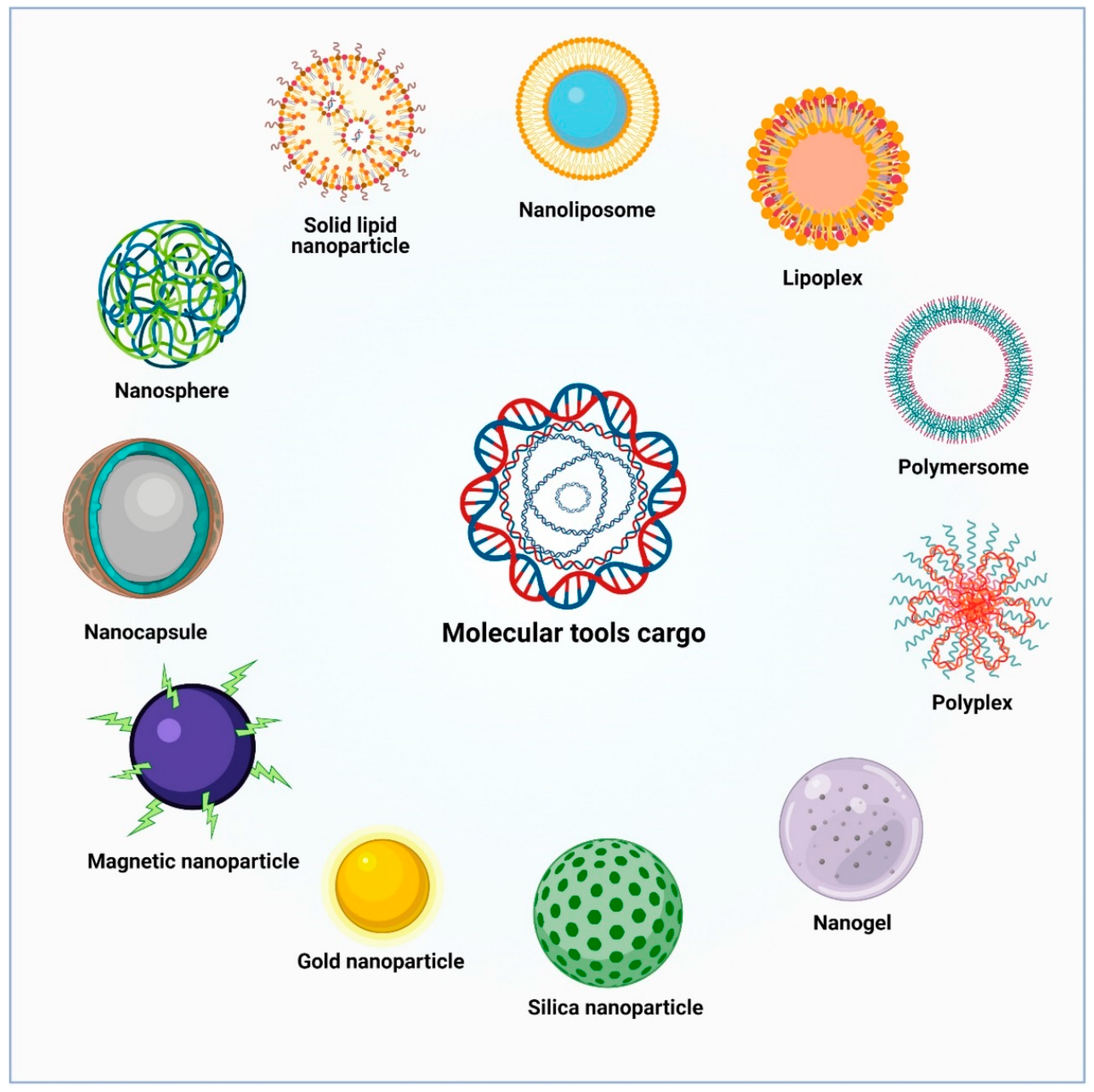
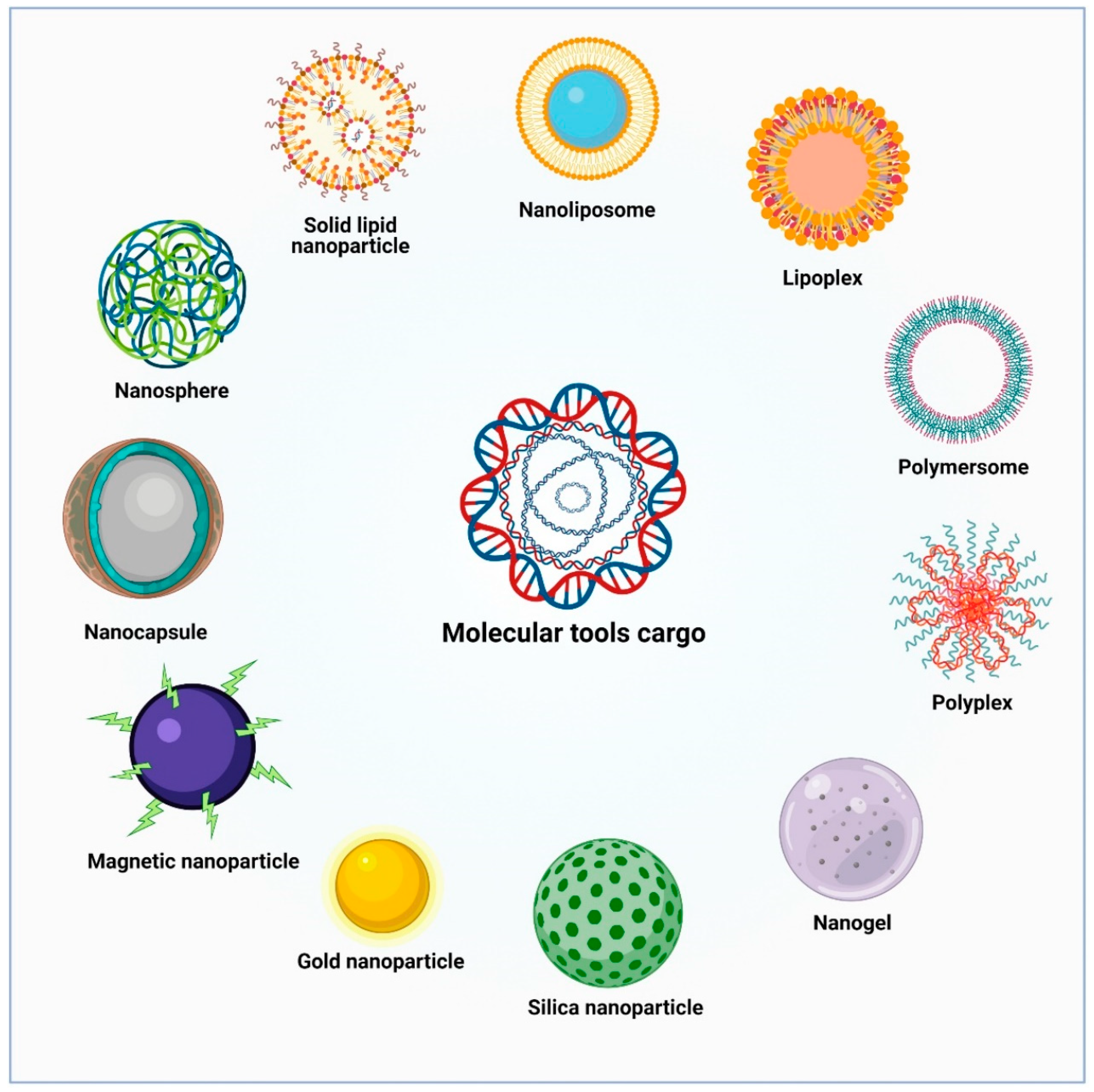
6. Novel Nanovector Tools for Brain Delivery of Therapeutic Molecules
6.1. Lipid-Based Nanoparticles
6.2. Polymeric Nanoparticles
6.3. Polyplexes
6.4. Metallic Nanoparticles
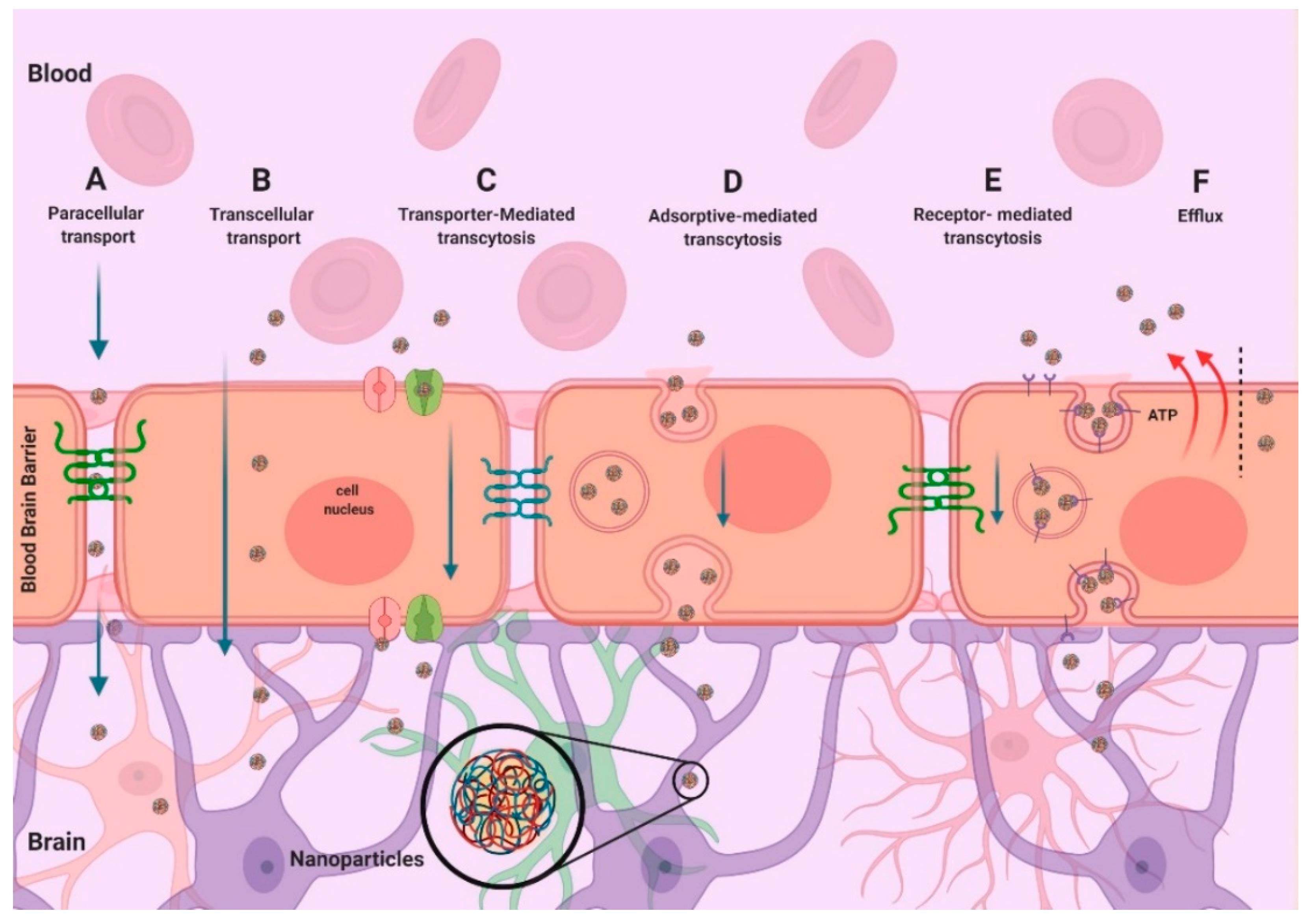
7. Targeting Strategies for Delivery of NPs into the Brain
7.1. Passive Targeting
7.2. Active Targeting
8. Transporter-Mediated Strategies for Drug Delivery into the Brain
8.1. Glucose
8.2. Transferrin
8.3. Lactoferrin
8.4. Insulin Receptor
8.5. Low-Density Lipoprotein Receptor-Related Protein
9. Proof of Concept of NPs Delivery on PolyQ Diseases
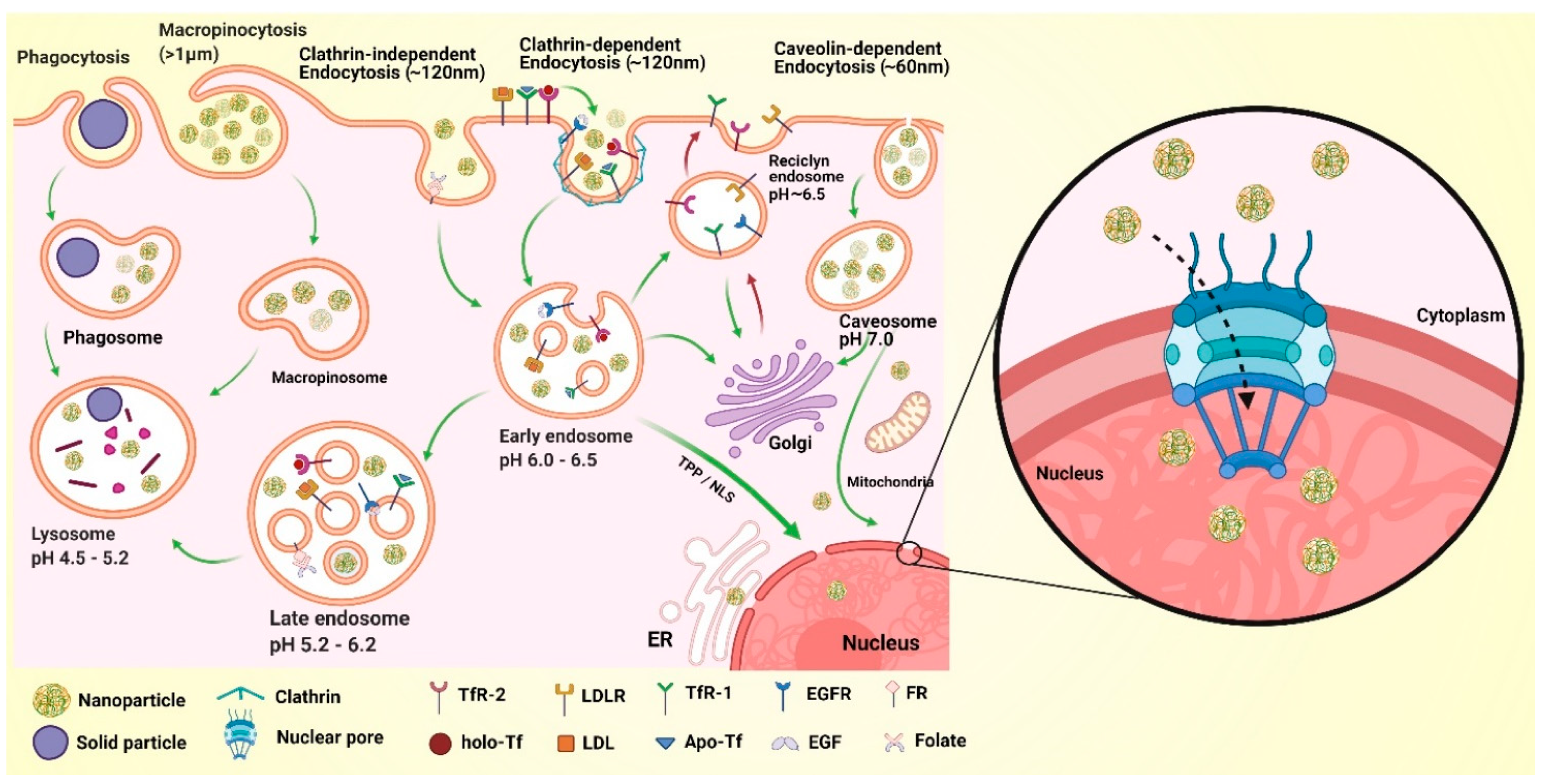
10. The Challenge of Cytoplasmic Transit and Nuclear Internalization of NPs
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matilla-Dueñas, A.; Sánchez, I.; Corral-Juan, M.; Dávalos, A.; Alvarez, R.; Latorre, P. Cellular and molecular pathways triggering neurodegeneration in the spinocerebellar ataxias. Cerebellum 2010, 9, 148–166. [Google Scholar] [CrossRef]
- Orr, H.T. Cell biology of spinocerebellar ataxia. J. Cell Biol. 2012, 197, 167–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teive, A.G.; Ashizawa, T. Primary and secondary ataxias. Curr. Opin. Neurol. 2015, 28, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Cho, J.W. Hereditary Cerebellar Ataxias: A Korean Perspective. J. Mov. Disord. 2015, 88, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.; Yan, W.; O’Connor, E.; Houlden, H. Spinocerebellar ataxia: An update. J. Neurol. 2019, 266, 533–544. [Google Scholar] [CrossRef] [Green Version]
- Harding, A.E. Classification of the hereditary ataxias and paraplegias. Lancet 1983, 321, 1151–1155. [Google Scholar] [CrossRef]
- Fujioka, S.; Sundal, C.; Wszolek, Z.K. Autosomal dominant cerebellar ataxia type III: A review of the phenotypic and genotypic characteristics. Orphanet J. Rare Dis. 2013, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.C.; Ashizawa, T.; Kuo, S.H. Collaborative Efforts for Spinocerebellar Ataxia Research in the United States: CRC-SCA and READISCA. Front. Neurol. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Ruano, L.; Melo, C.; Silva, M.C.; Coutinho, P. The global epidemiology of hereditary ataxia and spastic paraplegia: A systematic review of prevalence studies. Neuroepidemiology 2014, 42, 174–183. [Google Scholar] [CrossRef]
- Rüb, U.; Schöls, L.; Paulson, H.; Auburger, G.; Kermer, P.; Jen, J.C.; Seidel, K.; Korf, H.-W.; Deller, T. Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog. Neurobiol. 2013, 104, 38–66. [Google Scholar] [CrossRef]
- Niewiadomska-Cimicka, A.; Hache, A.; Trottier, Y. Gene Deregulation and Underlying Mechanisms in Spinocerebellar Ataxias with Polyglutamine Expansion. Front. Neurosci. 2020, 14, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Orr, H.T.; Zoghbi, H.Y. Trinucleotide Repeat Disorders. Annu. Rev. Neurosci. 2007, 30, 575–623. [Google Scholar] [CrossRef] [PubMed]
- Fiszer, A.; Krzyzosiak, W.J. Oligonucleotide-based strategies to combat polyglutamine diseases. Nucleic Acids Res. 2014, 42, 6787–6810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurray, C.T. Mechanisms of trinucleotide repeat instability during human development. Nat. Rev. Genet. 2010, 11, 786–799. [Google Scholar] [CrossRef] [Green Version]
- Orr, H.T. SCA1—Phosphorylation, a regulator of Ataxin-1 function and pathogenesis. Prog. Neurobiol. 2012, 99, 179–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matilla-Dueñas, A.; Goold, R.; Giunti, P. Clinical, geneti, molecular, and pathophysiological insights into spinocerebellar ataxia type 1. Cerebellum 2008, 7, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Rüb, U.; Auburger, G. Spinocerebellar ataxia 2 (SCA2). Cerebellum 2008, 2, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Riess, O.; Rüb, U.; Pastore, A.; Bauer, P.; Schöls, L. SCA3: Neurological features, pathogenesis and animal models. Cerebellum 2008, 7, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Gomez, C.M. Spinocerebellum Ataxia Type 6: Molecular Mechanisms and Calcium Channel Genetics. In Polyglutamine Disorders; Springer: Cham, Switzerland, 2018; pp. 147–173. ISBN 9783319717791. [Google Scholar]
- Garden, G.A.; La Spada, A.R. Molecular pathogenesis and cellular pathology of spinocerebellar ataxia type 7 neurodegeneration. Cerebellum 2008, 7, 138–149. [Google Scholar] [CrossRef]
- Stevanin, G.; Brice, A.; Curie, M.; Umr, S.; Fe, I. Spinocerebellar ataxia 17 (SCA17) and Huntington’s disease-like 4 (HDL4). Cerebellum 2008, 17, 170–178. [Google Scholar] [CrossRef]
- Tsuji, S. Dentatorubral-pallidoluysian atrophy: Clinical aspects and molecular genetics. Adv. Neurol. 2002, 89, 231–239. [Google Scholar]
- Matilla-Dueñas, A.; Ashizawa, T.; Magri, S.; McFarland, K.N.; Pandolfo, M.; Pulst, S.M.; Riess, O.; Rubinsztein, D.C. Consensus Paper: Pathological Mechanisms Underlying Neurodegeneration in Spinocerebellar Ataxias. Cerebellum 2014, 13, 269–302. [Google Scholar] [CrossRef] [Green Version]
- Nagai, Y.; Popiel, H.A. Conformational Changes and Aggregation of Expanded Polyglutamine Proteins as Therapeutic Targets of the Polyglutamine Diseases: Exposed-Sheet Hypothesis. Curr. Pharm. Des. 2008, 14, 3267–3279. [Google Scholar] [CrossRef]
- Weber, J.J.; Sowa, A.S.; Binder, T.; Hübener, J. From Pathways to Targets: Understanding the Mechanisms behind Polyglutamine Disease. Biomed Res. Int. 2014, 2014, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Gatchel, J.R.; Zoghbi, H.Y. Diseases of unstable repeat expansion: Mechanisms and common principles. Nat. Rev. Genet. 2005, 6, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Fiszer, A.; Krzyzosiak, W.J. RNA toxicity in polyglutamine disorders: Concepts, model, and progress of research. J. Mol. Med. 2013, 91, 683–691. [Google Scholar] [CrossRef] [Green Version]
- Mcintosh, C.S.; Aung-Htut, M.T.; Fletcher, S.; Wilton, S.D. Polyglutamine ataxias: From Clinical and Molecular Features to Current Therapeutic Strategies. J. Genet. Syndr. Gene Ther. 2017, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Niewiadomska-Cimicka, A.; Trottier, Y. Molecular Targets and Therapeutic Strategies in Spinocerebellar Ataxia Type 7. Neurotherapeutics 2019, 16, 1074–1096. [Google Scholar] [CrossRef] [PubMed]
- Chanu, S.I.; Singh, M.D.; Sarkar, S. Transcriptional up-regulation and its impact on poly(Q) disorders. Ther. Targets Neurol. Dis. 2014, 1, 1–16. [Google Scholar] [CrossRef]
- Helmlinger, D.; Hardy, S.; Abou-Sleymane, G.; Eberlin, A.; Bowman, A.B.; Gansmüller, A.; Picaud, S.; Zoghbi, H.Y.; Trottier, Y.; Tora, L.; et al. Glutamine-expanded ataxin-7 alters TFTC/STAGA recruitment and chromatin structure leading to photoreceptor dysfunction. PLoS Biol. 2006, 4, 432–445. [Google Scholar] [CrossRef] [Green Version]
- Mohan, R.D.; Abmayr, S.M.; Workman, J.L. The expanding role for chromatin and transcription in polyglutamine disease. Curr. Opin. Genet. Dev. 2014, 26, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Carmona, V.; Cunha-Santos, J.; Onofre, I.; Simões, A.T.; Vijayakumar, U.; Davidson, B.L.; Pereira de Almeida, L. Unravelling Endogenous MicroRNA System Dysfunction as a New Pathophysiological Mechanism in Machado-Joseph Disease. Mol. Ther. 2017, 25, 1038–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgonio-Cuadra, V.M.; Valdez-Vargas, C.; Romero-Córdoba, S.; Hidalgo-Miranda, A.; Tapia-Guerrero, Y.; Cerecedo-Zapata, C.M.; Hernández-Hernández, O.; Cisneros, B. Wide Profiling of Circulating MicroRNAs in Spinocerebellar Ataxia Type 7. Mol. Neurobiol. 2019, 56, 6106–6120. [Google Scholar] [CrossRef] [PubMed]
- Raposo, M.; Bettencourt, C.; Maciel, P.; Gao, F.; Ramos, A.; Kazachkova, N.; Vasconcelos, J.; Kay, T.; Rodrigues, A.J.; Bettencourt, B.; et al. Novel candidate blood-based transcriptional biomarkers of machado-joseph disease. Mov. Disord. 2015, 30, 968–975. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Zhang, L.; Long, Z.; Chen, Z.; Hou, X.; Wang, C.; Peng, H.; Wang, J.; Li, J.; Duan, R.; et al. MiR-25 alleviates polyQ-mediated cytotoxicity by silencing ATXN3. FEBS Lett. 2014, 588, 4791–4798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalavade, R.; Griesche, N.; Ryan, D.P.; Hildebrand, S.; Krauß, S. Mechanisms of RNA-induced toxicity in CAG repeat disorders. Cell Death Dis. 2013, 4, e752. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Ashizawa, T. RNA toxicity and foci formation in microsatellite expansion diseases. Curr. Opin. Genet. Dev. 2017, 44, 17–29. [Google Scholar] [CrossRef]
