
Mannitol Polymorphs as Carrier in DPIs Formulations: Isolation Characterization and Performance

 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Crystallization Techniques of D-Mannitol
α Form Recrystallization
Hydrate Form Recrystallization
β Form Recrystallization
δ Form Recrystallization
2.1.2. Solid State Characterization
X-ray Diffraction on Powders
Particle Size Distribution
Differential Scanning Calorimetry
Thermogravimetric Calorimetry
Dynamic Vapor Sorption
Scanning Electron Microscopy
2.1.3. Preparation of Adhesive Mixtures
2.1.4. In Vitro Aerodynamic Property Assessment
2.1.5. High Performance Liquid Chromatography (HPLC)
2.1.6. Preliminary Cell Toxicity
3. Results
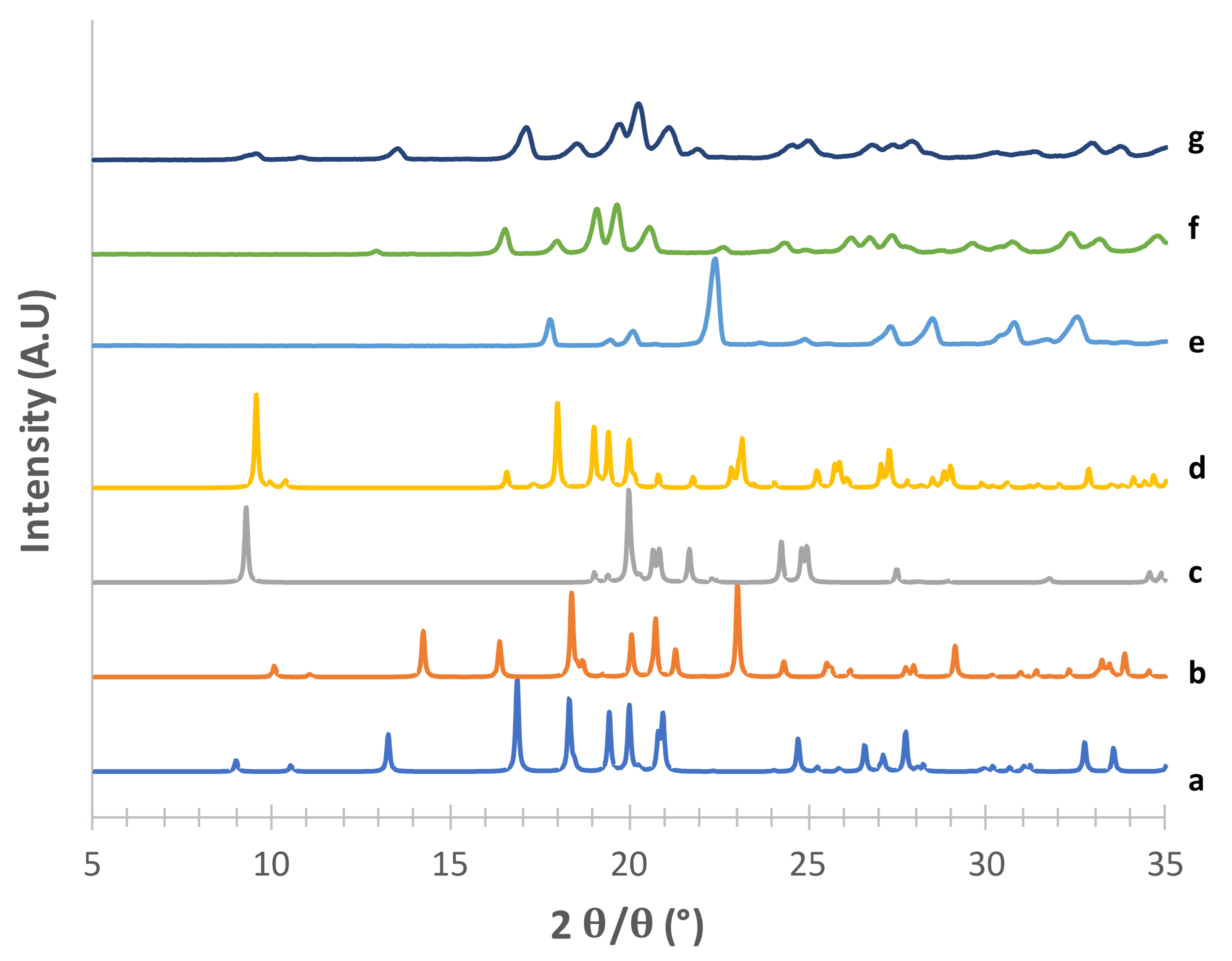
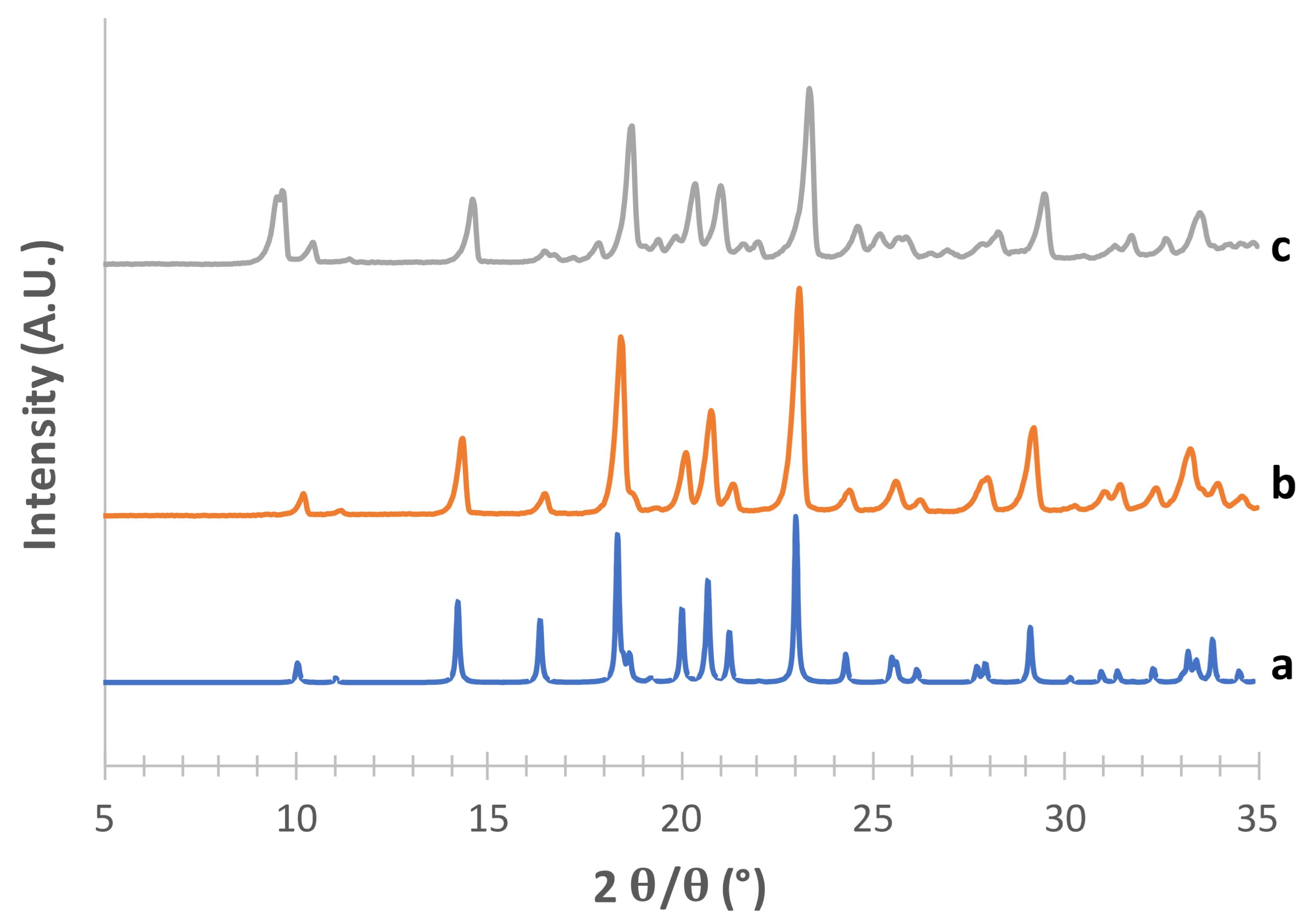
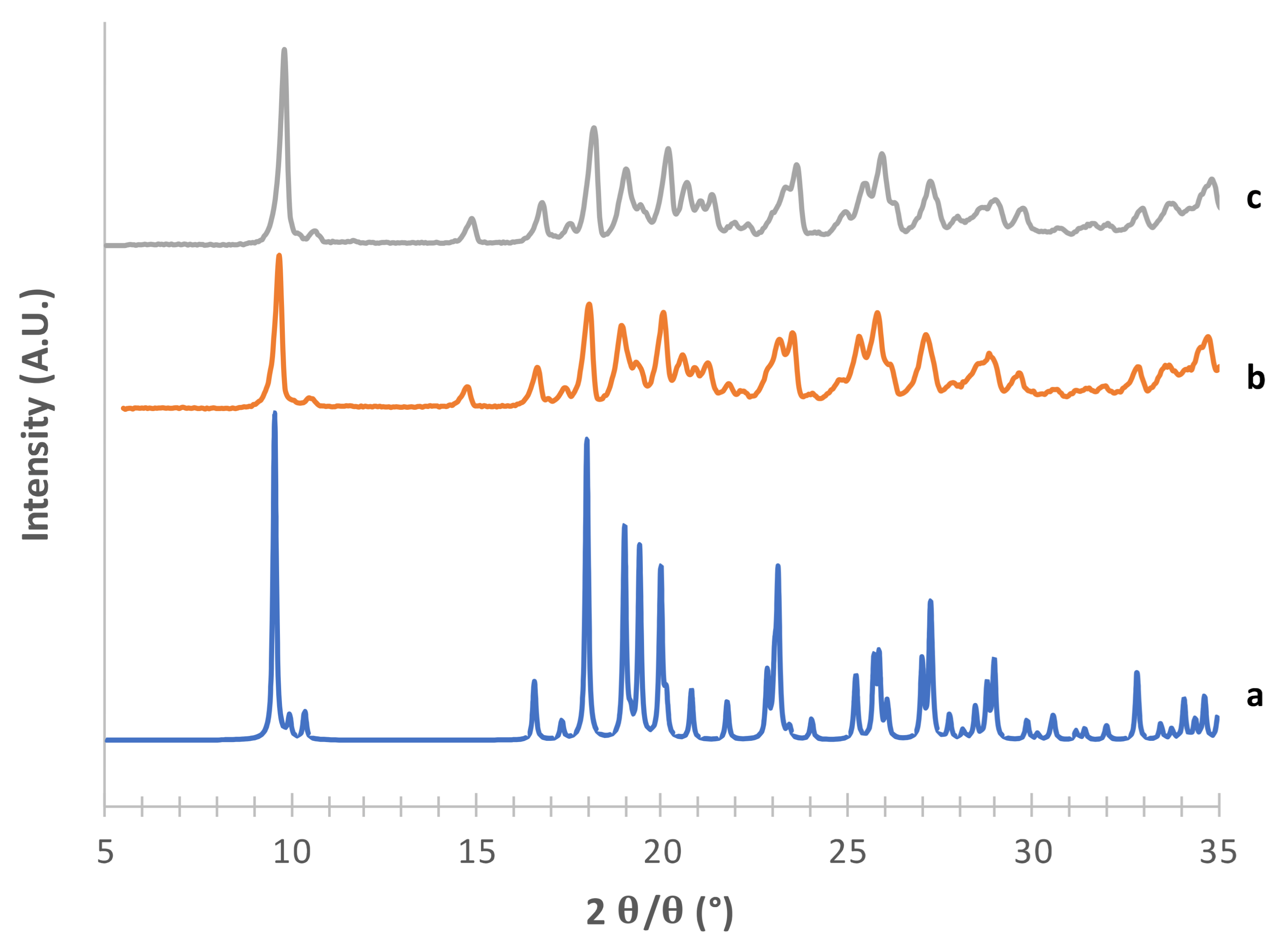
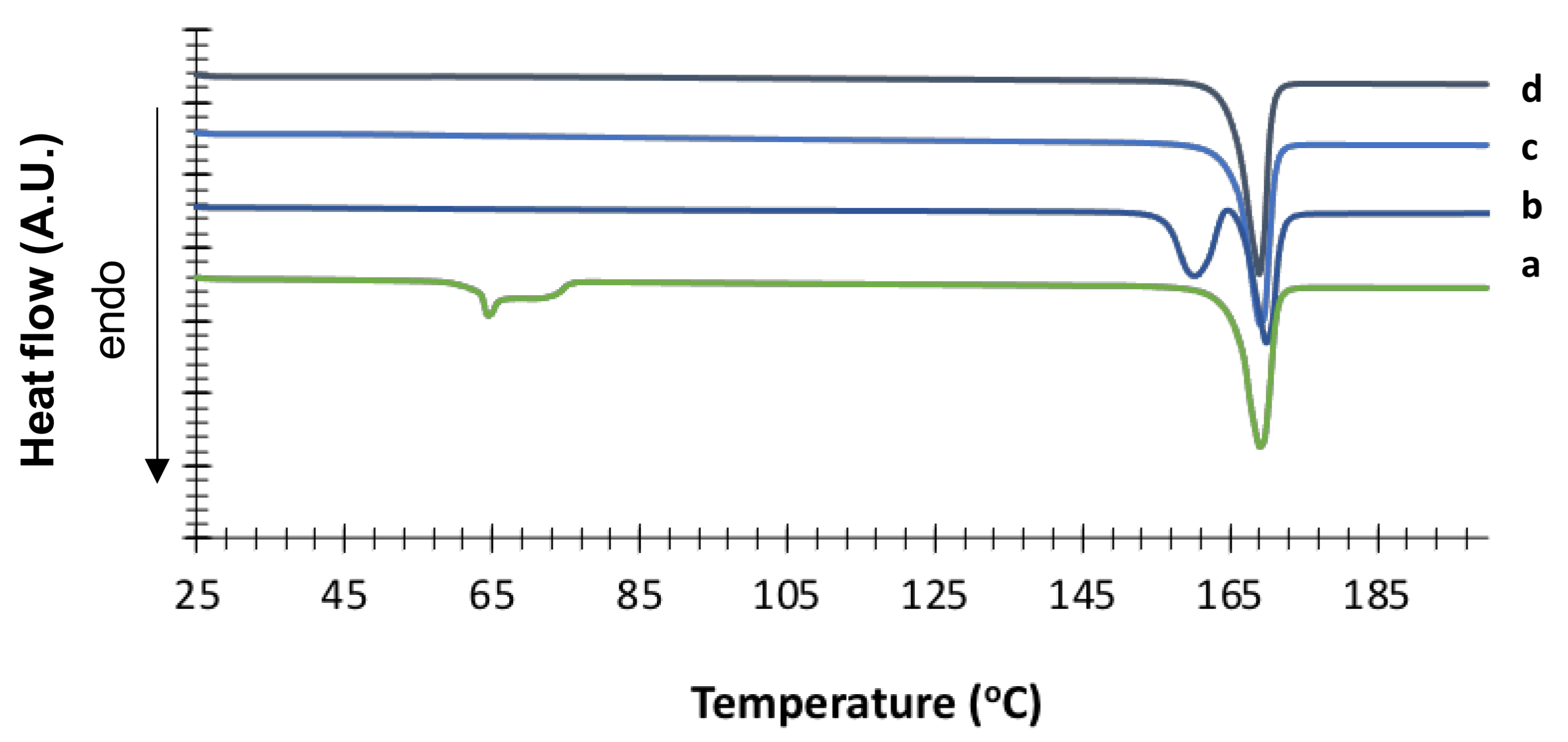
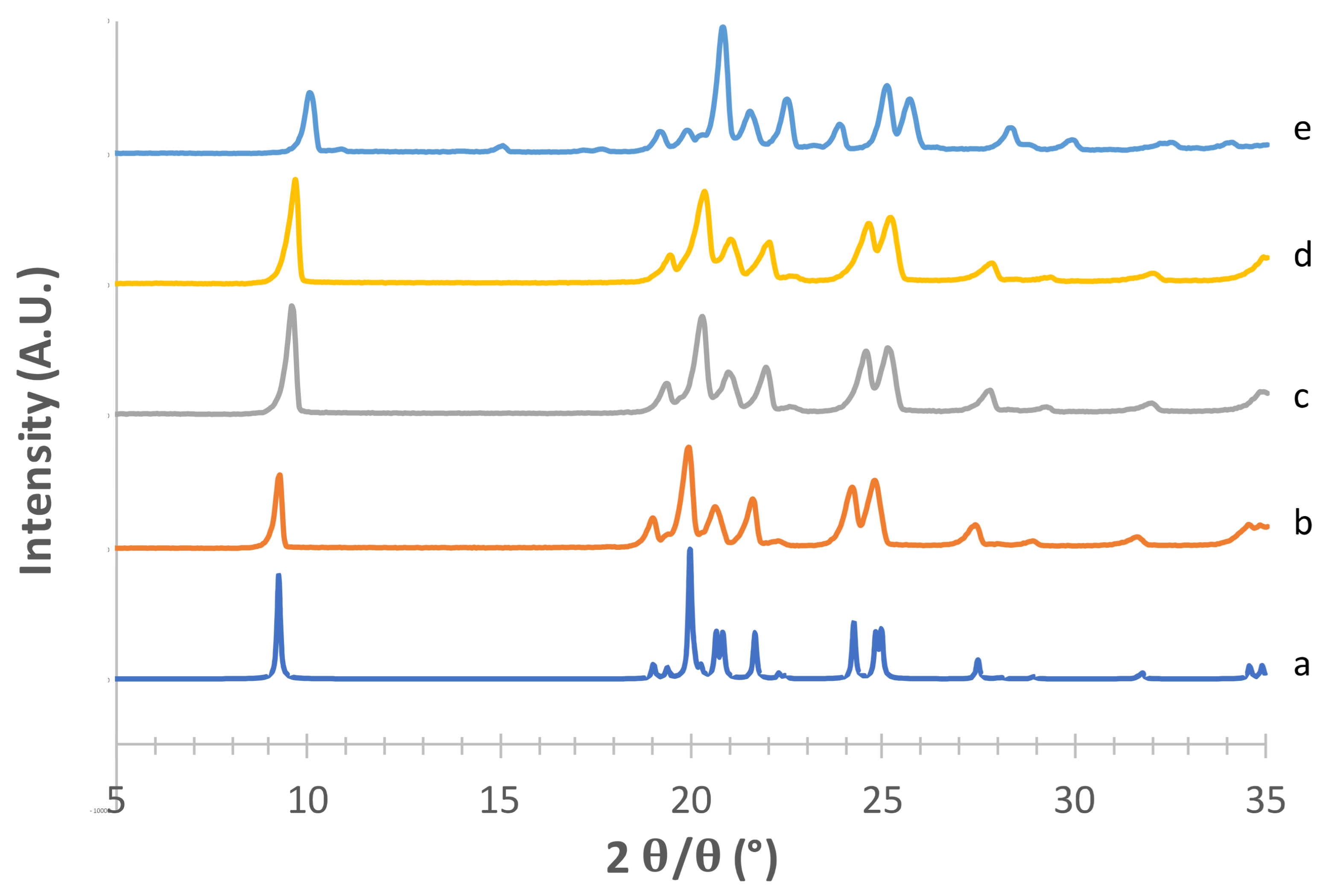
3.1. Solid-State and Physical Characteristics Assessment of Mannitol Crystallized Forms
3.2. Stability of Crystallized α and δ Mannitol
3.3. Particle Size Distribution and Morphology
3.4. Adhesive Mixtures
3.5. Salbutamol Sulphate Deposition
3.6. Budesonide Deposition
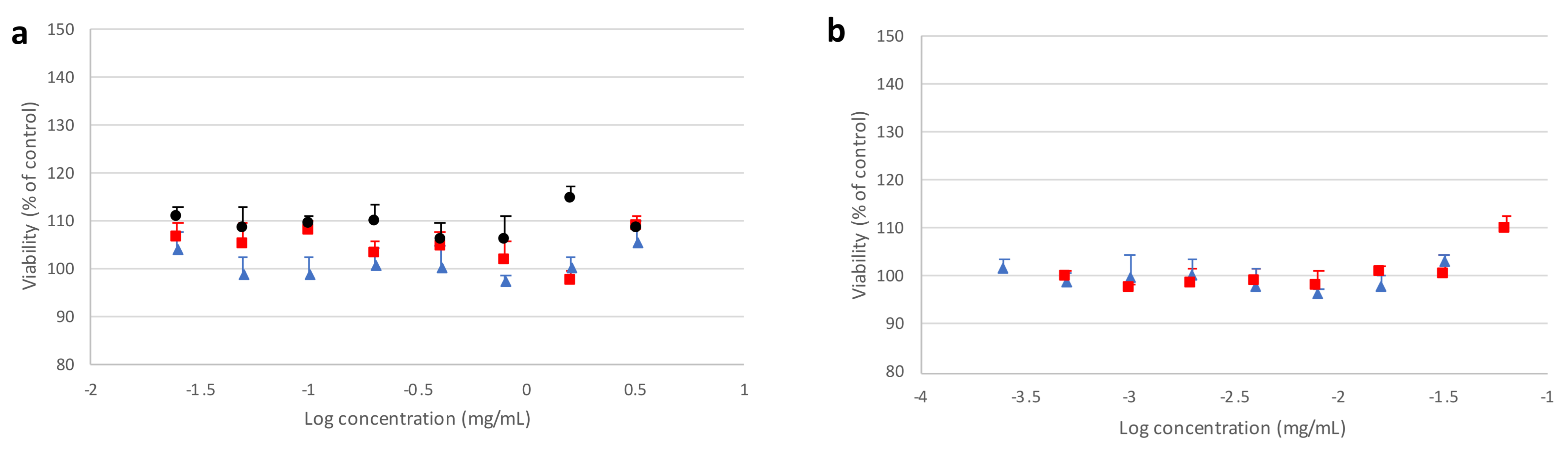
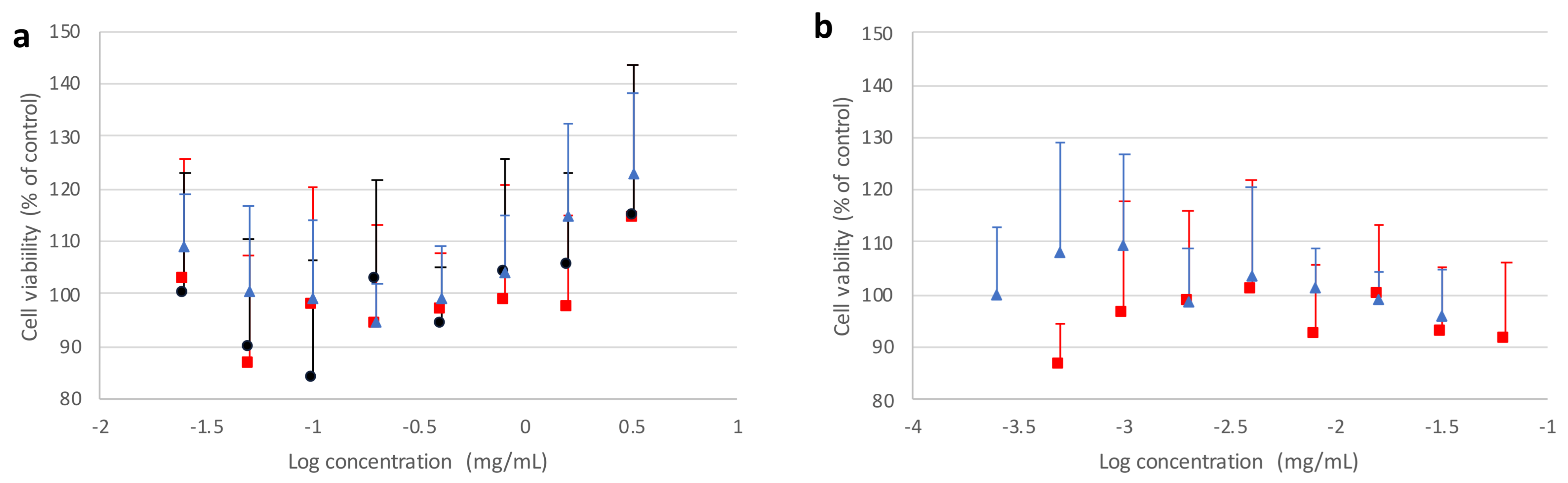
3.7. Preliminary Cell Toxicity Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ticehurst, M.D.; Marziano, I. Integration of Active Pharmaceutical Ingredient Solid Form Selection and Particle Engineering into Drug Product Design. J. Pharm. Pharmacol. 2015, 67, 782–802. [Google Scholar] [CrossRef]
- Pilcer, G.; De Bueger, V.; Traina, K.; Traore, H.; Sebti, T.; Vanderbist, F.; Amighi, K. Carrier-Free Combination for Dry Powder Inhalation of Antibiotics in the Treatment of Lung Infections in Cystic Fibrosis. Int. J. Pharm. 2013, 451, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, P.; Hayes, D.; Mansour, H.M. Dry Powder Inhalers in COPD, Lung Inflammation and Pulmonary Infections. Expert Opin. Drug Deliv. 