
A Biodegradable Copolyester, Poly(butylene succinate-co-ε-caprolactone), as a High Efficiency Matrix Former for Controlled Release of Drugs

 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Synthesis and Characterization of Poly(butylene succinate-co-ε-caprolactone)
2.2.2. Rheological Characterization of PBS_CL by SeDeM Method
2.2.3. Preparation of Matrix Systems
2.2.4. Matrix Systems Characterization
3. Results and Discussion
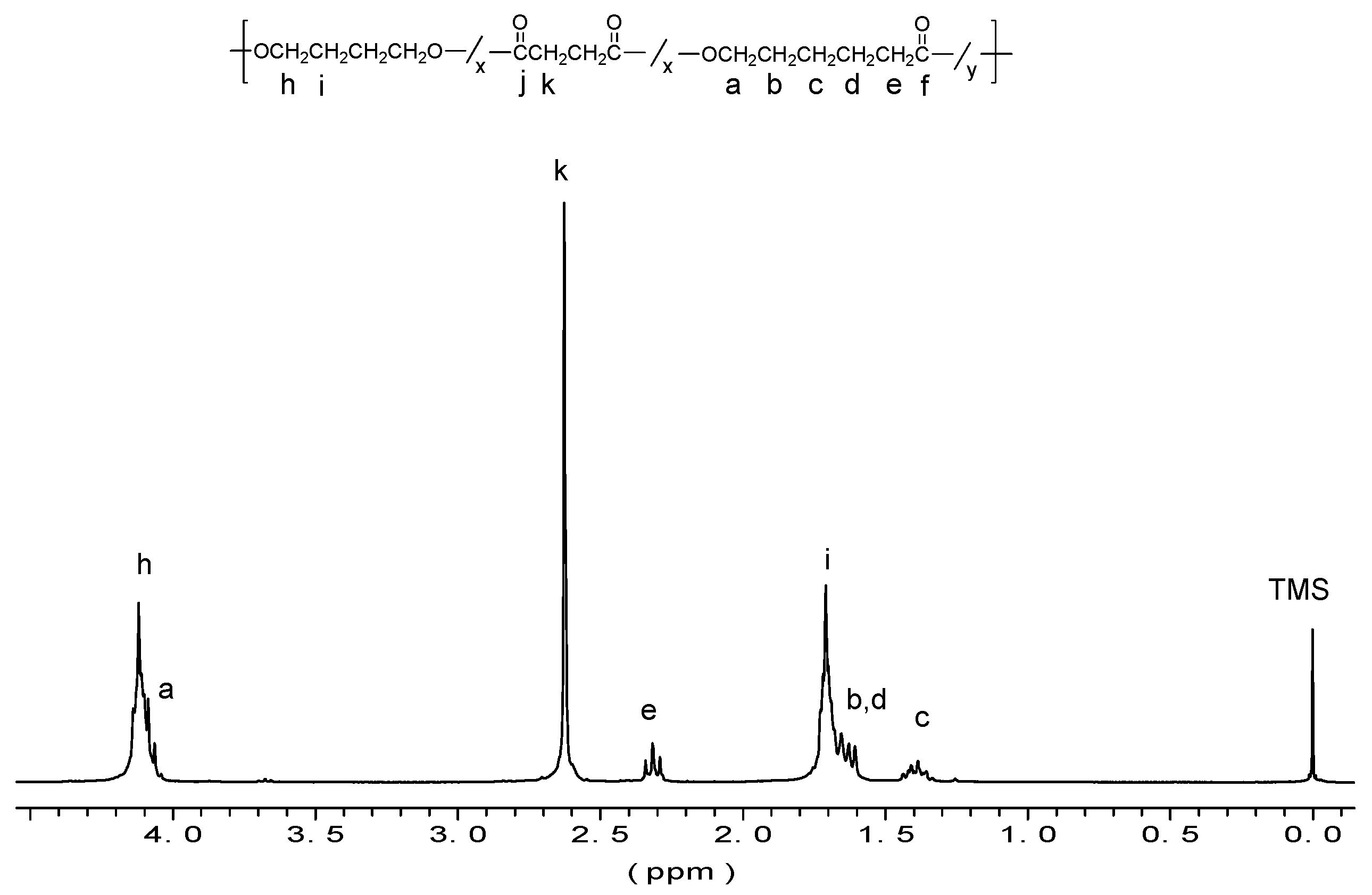
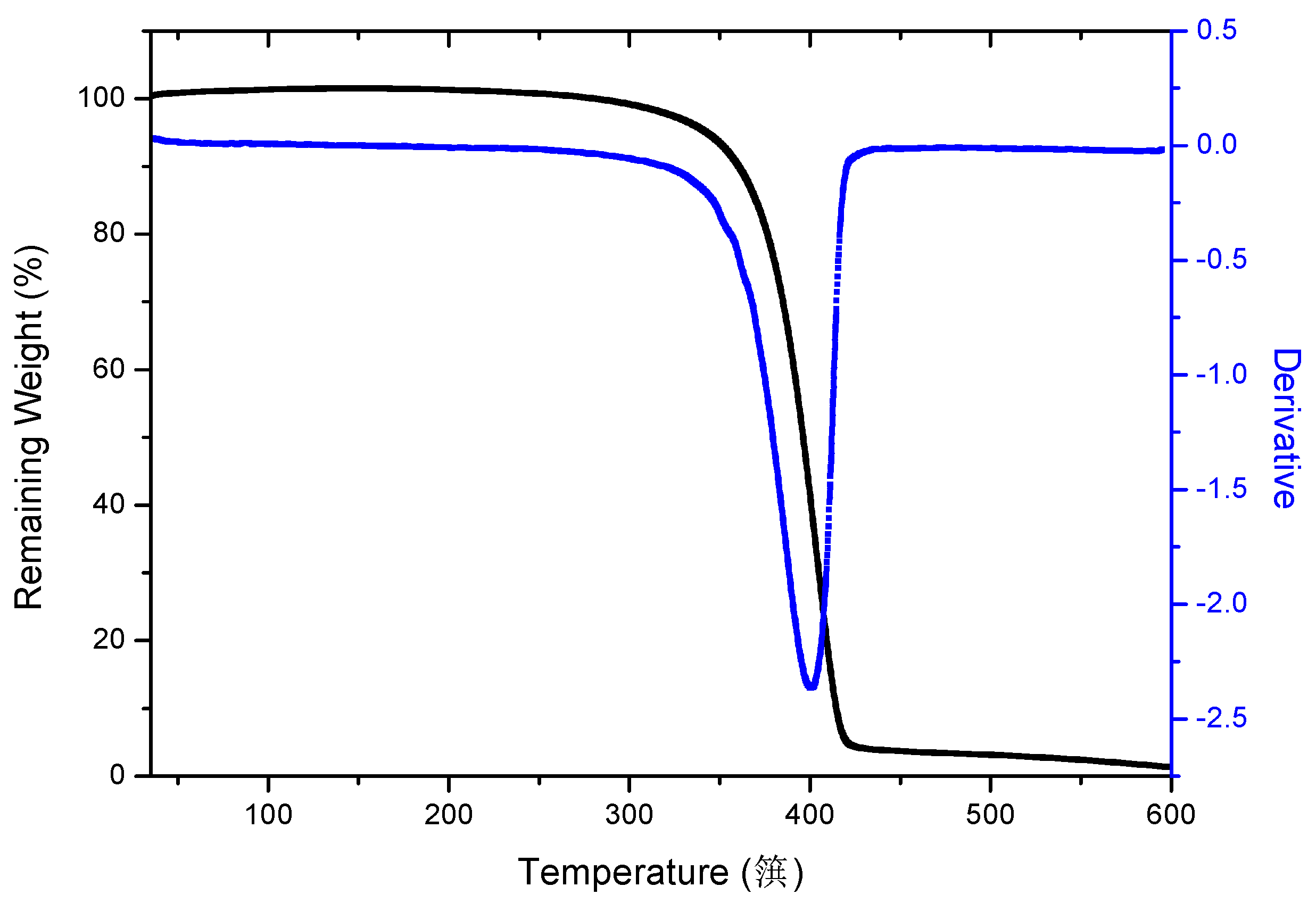
3.1. PBS_CL Characterization
3.2. Matrix Systems Characterization
3.3. Rheological Results According to SeDeM Method
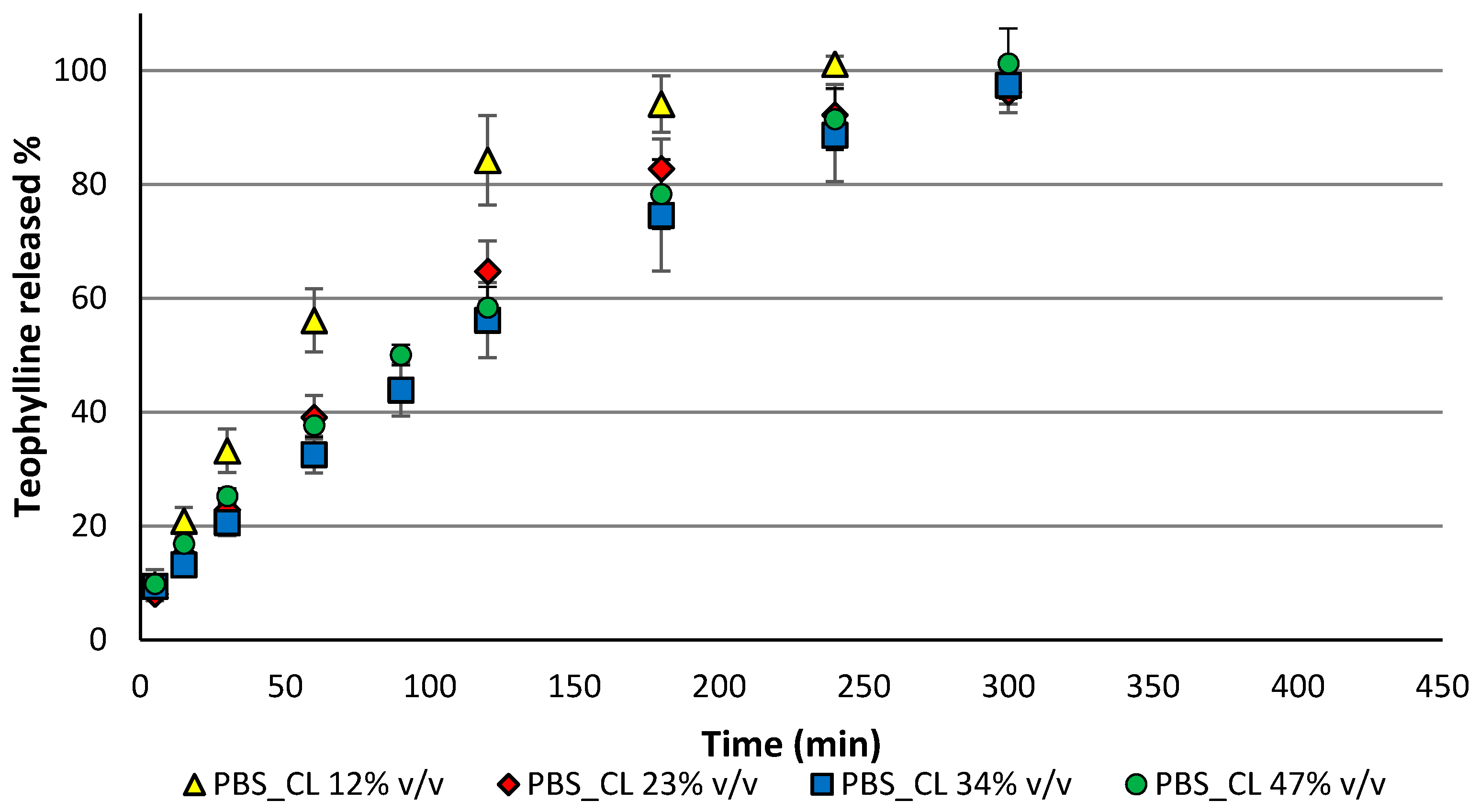
3.4. Drug Release Results
3.5. Kinetic Analysis
3.6. Internal Structure Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anselmo, A.C.; Mitragotri, S. An overview of clinical and commercial impact of drug delivery systems. J. Control. Release 2014, 190, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Englert, C.; Brendel, J.C.; Majdanski, T.C.; Yildirim, T.; Schubert, S.; Gottschaldt, M.; Windhab, N.; Schubert, U.S. Pharmapolymers in the 21st century: Synthetic polymers in drug delivery applications. Prog. Polym. Sci. 2018, 87, 107–164. [Google Scholar] [CrossRef]
- Fu, Y.; Kao, W.J. Drug release kinetics and transport mechanisms of non-degradable and degradable polymeric delivery systems. Expert Opin. Drug Deliv. 2010, 7, 429–444. [Google Scholar] [CrossRef]
- Mozafari, M.; Chauhan, N.P.S. Advanced Functional Polymers for Biomedical Applications; Elsevier: Amsterdam, The Netherlands, 2019; ISBN 9780128163498. [Google Scholar]