- Evers, M.M.; Toonen, L.J.A.; Van Roon-Mom, W.M.C. Ataxin-3 protein and RNA toxicity in spinocerebellar ataxia type 3: Current insights and emerging therapeutic strategies. Mol. Neurobiol. 2014, 49, 1513–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meera, P.; Pulst, S.M.; Otis, T.S. Cellular and circuit mechanisms underlying spinocerebellar ataxias. J. Physiol. 2016, 594, 4653–4660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magaña, J.J.; Velázquez-Pérez, L.; Cisneros, B. Spinocerebellar ataxia type 2: Clinical presentation, molecular mechanisms, and therapeutic perspectives. Mol. Neurobiol. 2013, 47, 90–104. [Google Scholar] [CrossRef] [PubMed]
- Mcloughlin, H.S.; Moore, L.R.; Paulson, H.L. Neurobiology of Disease Pathogenesis of SCA3 and implications for other polyglutamine diseases. Neurobiol. Dis. 2020, 134, 104635. [Google Scholar] [CrossRef]
- Srinivasan, S.R.; Shakkottai, V.G. Moving Towards Therapy in SCA1: Insights from Molecular Mechanisms, Identification of Novel Targets, and Planning for Human Trials. Neurotherapeutics 2019, 16, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Milne, S.C.; Corben, L.A.; Roberts, M.; Szmulewicz, D.; Burns, J.; Grobler, A.C.; Williams, S.; Chua, J.; Liang, C.; Lamont, P.J.; et al. Rehabilitation for ataxia study: Protocol for a randomised controlled trial of an outpatient and supported home-based physiotherapy programme for people with hereditary cerebellar ataxia. BMJ Open 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Tercero-Pérez, K.; Cortés, H.; Torres-Ramos, Y.; Rodríguez-Labrada, R.; Cerecedo-Zapata, C.; Hernández-Hernández, O.; Pérez-González, N.; González-Piña, R.; Leyva-García, N.; Cisneros, B.; et al. Effects of Physical Rehabilitation in Patients with Spinocerebellar Ataxia type 7. Cerebellum 2019, 18, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Velázquez-Pérez, L.; Rodríguez-Diaz, J.C.; Rodríguez-Labrada, R.; Medrano-Montero, J.; Aguilera Cruz, A.B.; Reynaldo-Cejas, L.; Góngora-Marrero, M.; Estupiñán-Rodríguez, A.; Vázquez-Mojena, Y.; Torres-Vega, R. Neurorehabilitation Improves the Motor Features in Prodromal SCA2: A Randomized, Controlled Trial. Mov. Disord. 2019, 34, 1060–1068. [Google Scholar] [CrossRef]
- Ashizawa, T.; Öz, G.; Paulson, H.L. Spinocerebellar ataxias: Prospects and challenges for therapy development. Nat. Rev. Neurol. 2018, 14, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Kim, H.-J.; Jeon, B.S. Parkinsonism in Spinocerebellar ataxia. Biomed Res. Int. 2015, 2015, 125273. [Google Scholar] [CrossRef] [Green Version]
- Bushart, D.D.; Chopra, R.; Singh, V.; Murphy, G.G.; Wulff, H.; Shakkottai, V.G. Targeting potassium channels to treat cerebellar ataxia. Ann. Clin. Transl. Neurol. 2018, 5, 297–314. [Google Scholar] [CrossRef]
- Egorova, P.; Popugaeva, E.; Bezprozvanny, I. Disturbed calcium signaling in spinocerebellar ataxias and Alzheimer’s disease. Semin. Cell Dev. Biol. 2015, 40, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Kasumu, A.W.; Hougaard, C.; Rode, F.; Jacobsen, T.A.; Sabatier, J.M.; Eriksen, B.L.; Strobæk, D.; Liang, X.; Egorova, P.; Vorontsova, D.; et al. Selective positive modulator of calcium-activated potassium channels exerts beneficial effects in a mouse model of spinocerebellar ataxia type 2. Chem. Biol. 2012, 19, 1340–1353. [Google Scholar] [CrossRef] [Green Version]
- Hourez, R.; Servais, L.; Orduz, D.; Gall, D.; Millard, I.; de Kerchove d’Exaerde, A.; Cheron, G.; Orr, H.T.; Pandolfo, M.; Schiffmann, S.N. Aminopyridines correct early dysfunction and delay neurodegeneration in a mouse model of spinocerebellar ataxia type. J. Neurosci. 2011, 31, 11795–11807. [Google Scholar] [CrossRef] [Green Version]
- Jayabal, S.; Ho, H.; Chang, V.; Cullen, K.E.; Watt, A.J. 4-aminopyridine reverses ataxia and cerebellar firing deficiency in a mouse model of spinocerebellar ataxia type 6. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Pirker, W.; Back, C. Chronic Thalamic Stimulation in a Patient with Spinocerebellar Ataxia Type 2. Mov. Disord. Off. J. Mov. Disord. Soc. 2003, 18, 222–225. [Google Scholar] [CrossRef]
- Freund, H.; Barnikol, U.B.; Nolte, D.; Treuer, H.; Auburger, G.; Tass, P.A.; Samii, M. Subthalamic-Thalamic DBS in a Case with Spinocerebellar Ataxia Type 2 and Severe Tremor—A Unusual Clinical Benefit. Mov. Disord. 2007, 22, 732–735. [Google Scholar] [CrossRef]
- Zesiewicz, T.A.; Greenstein, P.E.; Sullivan, K.L.; Wecker, L.; Miller, A.; Jahan, I.; Chen, R.; Perlman, S.L. A randomized trial of varenicline (Chantix) for the treatment of spinocerebellar ataxia type 3. Neurology 2012, 78, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Chort, A.; Alves, S.; Marinello, M.; Dufresnois, B.A.; Dornbierer, J.-G.; Tesson, C.; Latouche, M.; Baker, D.P.; Barkats, M.; El Hachimi, K.H.; et al. Interferon beta induces clearance of mutant ataxin 7 and improves locomotion in SCA7 knock-in mice. Brain 2013, 136, 1732–1745. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Cui, Y.; Liu, Q.; Yang, Y.; Li, Y.; Weng, L.; Tang, B.; Jin, P.; Li, X.J.; Yang, S.; et al. Piperine ameliorates SCA17 neuropathology by reducing ER stress. Mol. Neurodegener. 2018, 13, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.M.; Chen, W.L.; Hung, C.T.; Lin, T.H.; Chao, C.Y.; Lin, C.H.; Wu, Y.R.; Chang, K.H.; Yao, C.F.; Lee-Chen, G.J.; et al. The indole compound NC009-1 inhibits aggregation and promotes neurite outgrowth through enhancement of HSPB1 in SCA17 cells and ameliorates the behavioral deficits in SCA17 mice. Neurotoxicology 2018, 67, 259–269. [Google Scholar] [CrossRef]
- Shiraishi, H.; Egawa, K.; Ito, T.; Kawano, O.; Asahina, N.; Kohsaka, S. Efficacy of perampanel for controlling seizures and improving neurological dysfunction in a patient with dentatorubral-pallidoluysian atrophy (DRPLA). Epilepsy Behav. Case Rep. 2017, 8, 44–46. [Google Scholar] [CrossRef] [PubMed]
- Neves-Carvalho, A.; Duarte-Silva, S.; Teixeira-Castro, A.; Maciel, P. Polyglutamine spinocerebellar ataxias: Emerging therapeutic targets. Expert Opin. Ther. Targets 2020, 24, 1099–1119. [Google Scholar] [CrossRef] [PubMed]
- Bonanomi, M.; Natalello, A.; Visentin, C.; Pastori, V.; Penco, A.; Cornelli, G.; Colombo, G.; Malabarba, M.G.; Doglia, S.M.; Relini, A.; et al. Epigallocatechin-3-gallate and tetracycline differently affect ataxin-3 fibrillogenesis and reduce toxicity in spinocerebellar ataxia type 3 model. Hum. Mol. Genet. 2014, 23, 6542–6552. [Google Scholar] [CrossRef] [Green Version]
- Minakawa, E.N.; Popiel, H.A.; Tada, M.; Takahashi, T.; Yamane, H.; Saitoh, Y.; Takahashi, Y.; Ozawa, D.; Takeda, A.; Takeuchi, T.; et al. Arginine is a disease modifier for polyQ disease models that stabilizes polyQ protein conformation. Brain 2020, 143, 1811–1825. [Google Scholar] [CrossRef]
- Popiel, H.A.; Nagai, Y.; Fujikake, N.; Toda, T. Protein transduction domain-mediated delivery of QBP1 suppresses polyglutamine-induced neurodegeneration in vivo. Mol. Ther. 2007, 15, 303–309. [Google Scholar] [CrossRef]
- Takeuchi, T.; Nagai, Y. Protein misfolding and aggregation as a therapeutic target for polyglutamine diseases. Brain Sci. 2017, 7, 128. [Google Scholar] [CrossRef] [Green Version]
- Silva-Fernandes, A.; Duarte-Silva, S.; Neves-Carvalho, A.; Amorim, M.; Soares-Cunha, C.; Oliveira, P.; Thirstrup, K.; Teixeira-Castro, A.; Maciel, P. Chronic Treatment with 17-DMAG Improves Balance and Coordination in A New Mouse Model of Machado-Joseph Disease. Neurotherapeutics 2014, 11, 433–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Adachi, H.; Katsuno, M.; Sahashi, K.; Kondo, N.; Iida, M.; Tohnai, G.; Nakatsuji, H.; Sobue, G. BIIB021, a synthetic Hsp90 inhibitor, induces mutant ataxin-1 degradation through the activation of Heat Shock Factor 1. Neuroscience 2016, 327, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Taldone, T.; Chiosis, G. Purine-scaffold Hsp90 inhibitors. IDrugs 2006, 9, 778–782. [Google Scholar] [CrossRef] [PubMed]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Oroz, L.G.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wullner, U.; Evert, B.O.; O’Kane, C.J.; et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2006, 15, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Huebener, J.; Renna, M.; Bonin, M.; Riess, O.; Rubinsztein, D.C. Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain 2010, 133, 93–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watase, K.; Gatchel, J.R.; Sun, Y.; Emamian, E.; Atkinson, R.; Richman, R.; Mizusawa, H.; Orr, H.T.; Shaw, C.; Zoghbi, H.Y. Lithium therapy improves neurological function and hippocampal dendritic arborization in a spinocerebellar ataxia type 1 mouse model. PLoS Med. 2007, 4, 836–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.-Z.; Wang, C.-M.; Lee, G.-C.; Hsu, H.-C.; Wu, T.-J.; Lin, C.W.; Ma, C.; Lee-Chen, G.; Huang, H.-J.; Hsieh-Li, H.M. Trehalose Attenuates the Gait Ataxia and Gliosis of Spinocerebellar Ataxia Type 17 Mice. Neurochem. Res. 2015, 40, 800–810. [Google Scholar] [CrossRef]