2015, 12, 947–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buttini, F.; Rozou, S.; Rossi, A.; Zoumpliou, V.; Rekkas, D.M. The Application of Quality by Design Framework in the Pharmaceutical Development of Dry Powder Inhalers. Eur. J. Pharm. Sci. 2018, 113, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Kaialy, W.; Nokhodchi, A. Dry Powder Inhalers: Physicochemical and Aerosolization Properties of Several Size-Fractions of a Promising Alterative Carrier, Freeze-Dried Mannitol. Eur. J. Pharm. Sci. 2015, 68, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Della Bella, A.; Salomi, E.; Buttini, F.; Bettini, R. The Role of the Solid State and Physical Properties of the Carrier in Adhesive Mixtures for Lung Delivery. Eur. J. Pharm. Sci. 2018, 15, 665–674. [Google Scholar] [CrossRef]
- Benassi, A.; Perazzi, I.; Bosi, R.; Cottini, C.; Bettini, R. Quantifying the Loading Capacity of a Carrier-Based DPI Formulation and Its Dependence on the Blending Process. Powder Technol. 2019, 356, 607–617. [Google Scholar] [CrossRef]
- Smith, P.L.; Wall, D.A.; Gochoco, C.H.; Wilson, G. (D) Routes of Delivery: Case Studies: (5) Oral Absorption of Peptides and Proteins. Adv. Drug Deliv. Rev. 1992, 8, 253–290. [Google Scholar] [CrossRef]
- Kaialy, W.; Momin, M.N.; Ticehurst, M.D.; Murphy, J.; Nokhodchi, A. Engineered Mannitol as an Alternative Carrier to Enhance Deep Lung Penetration of Salbutamol Sulphate from Dry Powder Inhaler. Colloids Surf. B Biointerfaces 2010, 79, 345–356. [Google Scholar] [CrossRef]
- Kaialy, W.; Nokhodchi, A. The Use of Freeze-Dried Mannitol to Enhance the in Vitro Aerosolization Behaviour of Budesonide from the Aerolizer®. Powder Technol. 2016, 288, 291–302. [Google Scholar] [CrossRef]
- Hertel, N.; Birk, G.; Scherließ, R. Particle Engineered Mannitol for Carrier-Based Inhalation—A Serious Alternative? Int. J. Pharm. 2020, 577, 118901. [Google Scholar] [CrossRef]