- Papadimitriou, S.A.; Papageorgiou, G.Z.; Bikiaris, D.N. Crystallization and enzymatic degradation of novel poly(ε-caprolactone-co-propylene succinate) copolymers. Eur. Polym. J. 2008, 44, 2356–2366. [Google Scholar] [CrossRef]
- Albertsson, A.C.; Varma, I.K. Aliphatic polyesters: Synthesis, properties and applications. Adv. Polym. Sci. 2002, 157, 1–40. [Google Scholar]
- Manavitehrani, I.; Fathi, A.; Badr, H.; Daly, S.; Shirazi, A.N.; Dehghani, F. Biomedical applications of biodegradable polyesters. Polymers 2016, 8, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gigli, M.; Fabbri, M.; Lotti, N.; Gamberini, R.; Rimini, B.; Munari, A. Poly(butylene succinate)-based polyesters for biomedical applications: A review in memory of our beloved colleague and friend Dr. Lara Finelli. Eur. Polym. J. 2016, 75, 431–460. [Google Scholar] [CrossRef]
- Malikmammadov, E.; Tanir, T.E.; Kiziltay, A.; Hasirci, V.; Hasirci, N. PCL and PCL-based materials in biomedical applications. J. Biomater. Sci. Polym. Ed. 2018, 29, 863–893. [Google Scholar] [CrossRef] [PubMed]
- Safari, M.; Martínez De Ilarduya, A.; Mugica, A.; Zubitur, M.; Muñoz-Guerra, S.; Müller, A.J. Tuning the Thermal Properties and Morphology of Isodimorphic Poly[(butylene succinate)- ran-(ϵ-caprolactone)] Copolyesters by Changing Composition, Molecular Weight, and Thermal History. Macromolecules 2018, 51, 9589–9601. [Google Scholar] [CrossRef]
- Campiñez, M.D.; Ferris, C.; de Paz, M.V.; Aguilar-de-Leyva, Á.; Galbis, J.A.; Caraballo, I. A new biodegradable polythiourethane as controlled release matrix polymer. Int. J. Pharm. 2015, 480, 63–72. [Google Scholar] [CrossRef]