- Marcelo, A.; Brito, F.; Carmo-Silva, S.; Matos, C.A.; Alves-Cruzeiro, J.; Vasconcelos-Ferreira, A.; Koppenol, R.; Mendonça, L.; De Almeida, L.P.; Nóbrega, C. Cordycepin activates autophagy through AMPK phosphorylation to reduce abnormalities in Machado-Joseph disease models. Hum. Mol. Genet. 2019, 28, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.D.; Teixeira-Castro, A.; Maciel, P. From Pathogenesis to Novel Therapeutics for Spinocerebellar Ataxia Type 3: Evading Potholes on the Way to Translation. Neurotherapeutics 2019, 16, 1009–1031. [Google Scholar] [CrossRef]
- Wang, H.-L.; Hu, S.-H.; Chou, A.-H.; Wang, S.-S.; Weng, Y.-H.; Yeh, T.-H. H1152 promotes the degradation of polyglutamine-expanded ataxin-3 or ataxin-7 independently of its ROCK-inhibiting effect and ameliorates mutant ataxin-3-induced neurodegeneration in the SCA3 transgenic mouse. Neuropharmacology 2013, 70, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.K.; Bauer, P.O.; Kurosawa, M.; Goswami, A.; Washizu, C.; Machida, Y.; Tosaki, A.; Yamada, M.; Knöpfel, T.; Nakamura, T.; et al. Blocking acid-sensing ion channel 1 alleviates Huntington’s disease pathology via an ubiquitin-proteasome system-dependent mechanism. Hum. Mol. Genet. 2008, 17, 3223–3235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollitt, S.K.; Pallos, J.; Shao, J.; Desai, U.A.; Ma, A.A.K.; Thompson, L.M.; Marsh, J.L.; Diamond, M.I. A rapid cellular FRET assay of polyglutamine aggregation identifies a novel inhibitor. Neuron 2003, 40, 685–694. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.C.; Chang, C.N.; Chen, W.L.; Lin, T.H.; Chao, C.Y.; Lin, C.H.; Lin, H.Y.; Cheng, M.L.; Chiang, M.C.; Lin, J.Y.; et al. Targeting Ubiquitin Proteasome Pathway with Traditional Chinese Medicine for Treatment of Spinocerebellar Ataxia Type 3. Am. J. Chin. Med. 2019, 47, 63–95. [Google Scholar] [CrossRef]
- Chou, A.H.; Chen, Y.L.; Chiu, C.C.; Yuan, S.J.; Weng, Y.H.; Yeh, T.H.; Lin, Y.L.; Fang, J.M.; Wang, H.L. T1-11 and JMF1907 ameliorate polyglutamine-expanded ataxin-3-induced neurodegeneration, transcriptional dysregulation and ataxic symptom in the SCA3 transgenic mouse. Neuropharmacology 2015, 99, 308–317. [Google Scholar] [CrossRef]
- Johnson, J.A.; Johnson, D.A.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Vargas, M.R.; Chen, P.C. The Nrf2-ARE pathway: An indicator and modulator of oxidative stress in neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 61–69. [Google Scholar] [CrossRef]
- Liu, Y.; Hettinger, C.L.; Zhang, D.; Rezvani, K.; Wang, X.; Wang, H. Sulforaphane enhances proteasomal and autophagic activities in mice and is a potential therapeutic reagent for Huntington’s disease. J. Neurochem. 2014, 129, 539–547. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.H.; Lee, M.J.; Park, S.; Oh, D.C.; Elsasser, S.; Chen, P.C.; Gartner, C.; Dimova, N.; Hanna, J.; Gygi, S.P.; et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 2010, 467, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez, O.J.M.; Mollema, N.; Toker, N.; Adamski, C.J.; O’Callaghan, B.; Duvick, L.; Friedrich, J.; Walters, M.A.; Strasser, J.; Hawkinson, J.E.; et al. Reduction of Protein Kinase A-mediated Phosphorylation of ATXN1-S776 in Purkinje Cells Delays Onset of Ataxia in a SCA1 Mouse Model. Neurobiol. Dis. 2018, 116, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Vig, P.J.S.; Shao, Q.; Subramony, S.H.; Lopez, M.E.; Safaya, E. Bergmann glial S100B activates Myo-inositol monophosphatase 1 and Co-localizes to Purkinje cell vacuoles in SCA1 transgenic mice. Cerebellum 2009, 8, 231–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escalona-Rayo, O.; Fuentes-Vázquez, P.; Leyva-Gómez, G.; Cisneros, B.; Villalobos, R.; Magaña, J.J.; Quintanar-Guerrero, D. Nanoparticulate strategies for the treatment of polyglutamine diseases by halting the protein aggregation process. Drug Dev. Ind. Pharm. 2017, 43, 871–888. [Google Scholar] [CrossRef] [PubMed]
- Kuijper, E.C.; Bergsma, A.J.; Pijnappel, W.W.M.P.; Aartsma-Rus, A. Opportunities and challenges for antisense oligonucleotide therapies. J. Inherit. Metab. Dis. 2021, 44, 72–87. [Google Scholar] [CrossRef]
- Southwell, A.L.; Kordasiewicz, H.B.; Langbehn, D.; Skotte, N.H.; Parsons, M.P.; Villanueva, E.B.; Caron, N.S.; Ostergaard, M.E.; Anderson, L.M.; Xie, Y.; et al. Huntingtin suppression restores cognitive function in a mouse model of Huntington’s disease. Sci. Transl. Med. 2018, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Toonen, L.J.A.; Schmidt, I.; Luijsterburg, M.S.; Van Attikum, H.; Van Roon-mom, W.M.C. Antisense oligonucleotide-mediated exon skipping as a strategy to reduce proteolytic cleavage of ataxin-3. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Keiser, M.S.; Monteys, A.M.; Corbau, R.; Gonzalez-Alegre, P.; Davidson, B.L. RNAi prevents and reverses phenotypes induced by mutant human ataxin-1. Ann. Neurol. 2016, 80, 754–765. [Google Scholar] [CrossRef]
- Kole, R.; Krainer, A.R.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Mollanoori, H.; Rahmati, Y.; Hassani, B.; Mehr, M.H.; Teimourian, S. Promising therapeutic approaches using CRISPR/Cas9 genome editing technology in the treatment of Duchenne muscular dystrophy. Genes Dis. 2021, 8, 146–156. [Google Scholar] [CrossRef]
- Saha, S.K.; Saikot, F.K.; Rahman, M.S.; Jamal, M.A.H.M.; Rahman, S.M.K.; Islam, S.M.R.; Kim, K.H. Programmable Molecular Scissors: Applications of a New Tool for Genome Editing in Biotech. Mol. Ther. Nucleic Acids 2019, 14, 212–238. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.I.E.; Zain, R. Therapeutic Oligonucleotides: State of the Art. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 605–630. [Google Scholar] [CrossRef] [PubMed]
- Scoles, D.R.; Meera, P.; Schneider, M.D.; Paul, S.; Dansithong, W.; Figueroa, K.P.; Hung, G.; Rigo, F.; Bennett, C.F.; Otis, T.S.; et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature 2017, 544, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.R.; Rajpal, G.; Dillingham, I.T.; Qutob, M.; Blumenstein, K.G.; Gattis, D.; Hung, G.; Kordasiewicz, H.B.; Paulson, H.L.; McLoughlin, H.S. Evaluation of Antisense Oligonucleotides Targeting ATXN3 in SCA3 Mouse Models. Mol. Ther. Acids 2017, 7, 200–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLoughlin, H.S.; Moore, L.R.; Chopra, R.; Komlo, R.; McKenzie, M.; Blumenstein, K.G.; Zhao, H.; Kordasiewicz, H.B.; Shakkottai, V.G.; Paulson, H.L. Oligonucleotide therapy mitigates disease in Spinocerebellar Ataxia Type 3 mice. Ann. Neurol. 2018, 84, 64–77. [Google Scholar] [CrossRef] [PubMed]
- De Mezer, M.; Wojciechowska, M.; Napierala, M.; Sobczak, K.; Krzyzosiak, W.J. Mutant CAG repeats of Huntingtin transcript fold into hairpins, form nuclear foci and are targets for RNA interference. Nucleic Acids Res. 2011, 39, 3852–3863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourkouta, E.; Weij, R.; González-barriga, A.; Mulder, M.; Verheul, R.; Bosgra, S.; Groenendaal, B.; Puoliväli, J.; Toivanen, J.; Van Deutekom, J.C.T.; et al. Suppression of Mutant Protein Expression in SCA3 and SCA1 Mice Using a CAG Repeat-Targeting Antisense Oligonucleotide. Mol. Ther.-Nucleic Acid 2019, 17, 601–614. [Google Scholar] [CrossRef] [Green Version]
- Niu, C.; Prakash, T.P.; Kim, A.; Quach, J.L.; Huryn, L.A.; Yang, Y.; Lopez, E.; Jazayeri, A.; Hung, G.; La Spada, A.R.; et al. Antisense oligonucleotides targeting mutant Ataxin-7 restore visual function in a mouse model of spinocerebellar ataxia type 7. Sci. Transl. Med. 2018, 10, eaap8677. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Marque, L.O.; Cordner, Z.; Pruitt, J.L.; Bhat, M.; Li, P.P.; Kannan, G.; Ladenheim, E.E.; Moran, T.H.; Margolis, R.L.; et al. Phosphorodiamidate morpholino oligomers suppress mutant huntingtin expression and attenuate neurotoxicity. Hum. Mol. Genet. 2014, 23, 6302–6317. [Google Scholar] [CrossRef] [Green Version]
- Relizani, K.; Grif, G.; Echevarría, L.; Zarrouki, F.; Facchinetti, P.; Vaillend, C.; Leumann, C.; Garcia, L.; Goyenvalle, A. Efficacy and Safety Profile of Tricyclo-DNA Antisense Oligonucleotides in Duchenne Muscular Dystrophy Mouse Model. Mol. Ther. Acids 2017, 8, 144–157. [Google Scholar] [CrossRef] [Green Version]
- Evers, M.M.; Tran, H.D.; Zalachoras, I.; Pepers, B.A.; Meijer, O.C.; den Dunnen, J.T.; van Ommen, G.J.B.; Aartsma-Rus, A.; van Roon-Mom, W.M.C. Ataxin-3 protein modification as a treatment strategy for spinocerebellar ataxia type 3: Removal of the CAG containing exon. Neurobiol. Dis. 2013, 58, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yokota, T.; Matsumura, R.; Taira, K.; Mizusawa, H. Sequence-dependent and independent inhibition specific for mutant ataxin-3 by small interfering RNA. Ann. Neurol. 2004, 56, 124–129. [Google Scholar] [CrossRef]