- Mönckedieck, M.; Kamplade, J.; Fakner, P.; Urbanetz, N.A.; Walzel, P.; Steckel, H.; Scherließ, R. Dry Powder Inhaler Performance of Spray Dried Mannitol with Tailored Surface Morphologies as Carrier and Salbutamol Sulphate. Int. J. Pharm. 2017, 524, 351–363. [Google Scholar] [CrossRef] [PubMed]
- D’Addio, S.M.; Chan, J.G.Y.; Kwok, P.C.L.; Benson, B.R.; Prud’homme, R.K.; Chan, H.-K. Aerosol Delivery of Nanoparticles in Uniform Mannitol Carriers Formulated by Ultrasonic Spray Freeze Drying. Pharm. Res. 2013, 30, 2891–2901. [Google Scholar] [CrossRef] [PubMed]
- Bilton, D.; Robinson, P.; Cooper, P.; Gallagher, C.G.; Kolbe, J.; Fox, H.; Jaques, A.; Charlton, B. Inhaled Dry Powder Mannitol in Cystic Fibrosis: An Efficacy and Safety Study. Eur. Respir. J. 2011, 38, 1071–1080. [Google Scholar] [CrossRef]
- Aitken, M.L.; Bellon, G.; De Boeck, K.; Flume, P.A.; Fox, H.G.; Geller, D.E.; Haarman, E.G.; Hebestreit, H.U.; Lapey, A.; Schou, I.M.; et al. Long-Term Inhaled Dry Powder Mannitol in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2012, 185, 645–652. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, F.; Cocconi, D.; Bettini, R.; Giordano, F.; Santi, P.; Tobyn, M.; Price, R.; Young, P.; Caramella, C.; Colombo, P. The Surface Roughness of Lactose Particles Can Be Modulated by Wet-Smoothing Using a High-Shear Mixer. AAPS PharmSciTech 2004, 5, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, P.M.; Cocconi, D.; Colombo, P.; Bettini, R.; Price, R.; Steele, D.F.; Tobyn, M.J. Characterization of a Surface Modified Dry Powder Inhalation Carrier Prepared by “Particle Smoothing”. J. Pharm. Pharmacol. 2002, 54, 1339–1344. [Google Scholar] [CrossRef]
- Donovan, M.J.; Smyth, H.D.C. Influence of Size and Surface Roughness of Large Lactose Carrier Particles in Dry Powder Inhaler Formulations. Int. J. Pharm. 2010, 402, 1–9. [Google Scholar] [CrossRef]
- Della Bella, A.; Müller, M.; Danani, A.; Soldati, L.; Bettini, R. Effect of Lactose Pseudopolymorphic Transition on the Aerosolization Performance of Drug/Carrier Mixtures. Pharmaceutics 2019, 11, 576. [Google Scholar] [CrossRef] [Green Version]