- Maderuelo, C.; Zarzuelo, A.; Lanao, J.M. Critical factors in the release of drugs from sustained release hydrophilic matrices. J. Control. Release 2011, 154, 2–19. [Google Scholar] [CrossRef]
- Galdón, E.; Casas, M.; Gayango, M.; Caraballo, I. First study of the evolution of the SeDeM expert system parameters based on percolation theory: Monitoring of their critical behavior. Eur. J. Pharm. Biopharm. 2016, 109, 158–164. [Google Scholar] [CrossRef]
- Bode, C.; Kranz, H.; Fivez, A.; Siepmann, F.; Siepmann, J. Often neglected: PLGA/PLA swelling orchestrates drug release: HME implants. J. Control. Release 2019, 306, 97–107. [Google Scholar] [CrossRef]
- Bisharat, L.; Alkhatib, H.S.; Abdelhafez, A.; Barqawi, A.; Aljaberi, A.; Qi, S.; Berardi, A. Hot melt extruded zein for controlled delivery of diclofenac sodium: Effect of drug loading and medium composition. Int. J. Pharm. 2020, 585, 119503. [Google Scholar] [CrossRef]
- Goyanes, A.; Robles Martinez, P.; Buanz, A.; Basit, A.W.; Gaisford, S. Effect of geometry on drug release from 3D printed tablets. Int. J. Pharm. 2015, 494, 657–663. [Google Scholar] [CrossRef]
- Kempin, W.; Domsta, V.; Brecht, I.; Semmling, B.; Tillmann, S.; Weitschies, W.; Seidlitz, A. Development of a dual extrusion printing technique for an acid- and thermo-labile drug. Eur. J. Pharm. Sci. 2018, 123, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Desai, D.; Sandhu, H.; Shah, N.; Malick, W.; Zia, H.; Phuapradit, W.; Vaka, S.R.K. Selection of solid-state plasticizers as processing aids for hot-melt extrusion. J. Pharm. Sci. 