
Pheophorbide A and Paclitaxel Bioresponsive Nanoparticles as Double-Punch Platform for Cancer Therapy

 , , , ,
, , , ,  and
and 
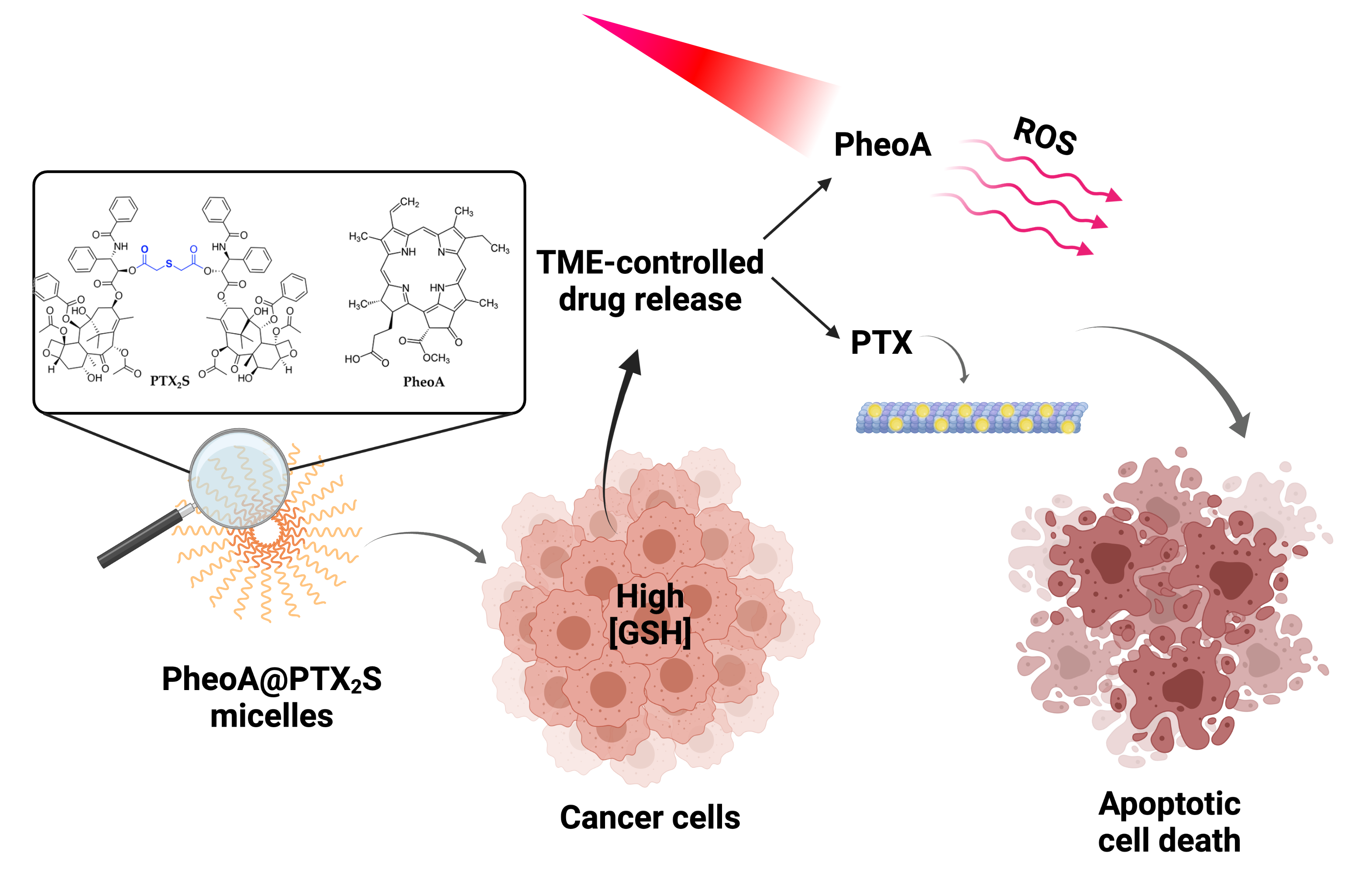
Abstract
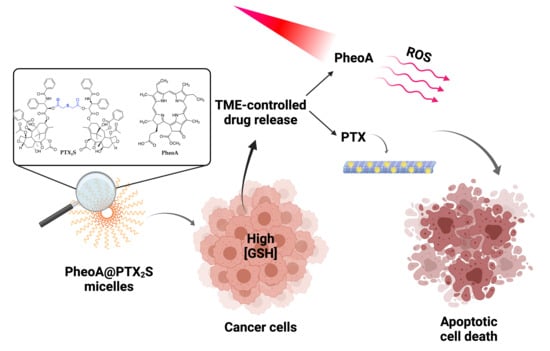
:
1. Introduction
2. Materials and Methods
2.1. Materials
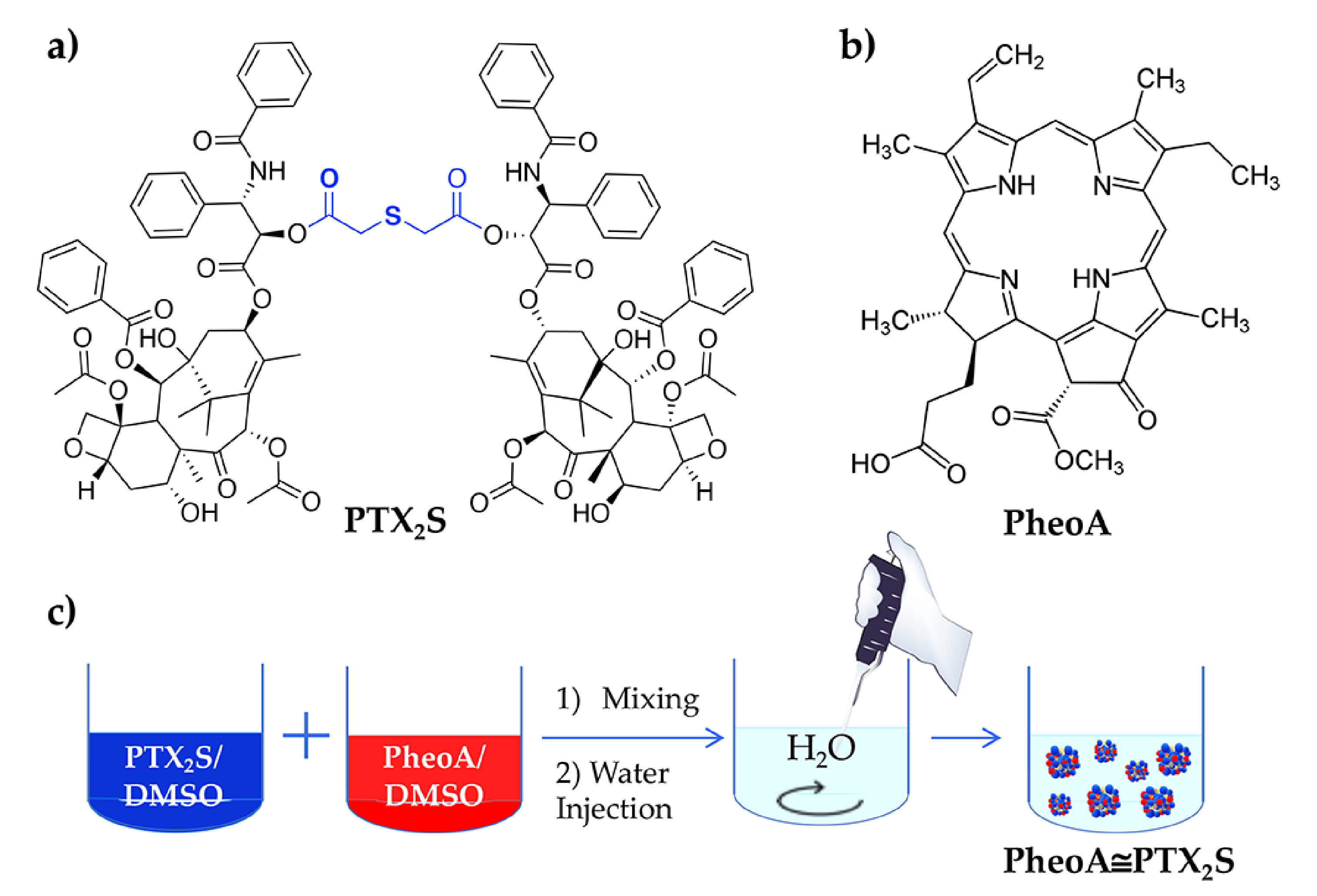
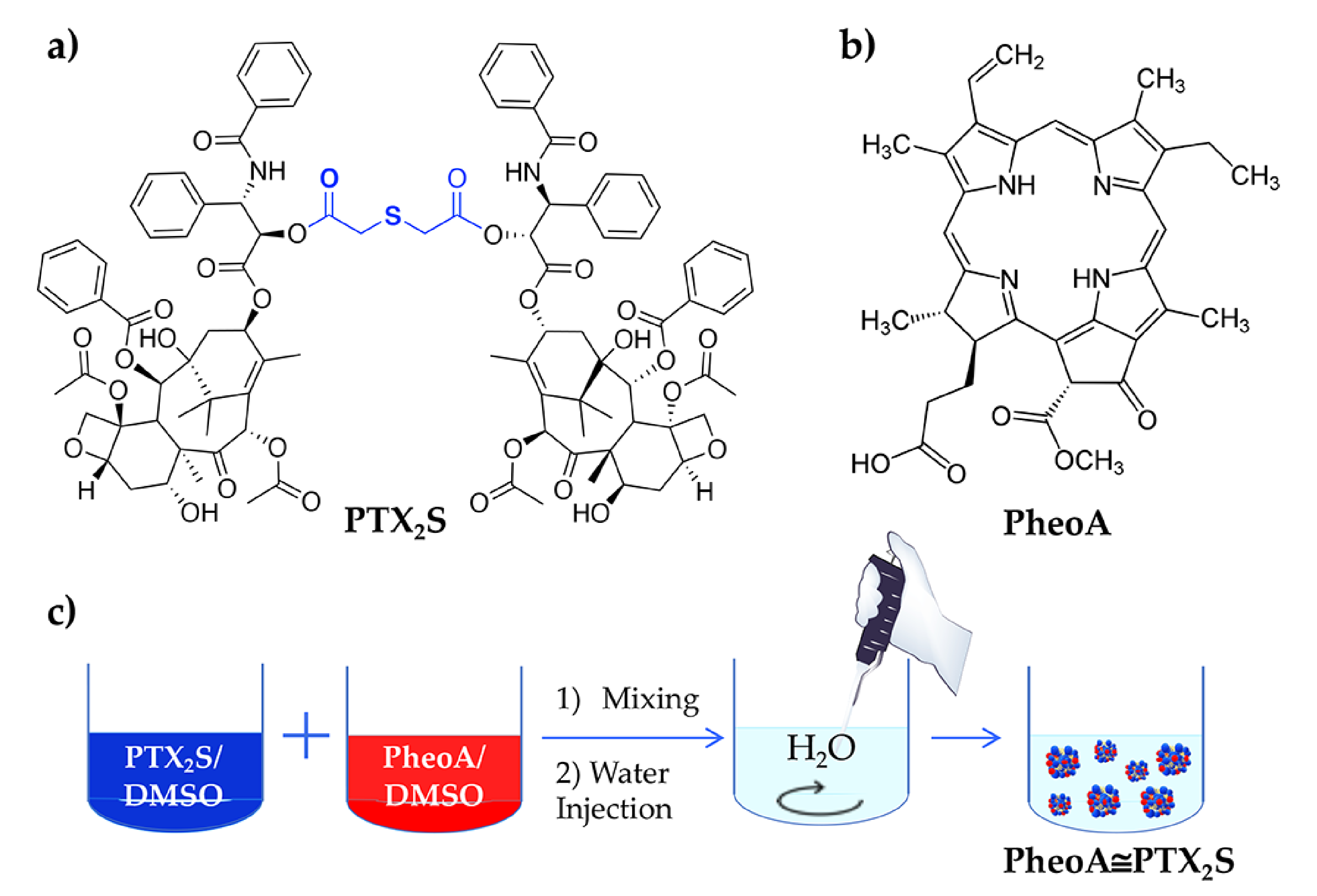
2.2. Synthesis of PTX2S
2.3. Preparation of PTX2S Nanoparticles (mPTX2S)
2.4. Preparation of PTX2S Nanoparticles Loaded with PheoA (PheoA≅PTX2S)
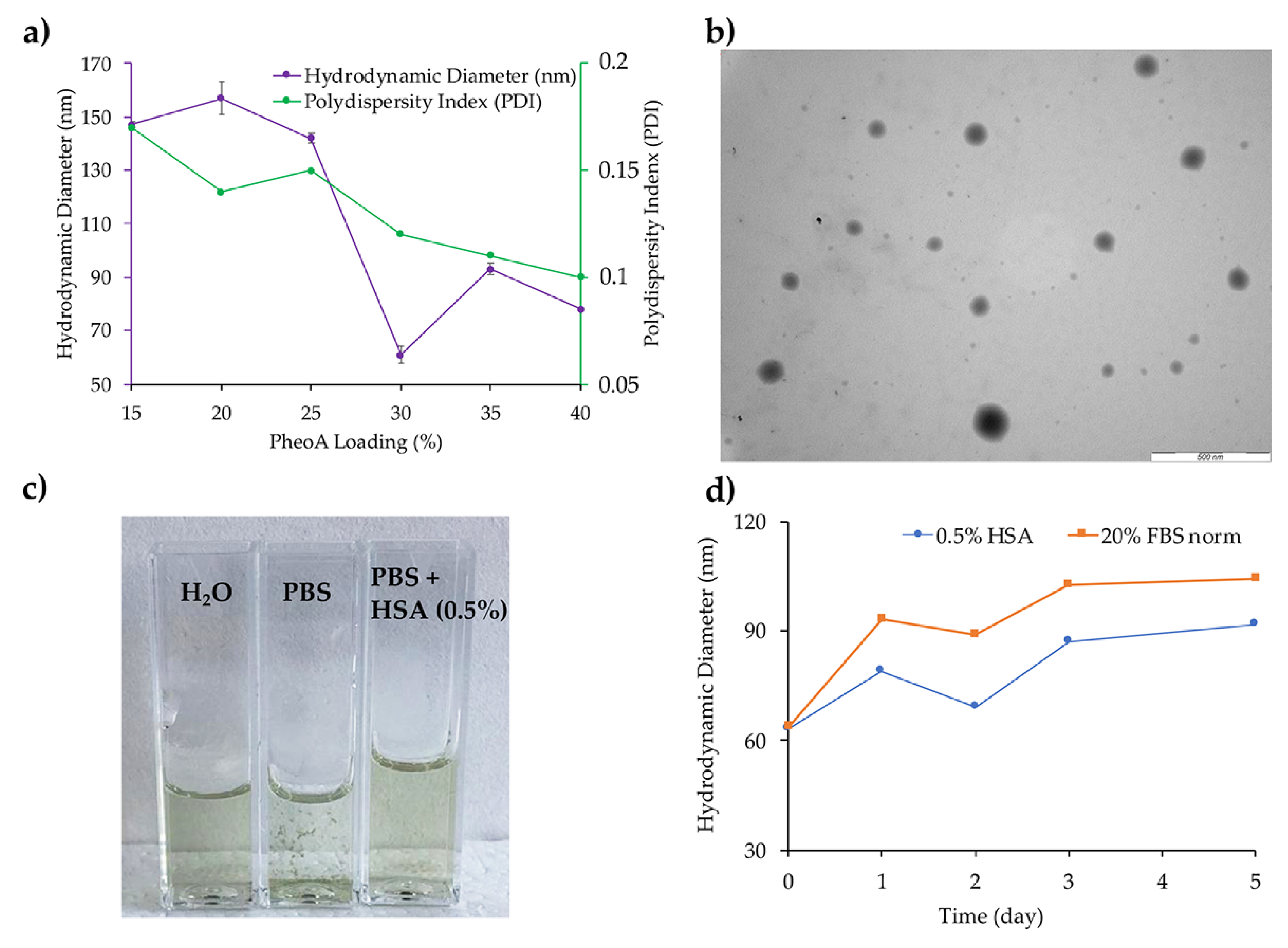
2.5. Characterization of Nanoparticles
2.6. In Vitro Stability of Nanoparticles
2.7. Reactive Oxygen Species and Singlet Oxygen Generation
2.8. GSH or H2O2-Triggered Nanoparticles Disassembly
2.9. Cell Lines
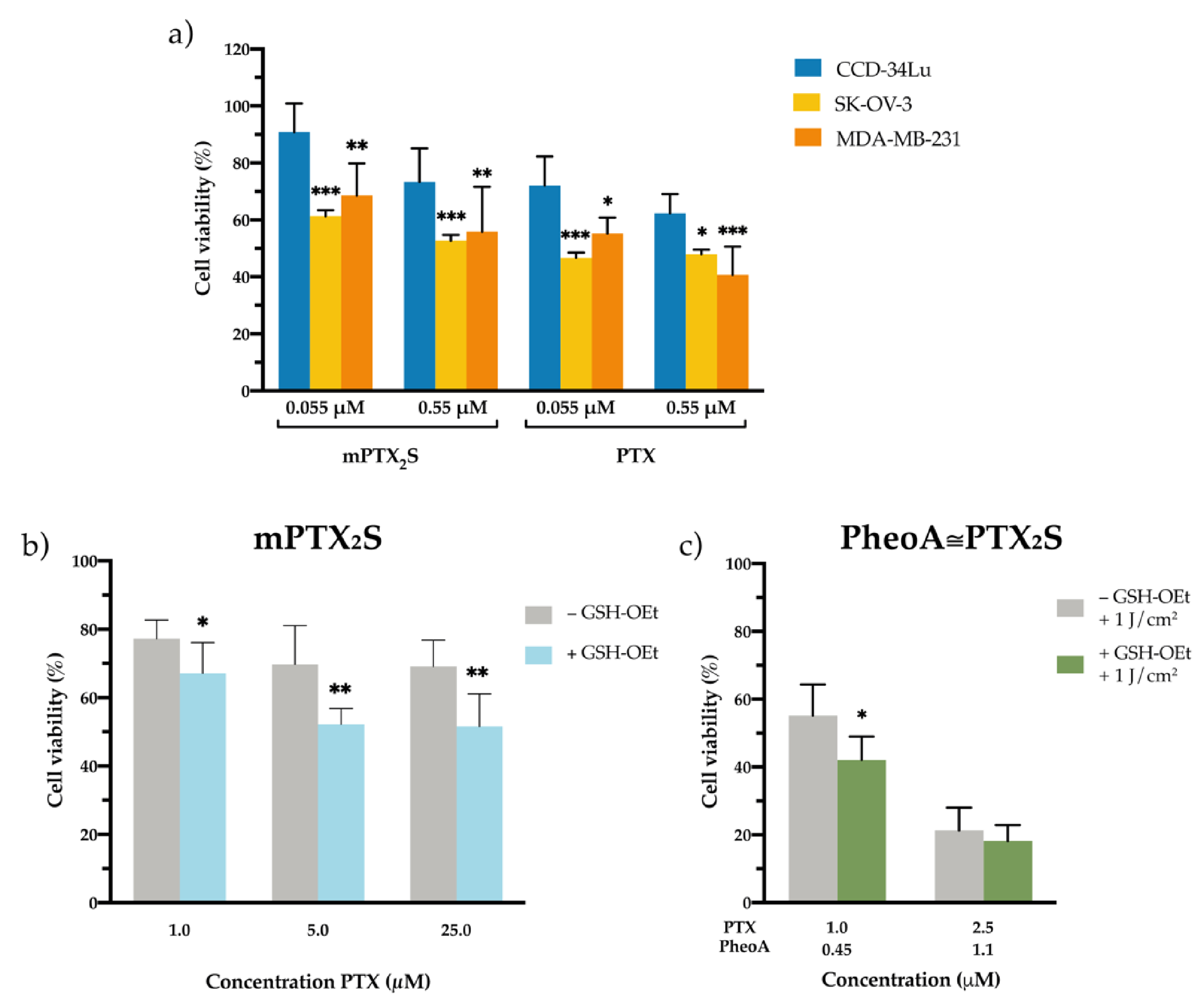
2.10. Cytotoxicity of Free PTX and mPTX2S toward Cancer and Normal Cells
2.11. In-Cell Simulation of Redox Environment (Experiment with GSH-OEt)
2.12. Combination Therapy Experiments
2.13. Cellular Uptake and Localization of PheoA and PheoA≅PTX2S
2.14. Annexin/PI Assay
3. Results and Discussion
3.1. Synthesis and Characterization of mPTX2S and PheoA≅PTX2S
3.2. Stability of mPTX2S and PheoA≅PTX2S
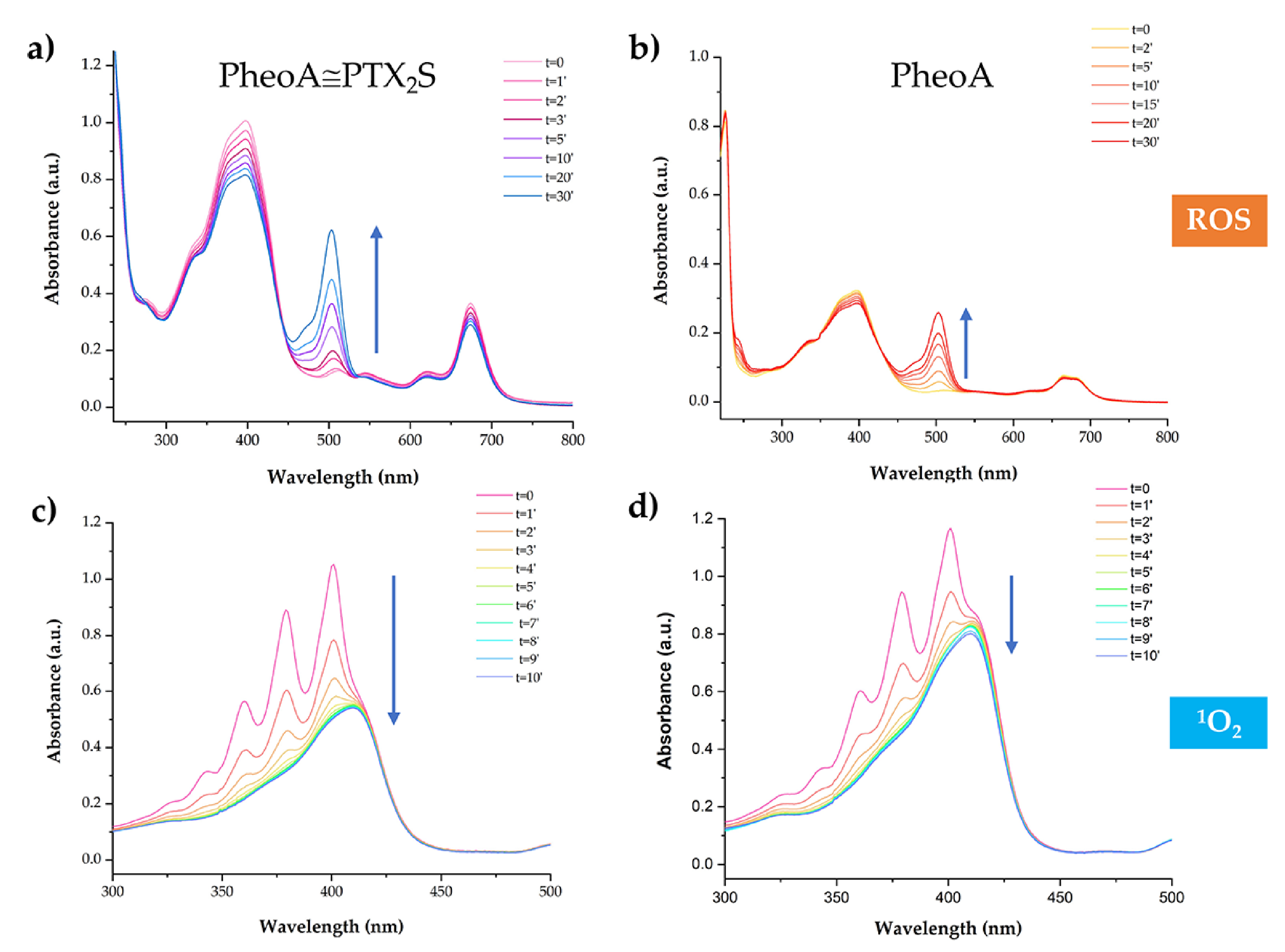
3.3. ROS and 1O2 Generation
3.4. GSH and H2O2 Triggered Disassembly of PheoA≅PTX2S Nanoparticles
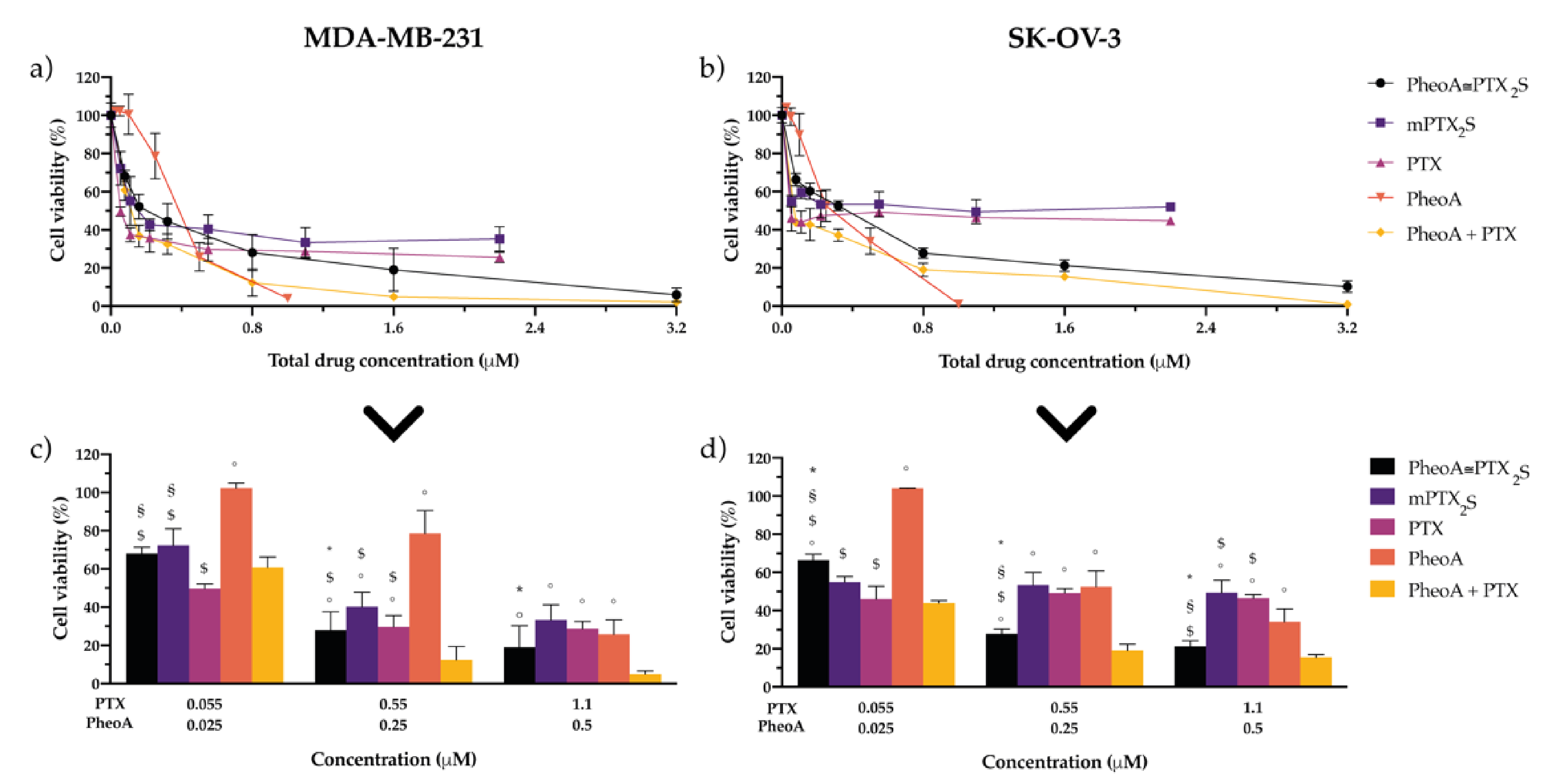
3.5. Cytotoxicity of Nanoparticles in Cancer and Normal Cells In Vitro
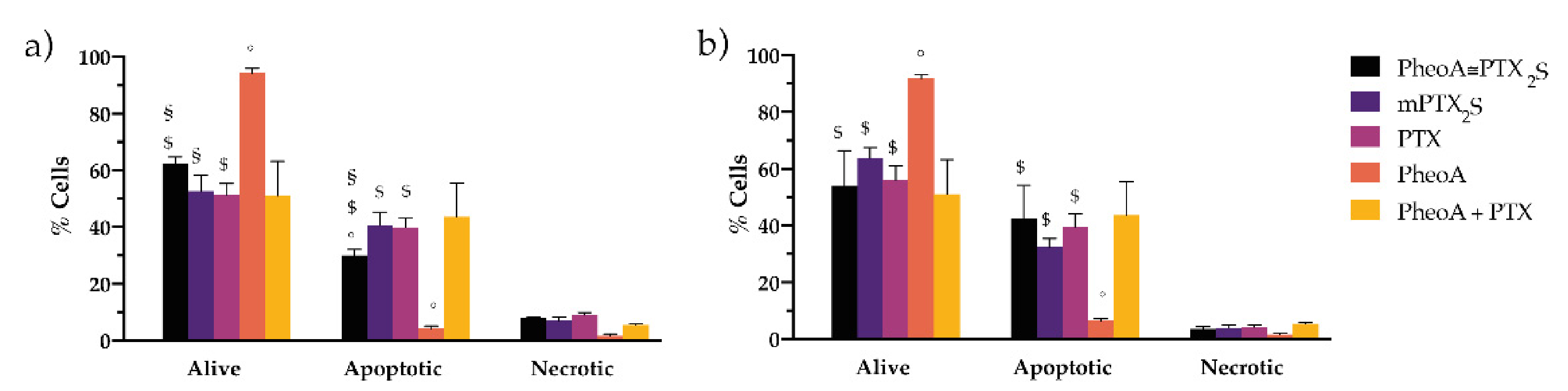