- Della Bella, A.; Müller, M.; Soldati, L.; Elviri, L.; Bettini, R. Quantitative Determination of Micronization-Induced Changes in the Solid State of Lactose. Int. J. Pharm. 2016, 505, 383–393. [Google Scholar] [CrossRef]
- Zeng, X.M.; Martin, G.P.; Marriott, C. Particulate Interactions in Dry Powder Formulation for Inhalation; CRC Press: Boca Raton, FL, USA, 2000; pp. 135–204. ISBN 9780367397975. [Google Scholar]
- Kaialy, W.; Alhalaweh, A.; Velaga, S.P.; Nokhodchi, A. Influence of Lactose Carrier Particle Size on the Aerosol Performance of Budesonide from a Dry Powder Inhaler. Powder Technol. 2012, 227, 74–85. [Google Scholar] [CrossRef]
- Fronczek, F.R.; Kamel, H.N.; Slattery, M. Three Polymorphs (α, β, and δ) of d-Mannitol at 100 K. Acta Cryst. Sect. C Cryst. Struct. Commun. 2003, 59, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Cares-Pacheco, M.G.; Vaca-Medina, G.; Calvet, R.; Espitalier, F.; Letourneau, J.J.; Rouilly, A.; Rodier, E. Physicochemical Characterization of D-Mannitol Polymorphs: The Challenging Surface Energy Determination by Inverse Gas Chromatography in the Infinite Dilution Region. Int. J. Pharm. 2014, 475, 69–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, R.R.; Shah, U.V.; Parambil, J.V.; Burnett, D.J.; Thielmann, F.; Heng, J.Y.Y. The Effect of Polymorphism on Surface Energetics of D-Mannitol Polymorphs. AAPS J. 2017, 19, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Nunes, C.; Suryanarayanan, R.; Botez, C.E.; Stephens, P.W. Characterization and Crystal Structure of D-Mannitol Hemihydrate. J. Pharm. Sci. 2004, 93, 2800–2809. [Google Scholar] [CrossRef] [PubMed]
- Vanhoorne, V.; Bekaert, B.; Peeters, E.; De Beer, T.; Remon, J.-P.; Vervaet, C. Improved Tabletability after a Polymorphic Transition of Delta-Mannitol during Twin Screw Granulation. Int. J. Pharm. 2016, 506, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhoorne, V.; Almey, R.; De Beer, T.; Vervaet, C. Delta-Mannitol to Enable Continuous Twin-Screw Granulation of a Highly Dosed, Poorly Compactable Formulation. Int. J. Pharm. 2020, 583, 119374. [Google Scholar] [CrossRef]