- Keiser, M.S.; Boudreau, R.L.; Davidson, B.L. Broad therapeutic benefit after RNAi expression vector delivery to deep cerebellar nuclei: Implications for spinocerebellar ataxia type 1 therapy. Mol. Ther. 2014, 22, 588–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, H.; Mao, Q.; Eliason, S.L.; Harper, S.Q.; Martins, I.H.; Orr, H.T.; Paulson, H.L.; Yang, L.; Kotin, R.M.; Davidson, B.L. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat. Med. 2004, 10, 816–820. [Google Scholar] [CrossRef]
- Keiser, M.S.; Geoghegan, J.C.; Boudreau, R.L.; Lennox, K.A.; Davidson, B.L. RNAi or overexpression: Alternative therapies for Spinocerebellar Ataxia Type 1. Neurobiol. Dis. 2013, 56, 6–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keiser, M.S.; Kordower, J.H.; Gonzalez-Alegre, P.; Davidson, B.L. Broad distribution of ataxin 1 silencing in rhesus cerebella for spinocerebellar ataxia type 1 therapy. Brain 2015, 138, 3555–3566. [Google Scholar] [CrossRef]
- Alves, S.; Nascimento-Ferreira, I.; Dufour, N.; Hassig, R.; Auregan, G.; Nóbrega, C.; Brouillet, E.; Hantraye, P.; de Lima, M.C.P.; Déglon, N.; et al. Silencing ataxin-3 mitigates degeneration in a rat model of Machado-Joseph disease: No role for wild-type ataxin-3? Hum. Mol. Genet. 2010, 19, 2380–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Lebrón, E.; Costa, M.D.; Luna-Cancalon, K.; Peron, T.M.; Fischer, S.; Boudreau, R.L.; Davidson, B.L.; Paulson, H.L. Silencing mutant ATXN3 expression resolves molecular phenotypes in SCA3 transgenic mice. Mol. Ther. 2013, 21, 1909–1918. [Google Scholar] [CrossRef] [Green Version]
- Do Carmo Costa, M.; Luna-Cancalon, K.; Fischer, S.; Ashraf, N.S.; Ouyang, M.; Dharia, R.M.; Martin-Fishman, L.; Yang, Y.; Shakkottai, V.G.; Davidson, B.L.; et al. Toward RNAi therapy for the polyglutamine disease Machado-Joseph disease. Mol. Ther. 2013, 21, 1898–1908. [Google Scholar] [CrossRef] [Green Version]
- Costa, C.; Paulson, H.L. Toward understanding Machado-Joseph disease. Prog. Neurobiol. 2012, 97, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Conceição, M.; Mendonça, L.; Nóbrega, C.; Gomes, C.; Costa, P.; Hirai, H.; Moreira, J.N.; Lima, M.C.; Manjunath, N.; de Almeida, L.P. Intravenous administration of brain-targeted stable nucleic acid lipid particles alleviates Machado-Joseph disease neurological phenotype. Biomaterials 2016, 82, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Huang, F.; Tang, B.; Li, J.; Wang, J.; Shen, L.; Xia, K.; Jiang, H. MicroRNA profiling in the serums of SCA3/MJD patients. Int. J. Neurosci. 2014, 124, 97–101. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Du, X.; Muramatsu, S. An miRNA-mediated therapy for SCA6 blocks IRES-driven translation of the CACNA1A second cistron. Sci. Transl. Med. 2016, 8, 347ra94. [Google Scholar] [CrossRef] [Green Version]
- Kubodera, T.; Yokota, T.; Ishikawa, K.; Mizusawa, H. New RNAi strategy for selective suppression of a mutant allele in polyglutamine disease. Oligonucleotides 2005, 15, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.S.; Bhattarai, S.; Singh, P.; Boudreau, R.L.; Thompson, S.; Laspada, A.R.; Drack, A.V.; Davidson, B.L. RNA Interference-Based Therapy for Spinocerebellar Ataxia Type 7 Retinal Degeneration. PLoS ONE 2014, 9, e95362. [Google Scholar] [CrossRef]
- Ramachandran, P.S.; Boudreau, R.L.; Schaefer, K.A.; La Spada, A.R.; Davidson, B.L. Nonallele specific silencing of ataxin-7 improves disease phenotypes in a mouse model of SCA7. Mol. Ther. 2014, 22, 1635–1642. [Google Scholar] [CrossRef] [Green Version]
- Fiszer, A.; Olejniczak, M.; Galka-Marciniak, P.; Mykowska, A.; Krzyzosiak, W.J. Self-duplexing CUG repeats selectively inhibit mutant huntingtin expression. Nucleic Acids Res. 2013, 41, 10426–10437. [Google Scholar] [CrossRef]
- Fiszer, A.; Ellison-Klimontowicz, M.E.; Krzyzosiak, W.J. Silencing of genes responsible for polyQ diseases using chemically modified single-stranded siRNAs. Acta Biochim. Pol. 2016, 63, 759–764. [Google Scholar] [CrossRef]
- Fiszer, A.; Wroblewska, J.P.; Nowak, B.M.; Krzyzosiak, W.J. Mutant CAG repeats effectively targeted by RNA interference in SCA7 cells. Genes 2016, 7, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, H.J.; Seow, Y.; Wood, M.J.A.; Varela, M.A. Knockdown and replacement therapy mediated by artificial mirtrons in spinocerebellar ataxia 7. Nucleic Acids Res. 2018, 45, 7870–7885. [Google Scholar] [CrossRef] [Green Version]
- Southwell, A.L.; Skotte, N.H.; Kordasiewicz, H.B.; Østergaard, M.E.; Watt, A.T.; Carroll, J.B.; Doty, C.N.; Villanueva, E.B.; Petoukhov, E.; Vaid, K.; et al. In vivo evaluation of candidate allele-specific mutant huntingtin gene silencing antisense oligonucleotides. Mol. Ther. 2014, 22, 2093–2106. [Google Scholar] [CrossRef] [Green Version]
- Stanek, L.M.; Sardi, S.P.; Mastis, B.; Richards, A.R.; Treleaven, C.M.; Taksir, T.; Misra, K.; Cheng, S.H.; Shihabuddin, L.S. Silencing mutant huntingtin by adeno-associated virus-mediated RNA interference ameliorates disease manifestations in the YAC128 mouse model of Huntington’s Disease. Hum. Gene Ther. 2014, 25, 461–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Liu, J.; Narayanannair, K.J.; Lackey, J.G.; Kuchimanchi, S.; Rajeev, K.G.; Manoharan, M.; Swayze, E.E.; Lima, W.F.; Prakash, T.P.; et al. Allele-selective inhibition of mutant atrophin-1 expression by duplex and single-stranded RNAs. Biochemistry 2014, 53, 4510–4518. [Google Scholar] [CrossRef]
- Magaña, J.J.; Gómez, R.; Maldonado-Rodríguez, M.; Velázquez-Pérez, L.; Tapia-Guerrero, Y.S.; Cortés, H.; Leyva-García, N.; Hernández-Hernández, O.; Cisneros, B. Origin of the spinocerebellar ataxia type 7 gene mutation in mexican population. Cerebellum 2013, 12, 902–905. [Google Scholar] [CrossRef]
- Greenberg, J.; Solomon, G.A.E.; Vorster, A.A.; Heckmann, J.; Bryer, A. Origin of the SCA7 gene mutation in South Africa: Implications for molecular diagnostics. Clin. Genet. 2006, 70, 415–417. [Google Scholar] [CrossRef]
- Scholefield, J.; Watson, L.; Smith, D.; Greenberg, J.; Wood, M.J. Allele-specific silencing of mutant Ataxin-7 in SCA7 patient-derived fibroblasts. Eur. J. Hum. Genet. 2014, 22, 1369–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailus, B.; Zhang, N.; Ellerby, L.M. Using genome engineering to understand huntington’s disease. In Genome Editing in Neurosciences. Research and Perspectives in Neurosciences; Jaenisch, R., Zhang, F., Gage, F., Eds.; Springer: Cham, Switzerland, 2017; pp. 87–101. ISBN 9783319601922. [Google Scholar]
- Malankhanova, T.B.; Malakhova, A.A.; Medvedev, S.P.; Zakian, S.M. Modern Genome Editing Technologies in Huntington’s Disease Research. J. Huntingt. Dis. 2017, 6, 19–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.W.; Kim, K.H.; Chao, M.J.; Atwal, R.S.; Gillis, T.; MacDonald, M.E.; Gusella, J.F.; Lee, J.M. Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum. Mol. Genet. 2016, 25, 4566–4576. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, S.; Xie, Y.; Xiong, Z.; Yang, Y.; Xian, Y.; Ou, Z.; Song, B.; Chen, Y.; Xie, Y.; Li, H.; et al. CRISPR/Cas9-Targeted deletion of polyglutamine in spinocerebellar ataxia type 3-derived induced pluripotent stem cells. Stem Cells Dev. 2018, 27, 756–770. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Tee, L.Y.; Wang, X.G.; Huang, Q.S.; Yang, S.H. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol. Ther.-Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef] [PubMed]
- Sazani, P.; Gemignani, F.; Kang, S.H.; Maier, M.A.; Manoharan, M.; Persmark, M.; Bortner, D.; Kole, R. Systemically delivered antisense oligomers upregulate gene expression in mouse tissues. Nat. Biotechnol. 2002, 20, 1228–1233. [Google Scholar] [CrossRef] [PubMed]
- Casaca-Carreira, J.; Temel, Y.; Larrakoetxea, I.; Jahanshahi, A. Distribution and Penetration of Intracerebroventricularly Administered 2′OMePS Oligonucleotide in the Mouse Brain. Nucleic Acid Ther. 2017, 27, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Song, E.; Zhu, P.; Lee, S.K.; Chowdhury, D.; Kussman, S.; Dykxhoorn, D.M.; Feng, Y.; Palliser, D.; Weiner, D.B.; Shankar, P.; et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat. Biotechnol. 2005, 23, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.P.; Youngblood, D.S.; Hassinger, J.N.; Lovejoy, C.E.; Nelson, M.H.; Iversen, P.L.; Moulton, H.M. Cell-penetrating peptides as transporters for morpholino oligomers: Effects of amino acid composition on intracellular delivery and cytotoxicity. Nucleic Acids Res. 2007, 35, 5182–5191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopalco, A.; Cutrignelli, A.; Denora, N.; Lopedota, A.; Franco, M.; Laquintana, V. Transferrin functionalized liposomes loading dopamine HCl: Development and permeability studies across an In vitro model of human blood-brain barrier. Nanomaterials 2018, 8, 178. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Moulton, H.M.; Betts, C.; Seow, Y.; Boutilier, J.; Iverson, P.L.; Wood, M.J.A. A fusion peptide directs enhanced systematic dystrophin exon skipping and functional restoration in dystrophin-deficient mdx mice. Hum. Mol. Genet. 2009, 18, 4405–4414. [Google Scholar] [CrossRef] [Green Version]