3.6. In Vitro Combination Therapy with PheoA≅PTX2S Nanoparticles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Misra, R.; Acharya, S.; Sahoo, S.K. Cancer nanotechnology: Application of nanotechnology in cancer therapy. Drug Discov. Today 2010, 15, 842–850. [Google Scholar] [CrossRef]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuluaga, M.-F.; Lange, N. Combination of Photodynamic Therapy with Anti-Cancer Agents. Curr. Med. Chem. 2008, 15, 1655–1673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Chen, Z.; Chi, J.; Sun, Y.; Sun, Y. Recent progress in synergistic chemotherapy and phototherapy by targeted drug delivery systems for cancer treatment. Artif. Cells Nanomed. Biotechnol. 2018, 46, 817–830. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.B.; Brown, E.A.; Walker, I. The present and future role of photodynamic therapy in cancer treatment. Lancet Oncol. 2004, 5, 497–508. [Google Scholar] [CrossRef]
- Celli, J.P.; Spring, B.Q.; Rizvi, I.; Evans, C.L.; Samkoe, K.S.; Verma, S.; Pogue, B.W.; Hasan, T. Imaging and Photodynamic Therapy: Mechanisms, Monitoring, and Optimization. Chem. Rev. 2010, 110, 2795–2838. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, A.F.; De Almeida, D.R.Q.; Terra, L.F.; Baptista, M.S.; Labriola, L. Photodynamic therapy in cancer treatment—An update review. J. Cancer Metastasis Treat. 2019, 2019, 25. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Jiang, C.; Figueiró Longo, J.P.; Azevedo, R.B.; Zhang, H.; Muehlmann, L.A. An updated overview on the development of new photosensitizers for anticancer photodynamic therapy. Acta Pharm. Sin. B 2018, 8, 137–146. [Google Scholar] [CrossRef]
- Ferroni, C.; Del Rio, A.; Martini, C.; Manoni, E.; Varchi, G. Light-Induced Therapies for Prostate Cancer Treatment. Front. Chem. 2019, 7, 719. [Google Scholar] [CrossRef] [PubMed]