- Wagner, C.M.; Pein, M.; Breitkreutz, J. Roll Compaction of Granulated Mannitol Grades and the Unprocessed Crystalline Delta-Polymorph. Powder Technol. 2015, 270, 470–475. [Google Scholar] [CrossRef]
- Lee, Y.-Y.; Wu, J.X.; Yang, M.; Young, P.M.; van den Berg, F.; Rantanen, J. Particle Size Dependence of Polymorphism in Spray-Dried Mannitol. Eur. J. Pharm. Sci. 2011, 44, 41–48. [Google Scholar] [CrossRef]
- Kaialy, W.; Nokhodchi, A. Treating Mannitol in a Saturated Solution of Mannitol: A Novel Approach to Modify Mannitol Crystals for Improved Drug Delivery to the Lungs. Int. J. Pharm. 2013, 448, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Milton, N.; Groleau, E.G.; Mishra, D.S.; Vansickle, R.E. Existence of a Mannitol Hydrate during Freeze-Drying and Practical Implications. Int. J. Pharm. 1999, 88, 196–198. [Google Scholar] [CrossRef]
- Foster, K.A.; Avery, M.L.; Yazdanian, M.; Audus, K.L. Characterization of the Calu-3 Cell Line as a Tool to Screen Pulmonary Drug Delivery. Int. J. Pharm. 2000, 208, 1–11. [Google Scholar] [CrossRef]
- Hastedt, J.E.; Bäckman, P.; Clark, A.R.; Doub, W.; Hickey, A.; Hochhaus, G.; Kuehl, P.J.; Lehr, C.-M.; Mauser, P.; McConville, J.; et al. Scope and Relevance of a Pulmonary Biopharmaceutical Classification System AAPS/FDA/USP Workshop March 16–17th, 2015 in Baltimore, MD. AAPS Open 2016, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Boshhiha, A.M.; Urbanetz, N.A. Influence of Carrier Surface Fines on Dry Powder Inhalation Formulations. Drug Dev. Ind. Pharm. 2009, 35, 904–916. [Google Scholar] [CrossRef]