- McBride, J.L.; Boudreau, R.L.; Harper, S.Q.; Staber, P.D.; Monteys, A.M.; Martins, I.; Gilmore, B.L.; Burstein, H.; Peluso, R.W.; Polisky, B.; et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: Implications for the therapeutic development of RNAi. Proc. Natl. Acad. Sci. USA 2008, 105, 5868–5873. [Google Scholar] [CrossRef] [Green Version]
- Ehlert, E.M.; Eggers, R.; Niclou, S.P.; Verhaagen, J. Cellular toxicity following application of adeno-associated viral vector-mediated RNA interference in the nervous system. BMC Neurosci. 2010, 11. [Google Scholar] [CrossRef] [Green Version]
- Conniot, J.; Talebian, S.; Simões, S.; Ferreira, L.; Conde, J. Revisiting gene delivery to the brain: Silencing and editing. Biomater. Sci. 2021, 9, 1065–1087. [Google Scholar] [CrossRef]
- Leyva-Gómez, G.; Cortés, H.; Magaña, J.J.; Leyva-García, N.; Quintanar-Guerrero, D.; Florán, B. Nanoparticle technology for treatment of Parkinson’s disease: The role of surface phenomena in reaching the brain. Drug Discov. Today 2015, 20, 824–837. [Google Scholar] [CrossRef]
- Singha, K.; Namgung, R.; Kim, W.J. Polymers in small-interfering RNA delivery. Nucleic Acid Ther. 2011, 21, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.J.; Martins, S.; Sarmento, B. SiRNA as a tool to improve the treatment of brain diseases: Mechanism, targets and delivery. Ageing Res. Rev. 2015, 21, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, S.; Archunan, G.; Sivakumar, M.; Selvan, S.T.; Fred, A.L.; Kumar, S.; Gulyás, B.; Padmanabhan, P. Theranostic applications of nanoparticles in neurodegenerative disorders. Int. J. Nanomed. 2018, 13, 5561–5576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreuter, J. Drug delivery to the central nervous system by polymeric nanoparticles: What do we know? Adv. Drug Deliv. Rev. 2014, 71, 2–14. [Google Scholar] [CrossRef]
- Kim, B.; Park, J.H.; Sailor, M.J. Rekindling RNAi Therapy: Materials Design Requirements for In Vivo siRNA Delivery. Adv. Mater. 2019, 31, 1903637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, R.; Singh, D.; Prakash, A.; Mishra, N. Development, characterization and nasal delivery of rosmarinic acid-loaded solid lipid nanoparticles for the effective management of Huntingtons disease. Drug Deliv. 2015, 22, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Rassu, G.; Soddu, E.; Maria, A.; Pintus, G.; Sarmento, B.; Giunchedi, P.; Gavini, E. Nose-to-brain delivery of BACE1 siRNA loaded in solid lipid nanoparticles for Alzheimer’s therapy. Colloids Surf. B Biointerfaces 2017, 152, 296–301. [Google Scholar] [CrossRef]
- Gomes, M.J.; Fernandes, C.; Martins, S.; Borges, F.; Sarmento, B. Tailoring Lipid and Polymeric Nanoparticles as siRNA Carriers towards the Blood-Brain Barrier—From Targeting to Safe Administration. J. Neuroimmune Pharmacol. 2017, 12, 107–119. [Google Scholar] [CrossRef]
- Li, J.; Xue, S.; Mao, Z.W. Nanoparticle delivery systems for siRNA-based therapeutics. J. Mater. Chem. B 2016, 4, 6620–6639. [Google Scholar] [CrossRef]
- Wang, J.; Lu, Z.; Wientjes, M.G.; Au, J.L.S. Delivery of siRNA therapeutics: Barriers and carriers. AAPS J. 2010, 12, 492–503. [Google Scholar] [CrossRef]
- Risnayanti, C.; Jang, Y.-S.; Lee, J.; Ahn, H.J. PLGA nanoparticles co-delivering MDR1 and BCL2 siRNA for overcoming resistance of paclitaxel and cisplatin in recurrent or advanced ovarian cancer. Sci. Rep. 2018, 8, 7498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do Carmo Costa, M.; Ashraf, N.S.; Fischer, S.; Yang, Y.; Schapka, E.; Joshi, G.; Mcquade, T.J.; Dharia, R.M.; Dulchavsky, M.; Ouyang, M.; et al. Unbiased screen identifies aripiprazole as a modulator of abundance of the polyglutamine disease protein, ataxin-3. Brain 2016, 139, 2891–2908. [Google Scholar] [CrossRef]
- Sawant, K.; Pandey, A.; Patel, S. Aripiprazole loaded poly(caprolactone) nanoparticles: Optimization and in vivo pharmacokinetics. Mater. Sci. Eng. C 2016, 66, 230–243. [Google Scholar] [CrossRef]
- Del Prado-Audelo, M.L.; Magaña, J.J.; Mejía-Contreras, B.A.; Borbolla-Jiménez, F.V.; Giraldo-Gomez, D.M.; Piña-Barba, M.C.; Quintanar-Guerrero, D.; Leyva-Gómez, G. In vitro cell uptake evaluation of curcumin-loaded PCL/F68 nanoparticles for potential application in neuronal diseases. J. Drug Deliv. Sci. Technol. 2019, 52, 905–914. [Google Scholar] [CrossRef]
- Gu, J.; Al-Bayati, K.; Ho, E.A. Development of antibody-modified chitosan nanoparticles for the targeted delivery of siRNA across the blood-brain barrier as a strategy for inhibiting HIV replication in astrocytes. Drug Deliv. Transl. Res. 2017, 7, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Wang, N.; Zeng, Z.; Huang, J.; Xiang, Z.; Guan, Y.Q. Neuroprotective effect of chitosan nanoparticle gene delivery system grafted with acteoside (ACT) in Parkinson’s disease models. J. Mater. Sci. Technol. 2020, 43, 197–207. [Google Scholar] [CrossRef]
- Godinho, B.M.D.C.; Ogier, J.R.; Darcy, R.; O’Driscoll, C.M.; Cryan, J.F. Self-assembling modified β-cyclodextrin nanoparticles as neuronal siRNA delivery vectors: Focus on huntington’s disease. Mol. Pharm. 2013, 10, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Riley, M.K.; Vermerris, W. Recent advances in nanomaterials for gene delivery—A review. Nanomaterials 2017, 7, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández, M.; Leyva, G.; Magaña, J.J.; Guzmán-Vargas, A.; Felipe, C.; Lara, V.; Lima, E. New copolymers as hosts of Ribosomal RNA. BMC Chem. 2019, 13, 1–12. [Google Scholar] [CrossRef]
- Sizovs, A.; Xue, L.; Tolstyka, Z.P.; Ingle, N.P.; Wu, Y.; Cortez, M.; Reineke, T.M. Poly(trehalose): Sugar-coated nanocomplexes promote stabilization and effective polyplex-mediated siRNA delivery. J. Am. Chem. Soc. 2013, 135, 15417–15424. [Google Scholar] [CrossRef]
- Morris, V.B.; Labhasetwar, V. Arginine-rich polyplexes for gene delivery to neuronal cells. Biomaterials 2015, 60, 151–160. [Google Scholar] [CrossRef] [Green Version]
- Koji, K.; Yoshinaga, N.; Mochida, Y.; Hong, T.; Miyazaki, T.; Kataoka, K.; Osada, K.; Cabral, H.; Uchida, S. Bundling of mRNA strands inside polyion complexes improves mRNA delivery efficiency in vitro and in vivo. Biomaterials 2020, 261, 120332. [Google Scholar] [CrossRef]
- Debnath, K.; Pradhan, N.; Singh, B.K.; Jana, N.R.; Jana, N.R. Poly(trehalose) Nanoparticles Prevent Amyloid Aggregation and Suppress Polyglutamine Aggregation in a Huntington’s Disease Model Mouse. ACS Appl. Mater. Interfaces 2017, 9, 24126–24139. [Google Scholar] [CrossRef] [PubMed]
- Lalatsa, A.; Schatzlein, A.G.; Uchegbu, I.F. Strategies to deliver peptide drugs to the brain. Mol. Pharm. 2014, 11, 1081–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhaveri, A.M.; Torchilin, V.P. Multifunctional polymeric micelles for delivery of drugs and siRNA. Front. Pharmacol. 2014, 5, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Zhang, Y.; Tan, Y.Z.; Hu, K.L.; Jiang, X.G.; Fu, S.K. Cationic albumin-conjugated pegylated nanoparticles as novel drug carrier for brain delivery. J. Control. Release 2005, 107, 428–448. [Google Scholar] [CrossRef] [PubMed]
- Parikh, T.; Bommana, M.M.; Squillante, E. Efficacy of surface charge in targeting pegylated nanoparticles of sulpiride to the brain. Eur. J. Pharm. Biopharm. 2010, 74, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.A.; Narang, A.S.; Kotb, M.; Gaber, A.O.; Miller, D.D.; Kim, S.W.; Mahato, R.I. Novel branched poly(ethylenimine)-Cholesterol water-soluble lipopolymers for gene delivery. Biomacromolecules 2002, 3, 1197–1207. [Google Scholar] [CrossRef]
- Kreuter, J.; Shamenkov, D.; Petrov, V.; Ramge, P.; Cychutek, K.; Koch-Brandt, C.; Alyautdin, R. Apolipoprotein-mediated transport of nanoparticle-bound drugs across the blood-brain barrier. J. Drug Target. 2002, 10, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Cao, S.; Yang, Z.; Zhang, S.; Zhang, Q.; Jiang, X. Preparation, characterization and anti-glioma effects of docetaxel-incorporated albumin-lipid nanoparticles. J. Biomed. Nanotechnol. 2015, 11, 2137–2147. [Google Scholar] [CrossRef]
- Yoo, J.; Park, C.; Yi, G.; Lee, D.; Koo, H. Active targeting strategies using biological ligands for nanoparticle drug delivery systems. Cancers 2019, 11, 640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadhvi, V.; Brijesh, K.; Gupta, A.; Roopchandani, K.; Patel, N. Nanoparticles for brain targeting. Res. J. Pharm. Technol. 2013, 6, 454–458. [Google Scholar]