2018, 107, 372–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, H.; Tiwari, R.V.; Repka, M.A. Hot-Melt Extrusion: From Theory to Application in Pharmaceutical Formulation. AAPS PharmSciTech 2016, 17, 20–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levina, M.; Rubinstein, M.H. The Effect of ultrasonic vibration on the compaction characteristics of Paracetamol. J. Pharm. Sci. 2000, 89, 705–723. [Google Scholar] [CrossRef]
- Millán-Jiménez, M.; Galdón, E.; Ferrero, C.; Caraballo, I. Application of ultrasound-assisted compression in pharmaceutical technology. Design and optimization of oral sustained-release dosage forms. J. Drug Deliv. Sci. Technol. 2017, 42, 119–125. [Google Scholar] [CrossRef]
- Caraballo, I.; Millán, M.; Fini, A.; Rodriguez, L.; Cavallari, C. Percolation thresholds in ultrasound compacted tablets. J. Control. Release 2000, 69, 345–355. [Google Scholar] [CrossRef]
- Millán, M.; Caraballo, I. Effect of drug particle size in ultrasound compacted tablets: Continuum percolation model approach. Int. J. Pharm. 2006, 310, 168–174. [Google Scholar] [CrossRef]
- Caraballo, I. Prices of the Controlled Release Society. In Proceedings of the 28th Symposium in San Diego, Controlled Release Society, San Diego, CA, USA, 23–24 June 2001. [Google Scholar]
- Theophylline | C7H8N4O2-PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/2153 (accessed on 23 June 2021).
- Yalkowsky, S.H.; He, Y.; Jain, P. Handbook of aqueous solubility data; CRC Press: Boca Raton, FL, USA, 2010; Volume 125, p. 395. ISBN 0-8493-1532-8. [Google Scholar]