- Vanhoorne, V.; Van Bockstal, P.-J.; Van Snick, B.; Peeters, E.; Monteyne, T.; Gomes, P.; De Beer, T.; Remon, J.P.; Vervaet, C. Continuous Manufacturing of Delta Mannitol by Cospray Drying with PVP. Int. J. Pharm. 2016, 501, 139–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, D.M.; Paleco, R.; Traini, D.; Sencadas, V. Development of Ciprofloxacin-Loaded Poly(Vinyl Alcohol) Dry Powder Formulations for Lung Delivery. Int. J. Pharm. 2018, 547, 114–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassoun, M.; Ho, S.; Muddle, J.; Buttini, F.; Parry, M.; Hammond, M.; Forbes, B. Formulating Powder—Device Combinations for Salmeterol Xinafoate Dry Powder Inhalers. Int. J. Pharm. 2015, 490, 360–367. [Google Scholar] [CrossRef]
- Momin, M.; Hedayati, A.; Nokhodchi, A. Investigation into Alternative Sugars as Potential Carriers for Dry Powder Formulation of Budesonide. BioImpacts 2011, 1, 105–111. [Google Scholar] [PubMed]
- Pilcer, G.; Wauthoz, N.; Amighi, K. Lactose Characteristics and the Generation of the Aerosol. Adv. Drug Deliv. Rev. 2011, 64, 233–256. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, N.; Hillaireau, H.; Vergnaud, J.; Santiago, L.A.; Kerdine-Romer, S.; Pallardy, M.; Tsapis, N.; Fattal, E. Toxicity of Surface-Modified PLGA Nanoparticles toward Lung Alveolar Epithelial Cells. Int. J. Pharm. 2013, 454, 686–694. [Google Scholar] [CrossRef]
- Madlova, M.; Jones, S.A.; Zwerschke, I.; Ma, Y.; Hider, R.C.; Forbes, B. Poly(Vinyl Alcohol) Nanoparticle Stability in Biological Media and Uptake in Respiratory Epithelial Cell Layers in Vitro. Eur. J. Pharm. Biopharm. 2009, 72, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Ahlberg, S.; Antonopulos, A.; Diendorf, J.; Dringen, R.; Epple, M.; Flöck, R.; Goedecke, W.; Graf, C.; Haberl, N.; Helmlinger, J.; et al. PVP-Coated, Negatively Charged Silver Nanoparticles: A Multi-Center Study of Their Physicochemical Characteristics, Cell Culture and in Vivo Experiments. Beilstein J. Nanotechnol. 2014, 5, 1944–1965. [Google Scholar] [CrossRef] [PubMed]
