
Gene Expression and Protein Abundance of Hepatic Drug Metabolizing Enzymes in Liver Pathology

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Liver Samples
2.2. mRNA Isolation and Quantitative Real-Time RT-PCR
2.3. Protein Quantification by LC−MC/MS
2.4. Genotyping
2.5. Statistical Analysis
3. Results
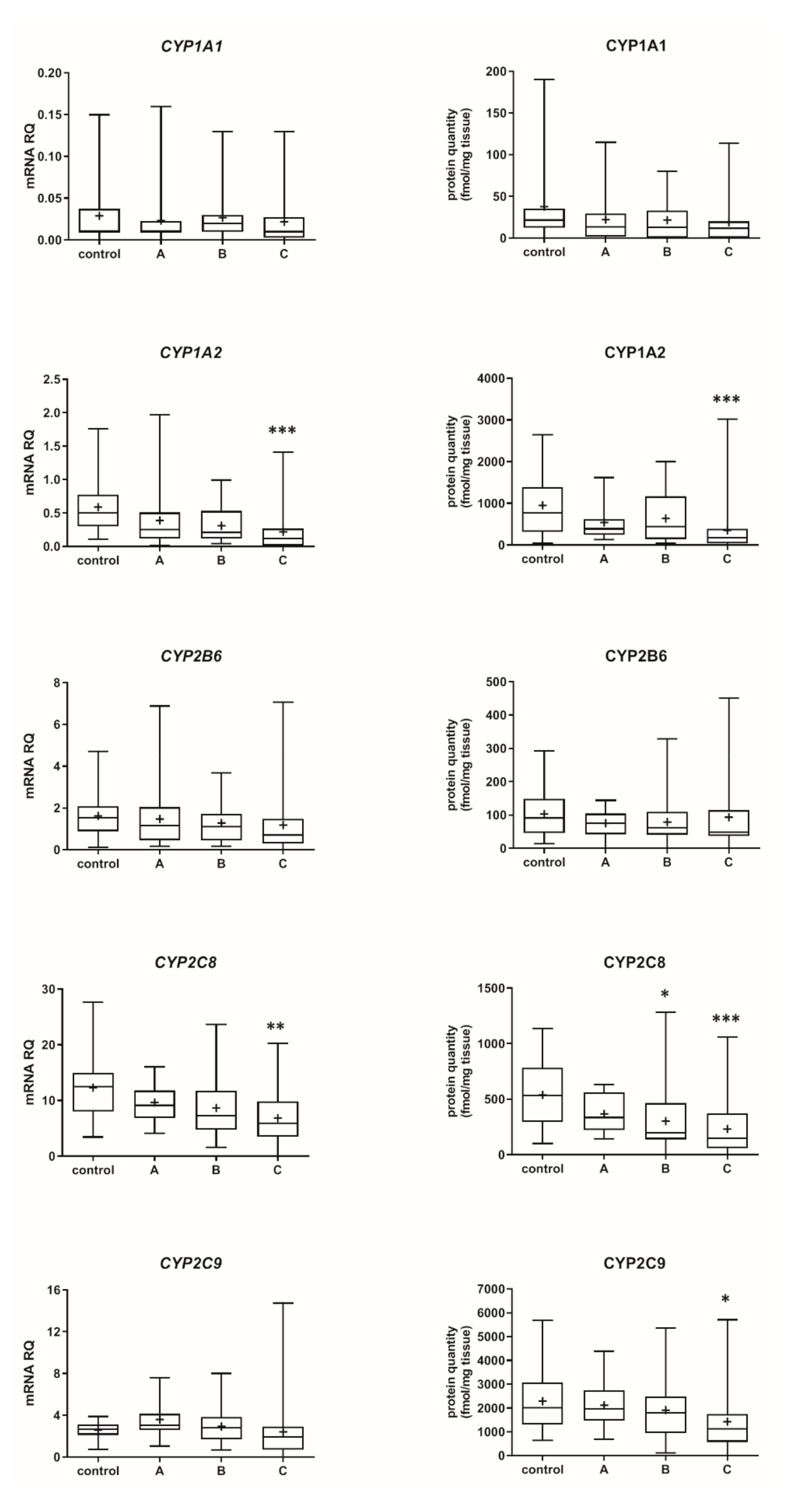
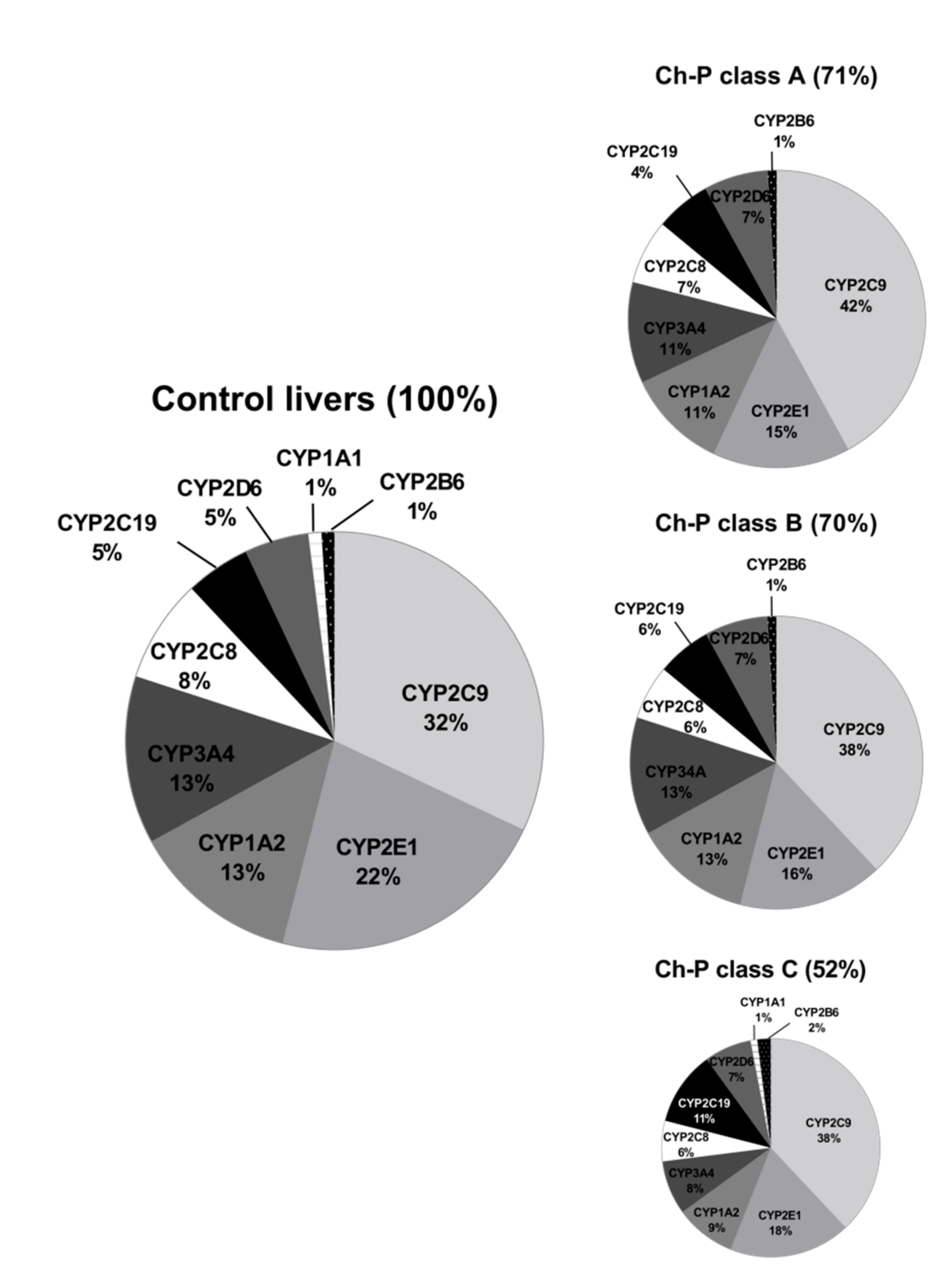
3.1. Hepatic Drug Metabolizing Enzymes Abundance According to Liver Functional State (The Child–Pugh Score)
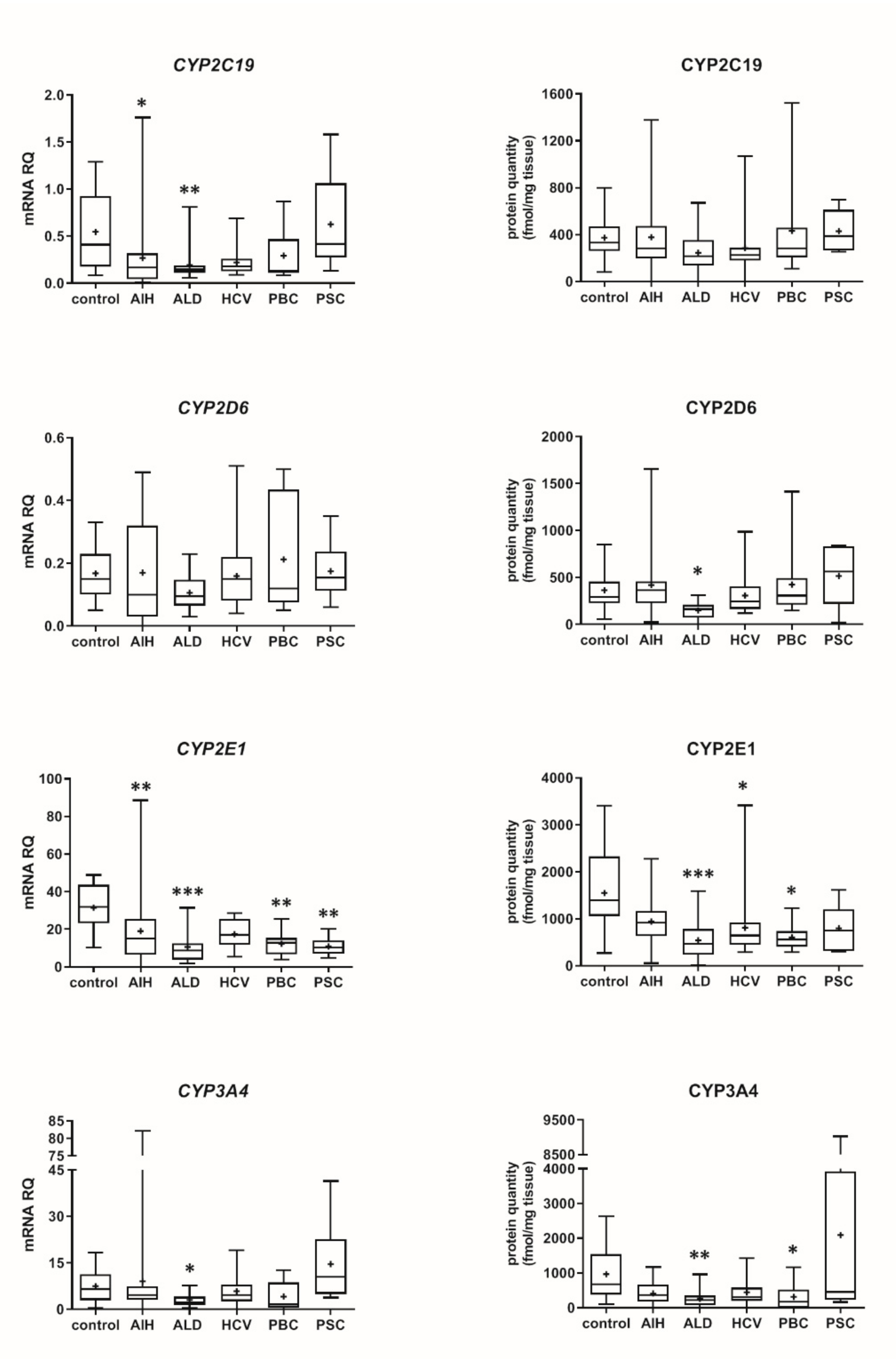
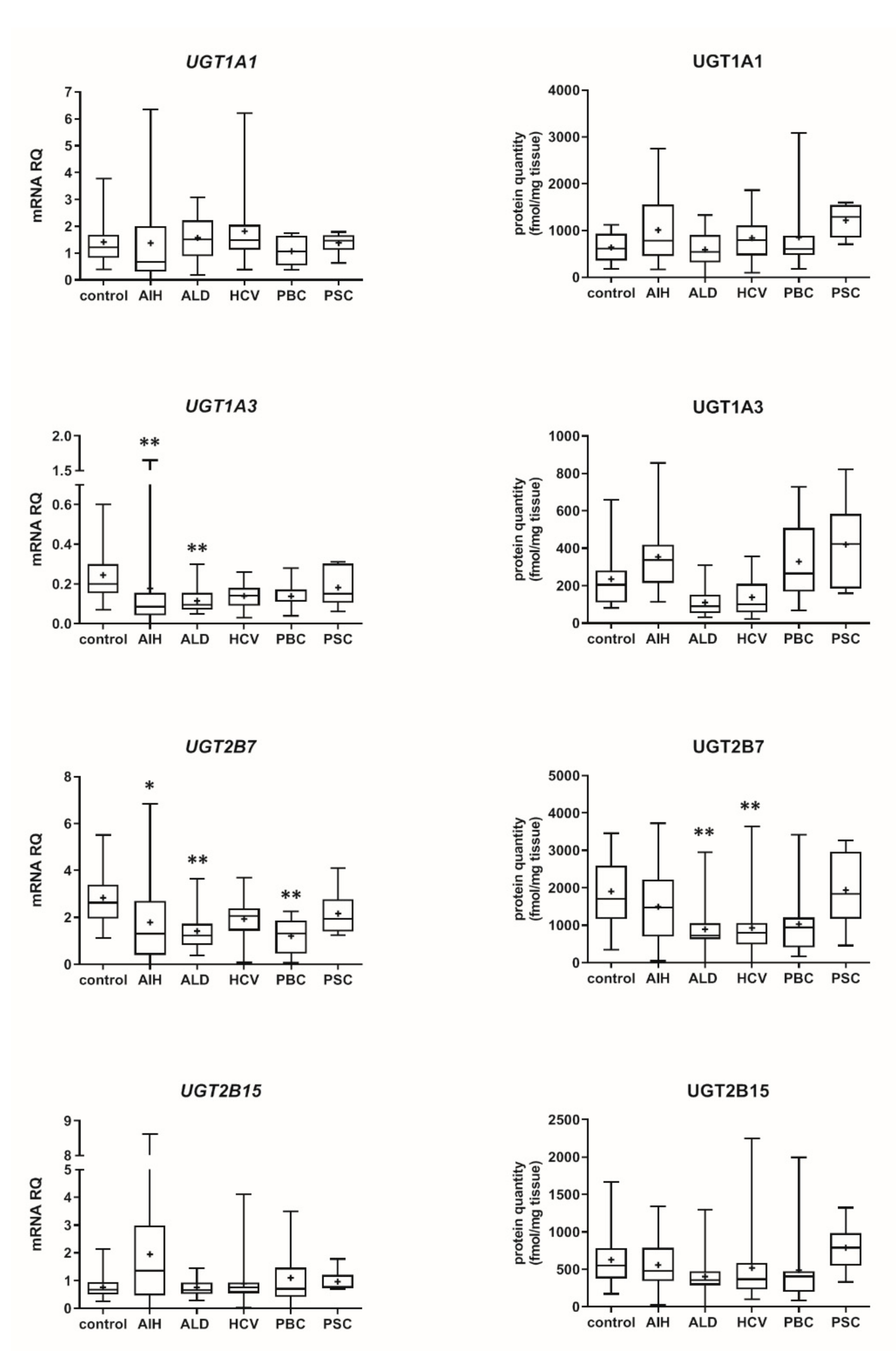
3.2. Hepatic Drug Metabolizing Enzymes in Different Forms of Liver Disease