- Avancini, G.; Guerrini, A.; Ferroni, C.; Tedesco, D.; Ballestri, M.; Columbaro, M.; Menilli, L.; Reddi, E.; Costa, R.; Leanza, L.; et al. Keratin nanoparticles and photodynamic therapy enhance the anticancer stem cells activity of salinomycin. Mater. Sci. Eng. C 2021, 122, 111899. [Google Scholar] [CrossRef] [PubMed]
- Khdair, A.; Chen, D.; Patil, Y.; Ma, L.; Dou, Q.P.; Shekhar, M.P.V.; Panyam, J. Nanoparticle-mediated combination chemotherapy and photodynamic therapy overcomes tumor drug resistance. J. Control. Release 2010, 141, 137–144. [Google Scholar] [CrossRef] [Green Version]
- Castano, A.P.; Mroz, P.; Wu, M.X.; Hamblin, M.R. Photodynamic therapy plus low-dose cyclophosphamide generates antitumor immunity in a mouse model. Proc. Natl. Acad. Sci. USA 2008, 105, 5495–5500. [Google Scholar] [CrossRef] [Green Version]
- Martella, E.; Ferroni, C.; Guerrini, A.; Ballestri, M.; Columbaro, M.; Santi, S.; Sotgiu, G.; Serra, M.; Donati, D.M.; Lucarelli, E.; et al. Functionalized Keratin as Nanotechnology-Based Drug Delivery System for the Pharmacological Treatment of Osteosarcoma. Int. J. Mol. Sci. 2018, 19, 3670. [Google Scholar] [CrossRef] [Green Version]
- Gaio, E.; Guerrini, A.; Ballestri, M.; Varchi, G.; Ferroni, C.; Martella, E.; Columbaro, M.; Moret, F.; Reddi, E. Keratin nanoparticles co-delivering Docetaxel and Chlorin e6 promote synergic interaction between chemo- and photo-dynamic therapies. J. Photochem. Photobiol. B. 2019, 199, 111598. [Google Scholar] [CrossRef]
- Chang, J.E.; Yoon, I.S.; Sun, P.L.; Yi, E.; Jheon, S.; Shim, C.K. Anticancer efficacy of photodynamic therapy with hematoporphyrin-modified, doxorubicin-loaded nanoparticles in liver cancer. J. Photochem. Photobiol. B Biol. 2014, 140, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Carter, K.A.; Miranda, D.; Lovell, J.F. Chemophototherapy: An Emerging Treatment Option for Solid Tumors. Adv. Sci. 2017, 4, 1600106. [Google Scholar] [CrossRef] [Green Version]