- Kurakula, M.; Rao, G.S.N.K. Pharmaceutical Assessment of Polyvinylpyrrolidone (PVP): As Excipient from Conventional to Controlled Delivery Systems with a Spotlight on COVID-19 Inhibition. J. Drug Deliv. Sci. Technol. 2020, 60, 102046. [Google Scholar] [CrossRef]
- Scherliess, R. The MTT Assay as Tool to Evaluate and Compare Excipient Toxicity in Vitro on Respiratory Epithelial Cells. Int. J. Pharm. 2011, 411, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Guillory, J.K. Generation of polymorphs, hydrates, solvates, and amorphous solids. In Polymorphism in Pharmaceutical Solids, 2nd ed.; Brittain, H.G., Ed.; CRC Press: Boca Raton, FL, USA, 1999; ISBN 978-0-429-14766-1. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mannitol | Dv10 | Dv50 | Dv90 | Span |
|---|---|---|---|---|
| β form | 2.90 ± 0.29 | 11.11 ± 1.75 | 28.87 ± 1.15 | 2.34 ± 0.17 |
| δ form | 6.21 ± 0.86 | 23.05 ± 1.85 | 57.93 ± 1.50 | 2.24 ± 0.18 |
| α form | 6.83 ± 0.42 | 22.68 ± 2.11 | 45.36 ± 3.32 | 2.98 ± 0.75 |
| Hydrate form | 6.83 ± 1.17 | 22.68 ± 2.72 | 58.14 ± 1.35 | 1.60 ± 0.07 |
| Carrier | Drug | Drug Content (%) | Loaded Powder Dose (mg) |
|---|---|---|---|
| MM50 | SS | 0.82 ± 0.03 (3.67) | 20.1 ± 0.2 (0.96) |
| MM50 | BUD | 0.70 ± 0.02 (2.86) | 20.3 ± 0.1 (0.41) |
| δ form | SS | 1.01 ± 0.04 (3.92) | 20.1 ± 0.1 (0.26) |
| δ form | BUD | 0.85 ± 0.03 (3.81) | 20.0 ± 0.1 (0.38) |
| β form | SS | 0.93 ± 0.03 (3.14) | 20.2 ± 0.2 (0.84) |
| β form | BUD | 0.99 ± 0.04 (4.04) | 20.0 ± 0.1 (0.32) |
| α form | SS | 0.83 ± 0.04 (4.83) | 20.1 ± 0.1 (0.24) |
| α form | BUD | 0.87 ± 0.01 (1.57) | 20.0 ± 0.1 (0.26) |
| Carrier | Emitted Dose (%) | FPD (µg) | FPF (%) | MMAD (µm) |
|---|---|---|---|---|
| MM50 | 80.1 ± 12.7 | 10.0 ± 2.5 | 8.4 ± 1.7 | 3.6 ± 0.1 |
| δ form | 90.4 ± 18.5 | 16.0 ± 1.6 | 9.3 ± 2.4 | 4.1 ± 0.0 |
| β form | 97.6 ± 3.2 | 29.8 ± 1.9 | 13.9 ± 0.7 | 4.7 ± 0.5 |
| α form | 81.7 ± 9.0 | 21.8 ± 4.8 | 13.4 ± 4.3 | 4.1 ± 0.1 |
| Carrier | Emitted Dose (%) | FPD (µg) | FPF (%) | MMAD (µm) |
|---|---|---|---|---|
| MM50 | 96.0 ± 6.2 | 14.6 ± 1.9 | 13.5 ± 3.3 | 3.9 ± 1.2 |