- Pérez, P.; Suñé-Negre, J.M.; Miñarro, M.; Roig, M.; Fuster, R.; García-Montoya, E.; Hernández, C.; Ruhí, R.; Ticó, J.R. A new expert systems (SeDeM diagram) for control batch powder formulation and preformulation drug products. Eur. J. Pharm. Biopharm. 2006, 64, 351–359. [Google Scholar] [CrossRef]
- Galdón, E.; Casas, M.; Caraballo, I. Achieving high excipient efficiency with elastic thermoplastic polyurethane by ultrasound assisted direct compression. Pharmaceutics 2019, 11, 157. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, T. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J. Pharm. Sci. 1963, 52, 1145–1149. [Google Scholar] [CrossRef] [PubMed]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Peppas, N.A.; Sahlin, J.J. A simple equation for the description of solute release. III. Coupling of diffusion and relaxation. Int. J. Pharm. 1989, 57, 169–172. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release I. Fickian and non-fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J. Control. Release 1987, 5, 23–36. [Google Scholar] [CrossRef]
- Caraballo, I. Newsletter Controlled Release Society. Control. Release Soc. 2001, 18, 12. [Google Scholar]
- Pubchem 1,4-Butanediol | HO(CH2)4OH-PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/8064#section=Toxicity&fullscreen=true (accessed on 28 June 2021).
- Hexanoic acid | C6H12O2-PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/8892#section=Toxicity&fullscreen=true (accessed on 28 June 2021).
- Succinic acid | C4H6O4-PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/1110#section=Toxicity&fullscreen=true (accessed on 28 June 2021).