- Georgieva, J.V.; Hoekstra, D.; Zuhorn, I.S. Smuggling drugs into the brain: An overview of ligands targeting transcytosis for drug delivery across the blood-brain barrier. Pharmaceutics 2014, 6, 557–583. [Google Scholar] [CrossRef] [Green Version]
- Pulgar, V.M. Transcytosis to cross the blood brain barrier, new advancements and challenges. Front. Neurosci. 2019, 13, 1–9. [Google Scholar] [CrossRef]
- Jiang, X.; Xin, H.; Ren, Q.; Gu, J.; Zhu, L.; Du, F.; Feng, C.; Xie, Y.; Sha, X.; Fang, X. Nanoparticles of 2-deoxy-d-glucose functionalized poly(ethylene glycol)-co-poly(trimethylene carbonate) for dual-targeted drug delivery in glioma treatment. Biomaterials 2014, 35, 518–529. [Google Scholar] [CrossRef]
- Tamba, B.I.; Streinu, V.; Foltea, G.; Neagu, A.N.; Dodi, G.; Zlei, M.; Tijani, A.; Stefanescu, C. Tailored surface silica nanoparticles for blood-brain barrier penetration: Preparation and in vivo investigation. Arab. J. Chem. 2018, 11, 981–990. [Google Scholar] [CrossRef]
- Aso, E.; Martinsson, I.; Appelhans, D.; Effenberg, C.; Benseny-cases, N.; Cladera, J.; Gouras, G.; Ferrer, I.; Klementieva, O. Poly (propylene imine) dendrimers with histidine-maltose shell as novel type of nanoparticles for synapse and memory protection. Nanomed. Nanotechnol. Biol. Med. 2019, 17, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Al-azzawi, S.; Masheta, D.; Guildford, A.L.; Phillips, G. Dendrimeric Poly (Epsilon-Lysine) Delivery Systems for the Enhanced Permeability of Flurbiprofen across the Blood-Brain Barrier in Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 3224. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Carter, D.; Liu, X.; Tockary, T.A.; Dirisala, A.; Toh, K. Targeting nanoparticles to the brain by exploiting the blood–brain barrier impermeability to selectively label the brain endothelium. Proc. Natl. Acad. Sci. USA 2020, 117, 19141–19250. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, H.; Zhang, Y.; Jiang, X.; Guo, Y.; An, S.; He, X.; Jiang, C. Choline Derivate-Modified Doxorubicin Loaded Micelle for Glioma Therapy. Appl. Mater. Interfaces 2015, 7, 21589–21601. [Google Scholar] [CrossRef] [PubMed]
- Tremmel, R.; Uhl, P.; Helm, F.; Wupperfeld, D.; Sauter, M.; Mier, W.; Stremmel, W.; Hofhaus, G.; Fricker, G. Delivery of Copper-chelating Trientine (TETA) to the central nervous system by surface modi fi ed liposomes. Int. J. Pharm. 2016, 512, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Ahlschwede, K.M.; Curran, G.L.; Rosenberg, J.T.; Grant, S.C.; Sarkar, G.; Jenkins, R.B.; Ramakrishnan, S.; Poduslo, J.F.; Kandimalla, K.K. Cationic carrier peptide enhances cerebrovascular targeting of nanoparticles in Alzheimer’s disease brain. Nanomed. Nanotechnol. Biol. Med. 2019, 16, 258–266. [Google Scholar] [CrossRef] [PubMed]
- de Castro, R.R.; do Carmo, F.A.; Martins, C.; Simon, A.; Ribeiro, R.; Castro, D.; Almada, F.; De Sousa, V.P.; Rodrigues, C.R.; Mendes, L.; et al. Clofazimine functionalized polymeric nanoparticles for brain delivery in the tuberculosis treatment. Int. J. Pharm. 2021, 602, 120655. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Wang, A.; Yan, X.; Chu, L.; Yang, X. Brain-targeted intranasal delivery of dopamine with borneol and lactoferrin co-modified nanoparticles for treating Parkinson’s disease. Drug Deliv. 2019, 26, 700–707. [Google Scholar] [CrossRef] [Green Version]
- Pandey, V.; Haider, T.; Chandak, A.R.; Chakraborty, A.; Banerjee, S.; Soni, V. Surface modified silk fibroin nanoparticles for improved delivery of doxorubicin: Development, characterization, in-vitro studies. Int. J. Biol. Macromol. 2020, 164, 2018–2027. [Google Scholar] [CrossRef]
- Kuo, Y.; Rajesh, R. Targeted delivery of rosmarinic acid across the blood–brain barrier for neuronal rescue using polyacrylamide-chitosan-poly (lactide-co-glycolide) nanoparticles with surface cross-reacting material 197 and apolipoprotein E. Int. J. Pharm. 2017, 528, 228–241. [Google Scholar] [CrossRef]
- Hu, Y.; Gaillard, P.J.; De Lange, E.C.M.; Hammarlund-udenaes, M. Targeted brain delivery of methotrexate by glutathione PEGylated liposomes: How can the formulation make a difference? Eur. J. Pharm. Biopharm. 2019, 139, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Navale, A.M.; Paranjape, A.N. Glucose transporters: Physiological and pathological roles. Biophys. Rev. 2016, 8, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Benarroch, E.E. Brain glucose transporters: Implications for neurologic disease. Neurology 2014, 82, 1374–1379. [Google Scholar] [CrossRef]
- Komuro, H.; Sasano, T.; Horiuchi, N.; Yamashita, K.; Nagai, A. The effect of glucose modification of hydroxyapatite nanoparticles on gene delivery. J. Biomed. Mater. Res. Part A 2019, 107, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Johnsen, K.B.; Burkhart, A.; Melander, F.; Kempen, P.J.; Vejlebo, J.B.; Siupka, P.; Nielsen, M.S.; Andresen, T.L.; Moos, T. Targeting transferrin receptors at the blood-brain barrier improves the uptake of immunoliposomes and subsequent cargo transport into the brain parenchyma. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Choudhury, H.; Pandey, M.; Chin, P.X.; Phang, Y.L.; Cheah, J.Y.; Ooi, S.C.; Mak, K.K.; Pichika, M.R.; Kesharwani, P.; Hussain, Z.; et al. Transferrin receptors-targeting nanocarriers for efficient targeted delivery and transcytosis of drugs into the brain tumors: A review of recent advancements and emerging trends. Drug Deliv. Transl. Res. 2018, 8, 1545–1563. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Ke, W.; Liu, Y.; Jiang, C.; Pei, Y. The use of lactoferrin as a ligand for targeting the polyamidoamine-based gene delivery system to the brain. Biomaterials 2008, 29, 238–246. [Google Scholar] [CrossRef]
- Wei, L.; Guo, X.Y.; Yang, T.; Yu, M.Z.; Chen, D.W.; Wang, J.C. Brain tumor-targeted therapy by systemic delivery of siRNA with Transferrin receptor-mediated core-shell nanoparticles. Int. J. Pharm. 2016, 510, 394–405. [Google Scholar] [CrossRef]
- dos Santos Rodrigues, B.; Kanekiyo, T.; Singh, J. ApoE-2 Brain-Targeted Gene Therapy Through Transferrin and Penetratin Tagged Liposomal Nanoparticles. Pharm. Res. 2019, 36, 161. [Google Scholar] [CrossRef]
- Kumari, S.; Ahsan, S.M.; Kumar, J.M.; Kondapi, A.K.; Rao, N.M. Overcoming blood brain barrier with a dual purpose Temozolomide loaded Lactoferrin nanoparticles for combating glioma (SERP-17-12433). Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Ke, W.; Liu, Y.; Wu, D.; Feng, L.; Jiang, C.; Pei, Y. Gene therapy using lactoferrin-modified nanoparticles in a rotenone-induced chronic Parkinson model. J. Neurol. Sci. 2010, 290, 123–130. [Google Scholar] [CrossRef]
- Huang, R.; Ke, W.; Han, L.; Liu, Y.; Shao, K.; Jiang, C.; Pei, Y. Lactoferrin-modified nanoparticles could mediate efficient gene delivery to the brain in vivo. Brain Res. Bull. 2010, 81, 600–604. [Google Scholar] [CrossRef]
- Liu, Z.; Jiang, M.; Kang, T.; Miao, D.; Gu, G.; Song, Q.; Yao, L.; Hu, Q.; Tu, Y.; Pang, Z.; et al. Lactoferrin-modified PEG-co-PCL nanoparticles for enhanced brain delivery of NAP peptide following intranasal administration. Biomaterials 2013, 34, 3870–3881. [Google Scholar] [CrossRef]
- Hu, K.; Li, J.; Shen, Y.; Lu, W.; Gao, X.; Zhang, Q.; Jiang, X. Lactoferrin-conjugated PEG-PLA nanoparticles with improved brain delivery: In vitro and in vivo evaluations. J. Control Release 2009, 134, 55–61. [Google Scholar] [CrossRef]
- Meng, Q.; Hua, H.; Jiang, Y.; Wang, Y.; Mu, H.; Wu, Z. Intranasal delivery of Huperzine A to the brain using lactoferrin-conjugated N-trimethylated chitosan surface-modified PLGA nanoparticles for Alzheimer’s disease. Int. J. Nanomed. 2018, 13, 705–718. [Google Scholar] [CrossRef] [Green Version]
- Lajoie, J.M.; Shusta, E.V. Targeting receptor-mediated transport for delivery of biologics across the blood-brain barrier. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 613–631. [Google Scholar] [CrossRef] [Green Version]
- Kuo, Y.C.; Shih-Huang, C.Y. Solid lipid nanoparticles carrying chemotherapeutic drug across the blood-brain barrier through insulin receptor-mediated pathway. J. Drug Target. 2013, 21, 730–738. [Google Scholar] [CrossRef]
- Ulbrich, K.; Knobloch, T.; Kreuter, J. Targeting the insulin receptor: Nanoparticles for drug delivery across the blood-brain barrier (BBB). J. Drug Target. 2011, 19, 125–132. [Google Scholar] [CrossRef]
- Wagner, S.; Zensi, A.; Wien, S.L.; Tschickardt, S.E.; Maier, W.; Vogel, T.; Worek, F.; Pietrzik, C.U.; Kreuter, J.; von Briesen, H. Uptake mechanism of ApoE-modified nanoparticles on brain capillary endothelial cells as a blood-brain barrier model. PLoS ONE 2012, 7, e32568. [Google Scholar] [CrossRef]