- Pedrosa, P.; Mendes, R.; Cabral, R.; Martins, L.M.D.R.S.; Baptista, P.V.; Fernandes, A.R. Combination of chemotherapy and Au-nanoparticle photothermy in the visible light to tackle doxorubicin resistance in cancer cells. Sci. Rep. 2018, 8, 11429. [Google Scholar] [CrossRef] [Green Version]
- Maeda, H.; Khatami, M. Analyses of repeated failures in cancer therapy for solid tumors: Poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin. Transl. Med. 2018, 7, 11. [Google Scholar] [CrossRef]
- van der Meel, R.; Sulheim, E.; Shi, Y.; Kiessling, F.; Mulder, W.J.M.; Lammers, T. Smart cancer nanomedicine. Nat. Nanotechnol. 2019, 14, 1007–1017. [Google Scholar] [CrossRef]
- Kennedy, L.; Sandhu, J.K.; Harper, M.-E.; Cuperlovic-Culf, M. Role of glutathione in cancer: From mechanisms to therapies. Biomolecules 2020, 10, 1429. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Cai, K.; Zhang, Y.; Lu, Y.; Guo, Q.; Zhang, Y.; Liu, L.; Ruan, C.; Chen, Q.; Chen, X.; et al. Dimeric Prodrug Self-Delivery Nanoparticles with Enhanced Drug Loading and Bioreduction Responsiveness for Targeted Cancer Therapy. ACS Appl. Mater. Interfaces 2018, 10, 39455–39467. [Google Scholar] [CrossRef]
- Li, S.; Shan, X.; Wang, Y.; Chen, Q.; Sun, J.; He, Z.; Sun, B.; Luo, C. Dimeric prodrug-based nanomedicines for cancer therapy. J. Control. Release 2020, 326, 510–522. [Google Scholar] [CrossRef]
- Singla, A.K.; Garg, A.; Aggarwal, D. Paclitaxel and its formulations. Int. J. Pharm. 2002, 235, 179–192. [Google Scholar] [CrossRef]