- Nardi-Ricart, A.; Nofrerias-Roig, I.; Suñé-Pou, M.; Pérez-Lozano, P.; Miñarro-Carmona, M.; García-Montoya, E.; Ticó-Grau, J.R.; Boronat, R.I.; Suñé-Negre, J.M. Formulation of sustained release hydrophilic matrix tablets of tolcapone with the application of sedem diagram: Influence of tolcapone’s particle size on sustained release. Pharmaceutics 2020, 12, 674. [Google Scholar] [CrossRef]
- Suñé-Negre, J.M.; Roig, M.; Fuster, R.; Hernández, C.; Ruhí, R.; García-Montoya, E.; Pérez-Lozano, P.; Miñarro, M.; Ticó, J.R. New classification of directly compressible (DC) excipients in function of the SeDeM Diagarm Expert System. Int. J. Pharm. 2014, 470, 15–27. [Google Scholar] [CrossRef]
- Crowley, M.M.; Schroeder, B.; Fredersdorf, A.; Obara, S.; Talarico, M.; Kucera, S.; McGinity, J.W. Physicochemical properties and mechanism of drug release from ethyl cellulose matrix tablets prepared by direct compression and hot-melt extrusion. Int. J. Pharm. 2004, 269, 509–522. [Google Scholar] [CrossRef]
- Casas, M.; Aguilar-de-Leyva, Á.; Caraballo, I. Towards a rational basis for selection of excipients: Excipient efficiency for controlled release. Int. J. Pharm. 2015, 494, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Ageitos, J.M.; Pulgar, A.; Csaba, N.; Garcia-Fuentes, M. Study of nanostructured fibroin/dextran matrixes for controlled protein release. Eur. Polym. J. 2019, 114, 197–205. [Google Scholar] [CrossRef]
- Zhou, J.; Han, S.; Dou, Y.; Lu, J.; Wang, C.; He, H.; Li, X.; Zhang, J. Nanostructured poly(l-lactide) matrix as novel platform for drug delivery. Int. J. Pharm. 2013, 448, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.-H.; Huang, S.; Wang, Y.-C.; Kuo, Y.-C. Novel nanostructured biodegradable polymer matrices fabricated by phase separation techniques for tissue regeneration. Acta Biomater. 2013, 9, 6915–6927. [Google Scholar] [CrossRef] [PubMed]
- ICH Expert Working Group. Pharmaceutical Development Q8 (R2); European Medicine Agency: London, UK, 2009; Volume 8. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Batch | PBS_CL Content (%vol) | Theophylline Content (%vol) | Total Weight (mg) | Diameter (mm) | Thickness (mm) | Volume (mm3) | Initial Porosity (ε0) |
|---|---|---|---|---|---|---|---|
| 12:88 DC | 12 | 88 | 246.1 ± 4.9 | 12.10 ± 0.01 | 2.001 ± 0.244 | 218.79 | 23.37 |
| 23:77 DC | 23 | 77 | 242.6 ± 2.1 | 12.13 ± 0.08 | 1.869 ± 0.224 | 205.26 | 17.77 |
| 34:66 DC | 34 | 66 | 240.4 ± 0.8 | 12.25 ± 0.23 | 1.764 ± 0.010 | 204.57 | 16.58 |
| 47:53 DC | 47 | 53 | 238.9 ± 1.6 | 12.14 ± 0.03 | 1.827 ± 0.023 | 211.39 | 18.09 |
| 12:88 USAC | 12 | 88 | 241.2 ± 1.7 | 11.15 ± 0.03 | 2.132 ± 0.022 | 208.89 | 21.33 |
| 23:77 USAC | 23 | 77 | 246.9 ± 0.2 | 11.25 ± 0.03 | 2.114 ± 0.019 | 210.81 | 18.52 |
| 34:66 USAC | 34 | 66 | 241.8 ± 2.7 | 11.21 ± 0.02 | 2.109 ± 0.028 | 208.24 | 17.86 |
| 47:53 USAC | 47 | 53 | 248.1 ± 3.0 | 11.19 ± 0.04 | 2.008 ± 0.009 | 197.33 | 9.18 |
| Lenght (mm) | Section (mm2) | ||||||
| 45:55 HME | 45 | 55 | 242.5 ± 6.1 | 47.22 ± 1.18 | 4.16 ± 0.00 | 196.48 | 10.92 |
| Incidence Factor | Parameter | Symbol | Unit | Normalized Result | Mean Incidence |