| δ form | 95.1 ± 5.6 | 13.2 ± 1.5 | 6.5 ± 0.1 | 5.7 ± 0.4 |
| β form | 90.3 ± 12.5 | 21.0 ± 3.0 | 11.1 ± 3.7 | 4.5 ± 0.0 |
| α form | 90.5 ± 13.4 | 18.0 ± 5.5 | 11.9 ± 6.0 | 4.0 ± 0.1 |
| Carrier | Emitted Dose (%) | FPD (µg) | FPF (%) | MMAD (µm) |
|---|---|---|---|---|
| MM50 | 87.3 ± 2.0 | 36.2 ± 0.2 | 26.6 ± 1.3 | 1.6 ± 0.2 |
| δ form | 78.4 ± 3.5 | 26.3 ± 8.2 | 19.6 ± 3.2 | 2.9 ± 0.4 |
| β form | 71.3 ± 7.6 | 104.7 ± 7.2 | 58.1 ± 3.9 | 1.8 ± 0.1 |
| α form | 76.5 ± 8.6 | 84.0 ± 14.6 | 53.5 ± 5.1 | 1.8 ± 0.1 |
| Carrier | Emitted Dose (%) | FPD (µg) | FPF (%) | MMAD (µm) |
|---|---|---|---|---|
| MM50 | 76.8 ± 9.7 | 48.0 ± 2.8 | 39.1 ± 3.4 | 2.1 ± 0.2 |
| δ form | 71.7 ± 5.0 | 47.1 ± 10.6 | 32.0 ± 9.2 | 1.7 ± 0.1 |
| β form | 85.0 ± 8.7 | 108.0 ± 18.0 | 57.7 ± 11.8 | 1.8 ± 0.1 |
| α form | 89.5 ± 1.7 | 87.3 ± 0.8 | 55.8 ± 1.5 | 2.0 ± 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altay Benetti, A.; Bianchera, A.; Buttini, F.; Bertocchi, L.; Bettini, R. Mannitol Polymorphs as Carrier in DPIs Formulations: Isolation Characterization and Performance. Pharmaceutics 2021, 13, 1113. https://doi.org/10.3390/pharmaceutics13081113
Altay Benetti A, Bianchera A, Buttini F, Bertocchi L, Bettini R. Mannitol Polymorphs as Carrier in DPIs Formulations: Isolation Characterization and Performance. Pharmaceutics. 2021; 13(8):1113. https://doi.org/10.3390/pharmaceutics13081113
Chicago/Turabian StyleAltay Benetti, Ayça, Annalisa Bianchera, Francesca Buttini, Laura Bertocchi, and Ruggero Bettini. 2021. "Mannitol Polymorphs as Carrier in DPIs Formulations: Isolation Characterization and Performance" Pharmaceutics 13, no. 8: 1113. https://doi.org/10.3390/pharmaceutics13081113
APA StyleAltay Benetti, A., Bianchera, A., Buttini, F., Bertocchi, L., & Bettini, R. (2021). Mannitol Polymorphs as Carrier in DPIs Formulations: Isolation Characterization and Performance. Pharmaceutics, 13(8), 1113. https://doi.org/10.3390/pharmaceutics13081113