- Gornstein, E.; Schwarz, T.L. The paradox of paclitaxel neurotoxicity: Mechanisms and unanswered questions. Neuropharmacology 2014, 76, 175–183. [Google Scholar] [CrossRef]
- Gelderblom, H.; Verweij, J.; Nooter, K.; Sparreboom, A. Cremophor EL: The drawbacks and advantages of vehicle selection for drug formulation. Eur. J. Cancer 2001, 37, 1590–1598. [Google Scholar] [CrossRef]
- Pei, Q.; Hu, X.; Zheng, X.; Xia, R.; Liu, S.; Xie, Z.; Jing, X. Albumin-bound paclitaxel dimeric prodrug nanoparticles with tumor redox heterogeneity-triggered drug release for synergistic photothermal/chemotherapy. Nano Res. 2019, 12, 877–887. [Google Scholar] [CrossRef]
- Meng, Z.; Lv, Q.; Lu, J.; Yao, H.; Lv, X.; Jiang, F.; Lu, A.; Zhang, G. Prodrug Strategies for Paclitaxel. Int. J. Mol. Sci. 2016, 17, 796. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Chen, J.; Jiang, M.; Zhang, N.; Na, K.; Luo, C.; Zhang, R.; Sun, M.; Lin, G.; Zhang, R.; et al. Paclitaxel-Paclitaxel Prodrug Nanoassembly as a Versatile Nanoplatform for Combinational Cancer Therapy. ACS Appl. Mater. Interfaces 2016, 8, 33506–33513. [Google Scholar] [CrossRef]
- Pei, Q.; Hu, X.; Zhou, J.; Liu, S.; Xie, Z. Glutathione-responsive paclitaxel dimer nanovesicles with high drug content. Biomater. Sci. 2017, 5, 1517–1521. [Google Scholar] [CrossRef]
- Luo, C.; Sun, J.; Liu, D.; Sun, B.; Miao, L.; Musetti, S.; Li, J.; Han, X.; Du, Y.; Li, L.; et al. Self-Assembled Redox Dual-Responsive Prodrug-Nanosystem Formed by Single Thioether-Bridged Paclitaxel-Fatty Acid Conjugate for Cancer Chemotherapy. Nano Lett. 2016, 16, 5401–5408. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Pei, Q.; Xia, R.; Liu, S.; Hu, X.; Xie, Z.; Jing, X. Comparison of Redox Responsiveness and Antitumor Capability of Paclitaxel Dimeric Nanoparticles with Different Linkers. Chem. Mater. 2020, 32, 10719–10727. [Google Scholar] [CrossRef]