3.3. Genotyping
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Frye, R.F.; Zgheib, N.K.; Matzke, G.R.; Chaves-Gnecco, D.; Rabinovitz, M.; Shaikh, O.S.; Branch, R.A. Liver disease selectively modulates cytochrome P450-mediated metabolism. Clin. Pharmacol. Ther. 2006, 80, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Adedoyin, A.; Arns, P.A.; Richards, W.O.; Wilkinson, G.R.; Branch, R.A. Selective effect of liver disease on the activities of specific metabolizing enzymes: Investigation of cytochromes P450 2C19 and 2D6. Clin. Pharmacol. Ther. 1998, 64, 8–17. [Google Scholar] [CrossRef]
- Pentikäinen, P.J.; Välisalmi, L.; Himberg, J.L.; Crevoisier, C. Pharmacokinetics of midazolam following intravenous and oral administration in patients with chronic liver disease and in healthy subjects. J. Clin. Pharmacol. 1989, 29, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Edginton, A.N.; Willmann, S. Physiology-based simulations of a pathological condition: Prediction of pharmacokinetics in patients with liver cirrhosis. Clin. Pharmacokinet. 2008, 47, 743–752. [Google Scholar] [CrossRef]
- Drozdzik, M.; Szelag-Pieniek, S.; Post, M.; Zeair, S.; Wrzesinski, M.; Kurzawski, M.; Prieto, J.; Oswald, S. Protein