|---|---|---|---|---|---|
| Dimension | Bulk density | ρbulk | g/mL | 4.19 | 4.70 |
| Tapped density | ρtapped | g/mL | 5.20 | ||
| Compressibility | Inter-particle Porosity | IP | - | 3.83 | 3.85 |
| Carr Index | C | % | 3.86 | ||
| Flowability/ powder flow | Hausner Ratio | HR | - | 2.42 | 4.37 |
| Angle of Repose | α | º | 2.88 | ||
| Powder flow | t” | s | 7.80 | ||
| Lubricity/ stability | Loss on Drying | %LOD | % | 5.50 | 5.50 |
| Lubricity/ dosage | Particles ≤45 μm | Pf | % | 9.98 | 6.50 |
| Homogeneity Index | Iθ | - | 3.25 | ||
| Parametric Index (PI) | 0.40 | ||||
| Parametric Profile Index (PPI) | 4.89 | ||||
| Good Compression Index (GCI) | 5.29 | ||||
| Batch | Zero Order | Higuchi | Korsmeyer | Peppas y Sahlin | EE | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| k0 | r2 | b (min−1) | r2 | n | r2 | kd (min−5) | kr (min−1) | r2 | (min1/2) | |
| 12:88 DC | 0.0083 | 0.9945 | 0.0844 | 0.9911 | 0.7065 | 0.9996 | 0.0844 | 0.1086 | 0.9911 | 12.23 |
| 23:77 DC | 0.0056 | 0.9982 | 0.0563 | 0.9832 | 0.6277 | 0.9941 | 0.0563 | 0.0596 | 0.9832 | |
| 34:66 DC | 0.0041 | 0.9992 | 0.0538 | 0.9742 | 0.5724 | 0.9724 | 0.0538 | 0.0637 | 0.9742 | |
| 47:53 DC | 0.0042 | 0.986 | 0.0566 | 0.9951 | 0.5672 | 0.9973 | 0.0566 | 0.0451 | 0.9951 | |
| 12:88 USAC | 0.0062 | 0.9894 | 0.0730 | 0.9477 | 0.8393 | 0.987 | 0.0758 | 0.1695 | 0.9682 | 25.47 |
| 23:77 USAC | 0.0016 | 0.8902 | 0.0330 | 0.9867 | 0.5699 | 0.971 | 0.039 | 0.0112 | 0.9859 | |
| 34:66 USAC | 0.0009 | 0.9468 | 0.0202 | 0.9885 | 0.4812 | 0.9921 | 0.0204 | 0.0041 | 0.9889 | |
| 47:53 USAC | 0.0007 | 0.9432 | 0.0185 | 0.9985 | 0.4116 | 0.9983 | 0.0185 | 0.0313 | 0.9985 | |
| 45:55 HME | 0.0008 | 0.9783 | 0.0167 | 0.9848 | 0.4886 | 0.9948 | 0.0205 | −0.0001 | 0.9904 | 27.43 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galdón, E.; Millán-Jiménez, M.; Mora-Castaño, G.; de Ilarduya, A.M.; Caraballo, I. A Biodegradable Copolyester, Poly(butylene succinate-co-ε-caprolactone), as a High Efficiency Matrix Former for Controlled Release of Drugs. Pharmaceutics 2021, 13, 1057. https://doi.org/10.3390/pharmaceutics13071057
Galdón E, Millán-Jiménez M, Mora-Castaño G, de Ilarduya AM, Caraballo I. A Biodegradable Copolyester, Poly(butylene succinate-co-ε-caprolactone), as a High Efficiency Matrix Former for Controlled Release of Drugs. Pharmaceutics. 2021; 13(7):1057. https://doi.org/10.3390/pharmaceutics13071057
Chicago/Turabian StyleGaldón, Eduardo, Mónica Millán-Jiménez, Gloria Mora-Castaño, Antxon Martínez de Ilarduya, and Isidoro Caraballo. 2021. "A Biodegradable Copolyester, Poly(butylene succinate-co-ε-caprolactone), as a High Efficiency Matrix Former for Controlled Release of Drugs" Pharmaceutics 13, no. 7: 1057. https://doi.org/10.3390/pharmaceutics13071057
APA StyleGaldón, E., Millán-Jiménez, M., Mora-Castaño, G., de Ilarduya, A. M., & Caraballo, I. (2021). A Biodegradable Copolyester, Poly(butylene succinate-co-ε-caprolactone), as a High Efficiency Matrix Former for Controlled Release of Drugs. Pharmaceutics, 13(7), 1057. https://doi.org/10.3390/pharmaceutics13071057