- Ferroni, C.; Sotgiu, G.; Sagnella, A.; Varchi, G.; Guerrini, A.; Giuri, D.; Polo, E.; Orlandi, V.T.; Marras, E.; Gariboldi, M.; et al. Wool Keratin 3D Scaffolds with Light-Triggered Antimicrobial Activity. Biomacromolecules 2016, 17, 2882–2890. [Google Scholar] [CrossRef] [PubMed]
- Ballestri, M.; Caruso, E.; Guerrini, A.; Ferroni, C.; Banfi, S.; Gariboldi, M.; Monti, E.; Sotgiu, G.; Varchi, G. Core–shell poly-methyl methacrylate nanoparticles covalently functionalized with a non-symmetric porphyrin for anticancer photodynamic therapy. J. Photochem. Photobiol. B Biol. 2018, 186, 169–177. [Google Scholar] [CrossRef]
- Yuan, L.; Chen, W.; Hu, J.; Zhang, J.Z.; Yang, D. Mechanistic Study of the Covalent Loading of Paclitaxel via Disulfide Linkers for Controlled Drug Release. Langmuir 2013, 29, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Gaio, E.; Conte, C.; Esposito, D.; Miotto, G.; Quaglia, F.; Moret, F.; Reddi, E. Co-delivery of Docetaxel and Disulfonate Tetraphenyl Chlorin in One Nanoparticle Produces Strong Synergism between Chemo- and Photodynamic Therapy in Drug-Sensitive and -Resistant Cancer Cells. Mol. Pharm. 2018, 15, 4599–4611. [Google Scholar] [CrossRef]
- Partikel, K.; Korte, R.; Mulac, D.; Humpf, H.-U.; Langer, K. Serum type and concentration both affect the protein-corona composition of PLGA nanoparticles. Beilstein J. Nanotechnol. 2019, 10, 1002–1015. [Google Scholar] [CrossRef] [Green Version]
- Rampado, R.; Crotti, S.; Caliceti, P.; Pucciarelli, S.; Agostini, M. Recent Advances in Understanding the Protein Corona of Nanoparticles and in the Formulation of “Stealthy” Nanomaterials. Front. Bioeng. Biotechnol. 2020, 8, 166. [Google Scholar] [CrossRef]