abundance of hepatic drug transporters in patients with different forms of liver damage. Clin. Pharmacol. Ther. 2020, 107, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Drozdzik, M.; Oswald, S.; Droździk, A. Extrahepatic drug transporters in liver failure: Focus on kidney and gastrointestinal tract. Int. J. Mol. Sci. 2020, 21, 5737. [Google Scholar] [CrossRef]
- Franz, C.C.; Hildbrand, C.; Born, C.; Egger, S.; Rätz, B.; Krähenbühl, S. Dose adjustment in patients with liver cirrhosis: Impact on adverse drug reactions and hospitalizations. Eur. J. Clin. Pharmacol. 2013, 69, 1565–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Medicines Agency 2005. Guideline on the Evaluation of the Pharmacokinetics of Medicinal Products in Patients with Impaired Hepatic Function. February 2005. Available online: https://www.ema.europa.eu/documents/scientific-guideline/guideline-evaluation-pharmacokinetics-medicinal-products-patients-impaired-hepatic-function_en.pdf (accessed on 14 March 2021).
- FDA. Guidance for Industry Pharmacokinetics in Patients with Impaired Hepatic Function: Study, Design, Data Analysis, and Impact on Dosing and Labeling. 2003. Available online: https://www.fda.gov/media/71311/download (accessed on 14 March 2021).