- Neves, A.R.; Queiroz, J.F.; Reis, S. Brain-targeted delivery of resveratrol using solid lipid nanoparticles functionalized with apolipoprotein E. J. Nanobiotechnol. 2016, 14, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Tamaru, M.; Akita, H.; Kajimoto, K.; Sato, Y.; Hatakeyama, H.; Harashima, H. An apolipoprotein e modified liposomal nanoparticle: Ligand dependent efficiency as a siRNA delivery carrier for mouse-derived brain endothelial cells. Int. J. Pharm. 2014, 465, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.S.; Singh, V.; Gahane, A.; Thakur, A.K. Biodegradable Nanoparticles Containing Mechanism Based Peptide Inhibitors Reduce Polyglutamine Aggregation in Cell Models and Alleviate Motor Symptoms in a Drosophila Model of Huntington’s Disease. ACS Chem. Neurosci. 2019, 10, 1603–1614. [Google Scholar] [CrossRef]
- Mandal, S.; Debnath, K.; Jana, N.R.; Jana, N.R. Trehalose-Conjugated, Catechin-Loaded Polylactide Nanoparticles for Improved Neuroprotection against Intracellular Polyglutamine Aggregates. Biomolecules 2020, 21, 1578–1586. [Google Scholar] [CrossRef]
- Hirunagi, T.; Sahashi, K.; Tachikawa, K.; Leu, A.I.; Nguyen, M.; Mukthavaram, R.; Karmali, P.P.; Chivukula, P.; Tohnai, G.; Iida, M.; et al. Selective suppression of polyglutamine-expanded protein by lipid nanoparticle-delivered siRNA targeting CAG expansions in the mouse CNS. Mol. Ther. Nucleic Acids 2021, 24, 1–10. [Google Scholar] [CrossRef]
- Cortés, H.; Alcalá-Alcalá, S.; Ávalos-Fuentes, A.; Mendoza-Muñoz, N.; Quintanar-Guerrero, D.; Leyva-Gómez, G.; Florán, B. Nanotechnology as potential tool for siRNA delivery in Parkinson’s disease. Curr. Drug Targets 2017, 18, 1866–1879. [Google Scholar] [CrossRef]
- Farkhani, S.M.; Valizadeh, A.; Karami, H.; Mohammadi, S.; Sohrabi, N.; Badrzadeh, F. Cell penetrating peptides: Efficient vectors for delivery of nanoparticles, nanocarriers, therapeutic and diagnostic molecules. Peptides 2014, 57, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Gessner, I.; Klimpel, A.; Klußmann, M.; Neundorf, I.; Mathur, S. Interdependence of charge and secondary structure on cellular uptake of cell penetrating peptide functionalized silica nanoparticles. Nanoscale Adv. 2020, 2, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Yameen, B.; Choi, W.I.; Vilos, C.; Swami, A.; Shi, J.; Farokhzad, O.C. Insight into nanoparticle cellular uptake and intracellular targeting. J. Control Release 2014, 190, 485–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Uertz, J.; Yohan, D.; Chithrani, B.D. Peptide modified gold nanoparticles for improved cellular uptake, nuclear transport, and intracellular retention. Nanoscale 2014, 6, 12026–12033. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Gene | Mutation (Localization) | Normal Alleles | Full Penetration Alleles | OMIM | Clinical Features | Neuropathological Findings | Ref. |
|---|---|---|---|---|---|---|---|---|
| SCA1 | ATXN1 | (CAG)n Exon 8 6p22-23 | 6–39 | >40 | 164400 | Ataxia, slurred speech, spasticity, cognitive impairment | Atrophy of cerebellum, pons and olives. Degeneration of lower cranial nerve nuclei, and atrophy of the dorsal columns, and spinocerebellar tracts. Loss of Purkinje cells, neurons of dentate gyrus, Bergmann’s gliosis, mesencephalic neurons in 3rd and 4th cranial nerves, variable loss of granule cells, atrophy of middle cerebellar peduncles. Intranuclear inclusions. | [15,16] |
| SCA2 | ATXN2 | (CAG)n Exon 1 12q23-24.12 | 14–31 | >34 | 183090 | Ataxia, slow saccades, decreased reflexes, polyneuropathy, motor neuropathy, infantile variant | Atrophy of cerebellum, pons, frontal lobe, medulla oblongata, cranial nerves, as well as pallor of the midbrain substantia nigra. Cytoplasmic inclusions. | [17] |
| SCA3 | ATXN3 | (CAG)n Exon 10 14q32.1 | 12–44 | >52 | 109150 | Ataxia, parkinsonism, severe spasticity | Loss of neurons and gliosis in the substantia nigra, pontine nuclei, nuclei of the vestibular and cranial nerves, columns of Clarke and anterior horns. The cerebellum is relatively spared, spinal cord with loss of myelinated fibers in the spinocerebellar tracts and posterior funiculi. Intranuclear and cytoplasmic inclusions. | [18] |
| SCA6 | CACNA1A | (CAG)n Exon 47 19p13 | 4–18 | >19 | 183086 | Ataxia, dysarthria, nystagmus, tremor | Selective atrophy of the cerebellum and extensive loss of PC in the cerebellar cortex. Numerous oval- or rod-shaped, not ubiquitinated aggregates are seen exclusively in the cytoplasm of PC. | [19] |
| SCA7 | ATXN7 | (CAG)n Exon 3 3p12-21.1 | 4–35 | >47 | 164500 | Ataxia, retinal degeneration | Neuronal intranuclear inclusions in multiple brain areas, although more frequent in the inferior olivary complex, the lateral geniculate body, the substantia nigra and the cerebral cortex. Olivopontocerebellar atrophy and thinning of the spinal cord. Retinal degeneration. | [20] |
| SCA17 | TBP | (CAG)n Exon 3 6q27 | 29–42 | >47 | 607136 | Ataxia, pyramidal and extrapyramidal signs, cognitive impairment, dementia, psychosis, bradykinesia and seizures | Mild neuronal loss with compaction of the neuropil in the cerebral cortex, striatum and moderate loss of PC. Nuclear inclusions. | [21] |
| DRPLA | ATN1 | (CAG)n 12p13.31 | 6–35 | >49 | 125370 | Ataxia, epilepsy, choreoathetosis, dementia | Atrophy and neuronal loss in the globus pallidus (particularly the lateral segment) and dentate nucleus, brainstem, cerebellar and cerebral white matter. Lipofuscin deposits. Nuclear and cytoplasmic inclusions. | [22] |
| PolyQ Disease | Current Treatment | Molecular Structure | Ref. |
|---|---|---|---|
| SCA 1 | 4-aminopyridine (4-AP) to ameliorate motor coordination deficiency of mouse model. | 4-AP: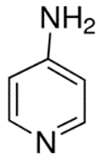 | [52] |
| SCA 2 | Levodopa to alleviate rigidity/ bradykinesia. Magnesium, quinine, mexiletine or vitamin B to ameliorate painful muscle contractions. Chlorzoxazone and riluzole (potassium channel modulators) to improve cerebellar electrophysiology. | Levodopa: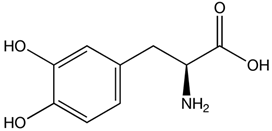 Chlorzoxazone:  Riluzole:  | [17,41,46,48,49,51] |
| SCA 3 | Varenicline (a partial agonist at α4β2 neuronal nicotinic acetylcholine receptors) to improve axial symptoms and rapid alternating movements. | Varenicline:  | [44,56] |
| SCA 6 | 4-aminopyridine (4-AP) to ameliorate motor coordination deficiency of mouse model. | 4-AP: | [53] |
| SCA 7 | Interferon beta to clear mutant ataxin-7 and improve Purkinje cell survival in SCA7 knock-in mice. | Interferon beta: C74H115N19O25 | [45,57] |
| SCA 17 | Piperine (alkaloid) alleviates toxicity caused by mutant TBP protein in mouse model. NC009-1 (C19H16N2O3) reduces polyQ aggregation in Purkinje cells and ameliorated behavioral deficits in mouse model. | Piperine: NC009-1:  | [58,59] |
| DRPLA | Perampanel stopped myoclonic seizures and helped to recover intellectual abilities in a 13-year-old male patient. | Perampanel: | [60] |
| Technology | Characteristics | Functions | Advantages | Limitations | Ref. |
|---|---|---|---|---|---|
| ASOs | -Short single or double strands of chemically modified oligonucleotides. -Selectively bind to complementary mRNA. | -May induce RNase H-mediated cleavage of the targeted mRNA. -May block translation of the corresponding protein. | -Useful to knock down a gene or protein expression from RNA levels. -ASOs have favorable properties, including good distribution throughout the brain after intracerebroventricular (ICV) injection, excellent uptake by neurons and other brain cells, and high stability with a half-life of several months. | -Do not discriminate between the wild-type and the mutant alleles. -Require continuous re-administration of ASOs to offer long-term alleviation. -Lack of selectivity entails a risk of having off-target effects. | [86] |
| Allele-specific ASO | -Can selectively target the CAG repeat expansion. | -Specifically knocks down the mutant allele. -Required the CAG tract expansion to be associated with single-nucleotide polymorphism (SNP), to target and lower mutant allele levels. | -Maintain the wild-type protein function. -ASOs delivered into cerebrospinal fluid distribute widely throughout the central nervous system of mammals. | -Requires continuous re-administration. -CAG repeats are ubiquitous in the human transcriptome, therefore challenging. -Not all patients have the same SNP, so it is limited to a reduced number of patients. | [87] |