- Roche, M.; Rondeau, P.; Singh, N.R.; Tarnus, E.; Bourdon, E. The antioxidant properties of serum albumin. FEBS Lett. 2008, 582, 1783–1787. [Google Scholar] [CrossRef]
- Yeo, E.L.L.; Cheah, J.U.J.; Thong, P.S.P.; Soo, K.C.; Kah, J.C.Y. Gold Nanorods Coated with Apolipoprotein E Protein Corona for Drug Delivery. ACS Appl. Nano Mater. 2019, 2, 6220–6229. [Google Scholar] [CrossRef]
- Yeo, E.L.L.; Cheah, J.U.J.; Lim, B.Y.; Thong, P.S.P.; Soo, K.C.; Kah, J.C.Y. Protein Corona around Gold Nanorods as a Drug Carrier for Multimodal Cancer Therapy. ACS Biomater. Sci. Eng. 2017, 3, 1039–1050. [Google Scholar] [CrossRef]
- Albiter, E.; Alfaro, S.; Valenzuela, M.A. Photosensitized oxidation of 9,10-dimethylanthracene with singlet oxygen by using a safranin O/silica composite under visible light. Photochem. Photobiol. Sci. 2015, 14, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhao, X.; Chen, J.; Chen, J.; Kuznetsova, L.; Wong, S.S.; Ojima, I. Mechanism-Based Tumor-Targeting Drug Delivery System. Validation of Efficient Vitamin Receptor-Mediated Endocytosis and Drug Release. Bioconjug. Chem. 2010, 21, 979–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, B.; Ryoo, I.; Kang, H.C.; Kwak, M.-K. The sensitivity of cancer cells to pheophorbide a-based photodynamic therapy is enhanced by Nrf2 silencing. PLoS ONE 2014, 9, e107158. [Google Scholar] [CrossRef] [Green Version]
- Tang, P.M.-K.; Liu, X.-Z.; Zhang, D.-M.; Fong, W.-P.; Fung, K.-P. Pheophorbide a based photodynamic therapy induces apoptosis via mitochondrial-mediated pathway in human uterine carcinosarcoma. Cancer Biol. Ther. 2009, 8, 533–539. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Formulation | IC50 (μM) | DRI | ||||