- Villeneuve, J.P.; Pichette, V. Cytochrome P450 and Liver Diseases. Curr. Drug Metab. 2004, 5, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.; Bhatt, D.K.; Johnson, K.; Chapa, R.; Chu, X.; Salphati, L.; Xiao, G.; Lee, C.; Hop, C.; Mathias, A.; et al. Abundance of phase 1 and 2 drug-metabolizing enzymes in alcoholic and hepatitis C cirrhotic livers: A quantitative targeted proteomics study. Drug Metab. Dispos. 2018, 46, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P.; Turvy, C.G. Comparison of levels of several human microsomal cytochrome P-450 enzymes and epoxide hydrolase in normal and disease states using immunochemical analysis of surgical liver samples. J. Pharmacol. Exp. Ther. 1991, 256, 1189–1194. [Google Scholar] [PubMed]
- George, J.; Murray, M.; Byth, K.; Farrell, G.C. Differential alterations of cytochrome P450 proteins in livers from patients with severe chronic liver disease. Hepatology 1995, 21, 120–128. [Google Scholar] [PubMed]
- Oswald, S.; Gröer, C.; Drozdzik, M.; Siegmund, W. Mass spectrometry-based targeted proteomics as a tool to elucidate the expression and function of intestinal drug transporters. AAPS J. 2013, 15, 1128–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drozdzik, M.; Busch, D.; Lapczuk, J.; Müller, J.; Ostrowski, M.; Kurzawski, M.; Oswald, S. Protein abundance of clinically relevant drug-metabolizing enzymes in the human liver and intestine: A comparative analysis in paired tissue specimens. Clin. Pharmacol. Ther. 2018, 104, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Gröer, C.; Busch, D.; Patrzyk, M.; Beyer, K.; Busemann, A.; Heidecke, C.D.; Drozdzik, M.; Siegmund, W.; Oswald, S. Absolute protein quantification of clinically relevant cytochrome P450 enzymes and UDP-glucuronosyltransferases by mass spectrometry-based targeted proteomics. J. Pharm. Biomed. Anal. 2014, 100, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.; Boussery, K.; Rowland-Yeo, K.; Tucker, G.; Rostami-Hodjegan, A. A semi-mechanistic model to predict the effects of liver cirrhosis on drug clearance. Clin. Pharmacokinet. 2010, 49, 189–206. [Google Scholar] [CrossRef] [PubMed]
- Lamba, J.; Hebert, J.M.; Schuetz, E.G.; Klein, T.E.; Altman, R.B. PharmGKB summary: Very important pharmacogene information for CYP3A5. Pharm. Genom. 2012, 22, 555–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Congiu, M.; Mashford, M.L.; Slavin, J.L.; Desmond, P.V. UDP glucuronosyltransferase mRNA levels in human liver disease. Drug Metab. Dispos. 2002, 30, 129–134. [Google Scholar] [CrossRef]
- Kamath, P.S.; Wiesner, R.H.; Malinchoc, M.; Kremers, W.; Therneau, T.M.; Kosberg, C.L.; D’Amico, G.; Dickson, E.R.; Kim, W.R. A model to predict survival in patients with end-stage liver disease. Hepatology 2001, 33, 464–470. [Google Scholar] [CrossRef]
- Peng, Y.; Qi, X.; Guo, X. Child-Pugh versus MELD score for the assessment of prognosis in liver cirrhosis: A systematic review and meta-analysis of observational studies. Medicine 2016, 95, e2877. [Google Scholar] [CrossRef]
- Talal, A.H.; Venuto, C.S.; Younis, I. Assessment of Hepatic Impairment and Implications for pharmacokinetics of Substance Use Treatment. Clin. Pharmacol. Drug Dev. 2017, 6, 206–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bock, L.; Boussery, K.; van Winckel, M.; de Paepe, P.; Rogiers, X.; Stephenne, X.; Sokal, E.; van Bocxlaer, J. In vitro cytochrome p450 activity decreases in children with high pediatric end-stage liver disease scores. Drug Metab. Dispos. 2013, 41, 390–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtsuki, S.; Schaefer, O.; Kawakami, H.; Inoue, T.; Liehner, S.; Saito, A.; Ishiguro, N.; Kishimoto, W.; Ludwig-Schwellinger, E.; Ebner, T.; et al. Simultaneous absolute protein quantification of transporters, cytochromes P450, and UDP-glucuronosyltransferases as a novel approach for the characterization of individual human liver: Comparison with mRNA levels and activities. Drug Metab. Dispos. 