| Exon skipping (by ASO) | -ASO-based strategy aimed to remove the expanded CAG tract through alternative splicing. | -ASOs can induce exon skipping by sterically blocking the binding of splicing factors to pre-mRNAs, maintaining the RNA reading frame and rendering a truncated but functional protein. | -Global protein levels are maintained. | -Internally truncated protein is obtained. -Previous knowledge about protein translation is needed. -Exon skipping might provide a low level of protein modification. | [88] |
| Interferent gene silencing | |||||
| Non-allele-specific interferent gene silencing (RNA interference—RNAi) | -Cellular mechanism that induces post-transcriptional gene silencing by promoting the cleavage of target RNAs. -Implicates small RNAs (21–23 nucleotides long) that can regulate gene expression in eukaryotic organisms. | -RNAi is an evolutionarily conserved process that induces post-transcriptional gene silencing, initiated by double-stranded RNA (dsRNA) sequences, whether small interfering RNAs (siRNAs) or derived from the expression of short hairpin RNAs (shRNAs). | Potential therapeutic tool aimed to reduce or silence pathogenic gene targets, including gain of function in CNS diseases. | -Can have low effectiveness of engineered constructs at the chromosomal target, time-consuming processing, and possible undesirable mutagenic effects. | [89] |
| Allele-specific small interfering RNA (siRNA) | -Degradation of complementary mRNA while selectively discriminating between wt and mutated alleles. | -Uses SNPs to discriminate between WT and mutant transcripts. -This is a promissory strategy against polyQ SCA disorders. | -May use RNA duplexes that contain mismatched bases respective to the CAG target. | -Off-target effects may occur. -Poor intracellular uptake and stability in plasma. - Allele-specific gene silencing is limited to the identification of gene-linked SNPs. | [90] |
| Genome editing nucleases | |||||
| CRISPR-Cas9 RNA-guided nucleases (CRISPR-Cas9) | -Are used to induce targeted double-strand breaks (DSBs) at the desired chromosomal locus. -Then, non-homologous end joining (NHEJ) or homology-directed repair (HDR) is used to repair the DSB. | -Technology that uses a guide strand and a protein (Cas9) to selectively bind to a DNA region and cut. Then, both ends can bind and inactivate the gene or introduce DNA templates to edit a gene. | -Can be used to remove duplicated exons, for precise correction of causative mutation and can induce the expression of compensatory proteins. -May bring long-term efficacy. -There is no need for repeated treatment. -The expression of the modified protein is under the control of a natural promoter. | -Need more in vivo studies monitoring the off-target effects. -More studies about the potential immune responses activated by viral delivery vectors. | [91] |
| Transcription activator-like effector nucleases (TALENs) | -Are simple modular codes for DNA recognition. -Can act as a versatile platform for programmable DNA-binding proteins. -A FokI nuclease domain is found in TALENs. | -TALENs are simpler to construct than ZFNs. -Any DNA sequence can be targeted by TALENs, including small DNA sequences. | -Single site targeting, the occurrence of nonspecific mutations and low efficiency. | [92] | |
| Zinc-finger nucleases (ZFNs) | -Each ZF is composed of approximately 30 aa in a conserved ββα configuration. -Then, each ZF is combined with DNA into the main channel of the DNA double helix and by a recognition of 3 to 4 bp sequence. -ZFNs are composed of 2 domains: the DNA-binding ZF protein (ZFP) domain and the FokI restriction enzyme site. | -Repair the gene sequence without the integration of any sequence into the genome. -Very high efficiency. | -Single site targeting, occurrence of nonspecific mutations and low efficiency. -Might have high immunogenic power. | [92] | |
| Type of Transport | Specific Target | Ligand | Examples of Nanosystems Using Ligands for Brain Delivery | Biological Effect | Ref. |
|---|---|---|---|---|---|
| Transporter-mediated transcytosis | Glucose receptors | Glucose Mannose | Paclitaxel-loaded PEG-co-poly(trimethylene carbonate) NPs modified with 2-deoxy-D-glucose | The glycosylated NPs were higher internalized compared to the NPs control. Modified NPs had high specificity and efficiency in intracranial tumor accumulation. | [177] |
| Silica NPs modified with glucose and glucose-PEG- methyl ether amine | Both NP systems exhibited a significant uptake in the brain region compared with the control NPs at 1 h post-administration. | [178] | |||
| Neutral amino acid transporter | Tyrosine Histidine Asparagine Phenylalanine threonine | Dendrimer of poly (propylene imine) coated with maltose-histidine | Maltose-histidine presence remarkably improved the biocompatibility and the ability to cross the blood–brain barrier in vivo in male wild-type mice. | [179] | |
| Cationic amino acid transporter | Arginine Lysine | Flurbiprofen-loaded poly (epsilon-lysine) dendrons | The penetration of the drug in bEnd.3 monolayer culture increased with the nanoformulation. | [180] | |
| Monocarboxylate transporter | Lactate Biotin Salicylic acid Valproic acid | Avidin-functionalized PEG- polypeptide [poly(α,β-aspartic acid) nanomicelles | Biotin targets were generated on EC surfaces. This selectively labeling promoted the targeting of avidin nanomicelles specifically to the brain microvasculature with minimal targeting into peripheral organs. | [181] | |
| Choline transporter | Choline Thiamine | Doxorubicin-loaded polymeric micelles modified with choline derivate | Nanocarriers treated with 20% of choline presented an enhancement in cellular uptake and antitumor activity. | [182] | |
| Adsorptive-mediated transcytosis | Cell-penetrating peptides | Penetrating/Albumin | Triethylenetetramine-loaded liposomes functionalized with albumin or penetratin | In vivo analysis showed that surface modification remarkably increased the drug uptake into the brain tissue compared with free drug or non-modification liposome behavior. | [183] |
| K16ApoE | PLGA/chitosan NPs conjugated with IgG4.1 or 125I-IgG4.1 and modified with K16ApoE by physical absorption | K16ApoE-targeted NPs were injected via femoral vein in DutchAβ40-treated WT mice. The results showed the accumulation of the NPs in various brain regions compared to non-modified NPs. | [184] | ||
| Receptor-mediated transcytosis | Transferrin receptor | Lactoferrin Transferrin | Clofazimine-loaded PLGA-PEG NPs modified with transferrin receptor-binding peptide | NPs presented an adequate cell interaction and high permeability across hCMEC/D3 cell monolayers. | [185] |
| Dopamine-loaded mPEG-PLGA NPs modified with lactoferrin | Cellular uptake of SH-SY5Y cells and 16HBE cells improved due to lactoferrin modification of NPs. | [186] | |||
| Endothelial LDL receptor | LDL ApoE | Doxorubicin-loaded silk fibroin/Tween 80 NPs | Tween-80 modification improved circulating time and facilitated their uptake by low-density lipoprotein. | [187] | |
| Rosmarinic acid-loaded polyacrylamide-chitosan-PLGA NPs functionalized with ApoE | A decrement in electrical resistance and increment in the ability to cross the BBB were observed with the concentration of ApoE increase. | [188] | |||
| Glutathione receptor | Glutathione | Liposomal formulations (hydrogenated soy phosphatidylcholine or egg yolk phosphatidylcholine) conjugated with glutathione for methotrexate delivery | Hydrogenated soy phosphatidylcholine-glutathione liposomal increased 4-fold the drug brain delivery. | [189] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borbolla-Jiménez, F.V.; Del Prado-Audelo, M.L.; Cisneros, B.; Caballero-Florán, I.H.; Leyva-Gómez, G.; Magaña, J.J. New Perspectives of Gene Therapy on Polyglutamine Spinocerebellar Ataxias: From Molecular Targets to Novel Nanovectors. Pharmaceutics 2021, 13, 1018. https://doi.org/10.3390/pharmaceutics13071018
Borbolla-Jiménez FV, Del Prado-Audelo ML, Cisneros B, Caballero-Florán IH, Leyva-Gómez G, Magaña JJ. New Perspectives of Gene Therapy on Polyglutamine Spinocerebellar Ataxias: From Molecular Targets to Novel Nanovectors. Pharmaceutics. 2021; 13(7):1018. https://doi.org/10.3390/pharmaceutics13071018
Chicago/Turabian StyleBorbolla-Jiménez, Fabiola V., María Luisa Del Prado-Audelo, Bulmaro Cisneros, Isaac H. Caballero-Florán, Gerardo Leyva-Gómez, and Jonathan J. Magaña. 2021. "New Perspectives of Gene Therapy on Polyglutamine Spinocerebellar Ataxias: From Molecular Targets to Novel Nanovectors" Pharmaceutics 13, no. 7: 1018. https://doi.org/10.3390/pharmaceutics13071018
APA StyleBorbolla-Jiménez, F. V., Del Prado-Audelo, M. L., Cisneros, B., Caballero-Florán, I. H., Leyva-Gómez, G., & Magaña, J. J. (2021). New Perspectives of Gene Therapy on Polyglutamine Spinocerebellar Ataxias: From Molecular Targets to Novel Nanovectors. Pharmaceutics, 13(7), 1018. https://doi.org/10.3390/pharmaceutics13071018