|---|---|---|---|---|---|---|
| MDA-MB-231 | SK-OV-3 | MDA-MB-231 | SK-OV-3 | |||
| PheoA | PTX | PheoA | PTX | |||
| mPTX2S | 0.15 | 5.72 | - | - | - | - |
| PTX | 0.028 | 0.053 | - | - | - | - |
| PheoA | 0.35 | 0.26 | - | - | - | - |
| PheoA + PTX | 0.12 | 0.12 | 9.06 | 0.32 | 6.88 | 0.63 |
| PheoA≅PTX2S | 0.21 | 0.26 | 5.30 | 1.02 | 3.19 | 31.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moret, F.; Menilli, L.; Battan, M.; Tedesco, D.; Columbaro, M.; Guerrini, A.; Avancini, G.; Ferroni, C.; Varchi, G. Pheophorbide A and Paclitaxel Bioresponsive Nanoparticles as Double-Punch Platform for Cancer Therapy. Pharmaceutics 2021, 13, 1130. https://doi.org/10.3390/pharmaceutics13081130
Moret F, Menilli L, Battan M, Tedesco D, Columbaro M, Guerrini A, Avancini G, Ferroni C, Varchi G. Pheophorbide A and Paclitaxel Bioresponsive Nanoparticles as Double-Punch Platform for Cancer Therapy. Pharmaceutics. 2021; 13(8):1130. https://doi.org/10.3390/pharmaceutics13081130
Chicago/Turabian StyleMoret, Francesca, Luca Menilli, Manuele Battan, Daniele Tedesco, Marta Columbaro, Andrea Guerrini, Greta Avancini, Claudia Ferroni, and Greta Varchi. 2021. "Pheophorbide A and Paclitaxel Bioresponsive Nanoparticles as Double-Punch Platform for Cancer Therapy" Pharmaceutics 13, no. 8: 1130. https://doi.org/10.3390/pharmaceutics13081130
APA StyleMoret, F., Menilli, L., Battan, M., Tedesco, D., Columbaro, M., Guerrini, A., Avancini, G., Ferroni, C., & Varchi, G. (2021). Pheophorbide A and Paclitaxel Bioresponsive Nanoparticles as Double-Punch Platform for Cancer Therapy. Pharmaceutics, 13(8), 1130. https://doi.org/10.3390/pharmaceutics13081130