2012, 40, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clermont, V.; Grangeon, A.; Barama, A.; Turgeon, J.; Lallier, M.; Malaise, J.; Michaud, V. Activity and mRNA expression levels of selected cytochromes P450 in various sections of the human small intestine. Br. J. Clin. Pharmacol. 2019, 85, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Achour, B.; Russell, M.R.; Barber, J.; Rostami-Hodjegan, A. Simultaneous quantification of the abundance of several cytochrome P450 and uridine 5-diphospho-glucuronosyltransferase enzymes in human liver microsomes using multiplexed targeted proteomics. Drug Metab. Dispos. 2014, 42, 500–510. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Recent advances in understanding liver fibrosis: Bridging basic science and individualized treatment concepts. F1000Research 2018, 7, 921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virgolini, I.; Müller, C.; Angelberger, P.; Höbart, J.; Bergmann, H.; Sinzinger, H. Functional liver imaging with 99Tcmgalactosylneoglycoalbumin (NGA) in alcoholic liver cirrhosis and liver fibrosis. Nucl. Med. Commun. 1991, 12, 507–517. [Google Scholar] [CrossRef]
- Kawakami, H.; Ohtsuki, S.; Kamiie, J.; Suzuki, T.; Abe, T.; Terasaki, T. Simultaneous absolute quantification of 11 cytochrome P450 isoforms in human liver microsomes by liquid chromatography tandem mass spectrometry with in silico target peptide selection. J. Pharm. Sci. 2011, 100, 341–352. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mRNA vs. Protein Correlation Coefficient | ||||||
|---|---|---|---|---|---|---|
| Protein | Control n = 20 a | AIH n = 20 b | ALD n = 20 | HCV n = 21 c | PBC n = 10 d | PSC n = 6 |
| CYP1A1 | 0.664 *** | 0.722 *** | 0.443 | 0.736 *** | 0.225 | −0.393 |
| CYP1A2 | 0.824 *** | 0.857 *** | 0.595 ** | 0.766 *** | 0.867 ** | 1.000 ** |
| CYP2B6 | 0.612 ** | 0.573 ** | −0.036 | 0.394 | 0.900 *** | −0.257 |
| CYP2C8 | 0.645 ** | 0.588 ** | 0.140 | 0.182 | 0.842 ** | −0.086 |
| CYP2C9 | 0.620 ** | 0.686 *** | 0.498 * | 0.347 | 0.867 ** | 0.829 * |
| CYP2C19 | 0.325 | −0.038 | −0.368 | 0.099 | 0.224 | 0.029 |
| CYP2D6 | 0.586 ** | 0.614 ** | 0.105 | 0.470 * | 0.600 | 0.086 |
| CYP2E1 | 0.352 | 0.580 ** | 0.499 * | 0.355 | 0.479 | 0.371 |
| CYP3A4 | 0.889 *** | 0.722 *** | 0.714 *** | 0.712 *** | 0.890 *** | 0.543 |
| UGT1A1 | 0.675 ** | 0.684 *** | 0.383 | 0.504 * | 0.927 *** | 0.143 |
| UGT1A3 | 0.699 *** | 0.453 * | −0.341 | 0.451 * | 0.612 | 0.771 |
| UGT2B15 | 0.800 *** | 0.453 * | 0.229 | 0.479 * | 0.467 | 0.371 |
| UGT2B7 | 0.725 *** | 0.740 *** | 0.505 * | 0.264 | 0.782 ** | 0.829 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drozdzik, M.; Lapczuk-Romanska, J.; Wenzel, C.; Szelag-Pieniek, S.; Post, M.; Skalski, Ł.; Kurzawski, M.; Oswald, S. Gene Expression and Protein Abundance of Hepatic Drug Metabolizing Enzymes in Liver Pathology. Pharmaceutics 2021, 13, 1334. https://doi.org/10.3390/pharmaceutics13091334
Drozdzik M, Lapczuk-Romanska J, Wenzel C, Szelag-Pieniek S, Post M, Skalski Ł, Kurzawski M, Oswald S. Gene Expression and Protein Abundance of Hepatic Drug Metabolizing Enzymes in Liver Pathology. Pharmaceutics. 2021; 13(9):1334. https://doi.org/10.3390/pharmaceutics13091334
Chicago/Turabian StyleDrozdzik, Marek, Joanna Lapczuk-Romanska, Christoph Wenzel, Sylwia Szelag-Pieniek, Mariola Post, Łukasz Skalski, Mateusz Kurzawski, and Stefan Oswald. 2021. "Gene Expression and Protein Abundance of Hepatic Drug Metabolizing Enzymes in Liver Pathology" Pharmaceutics 13, no. 9: 1334. https://doi.org/10.3390/pharmaceutics13091334