
Pharma 4.0 Continuous mRNA Drug Products Manufacturing †

 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
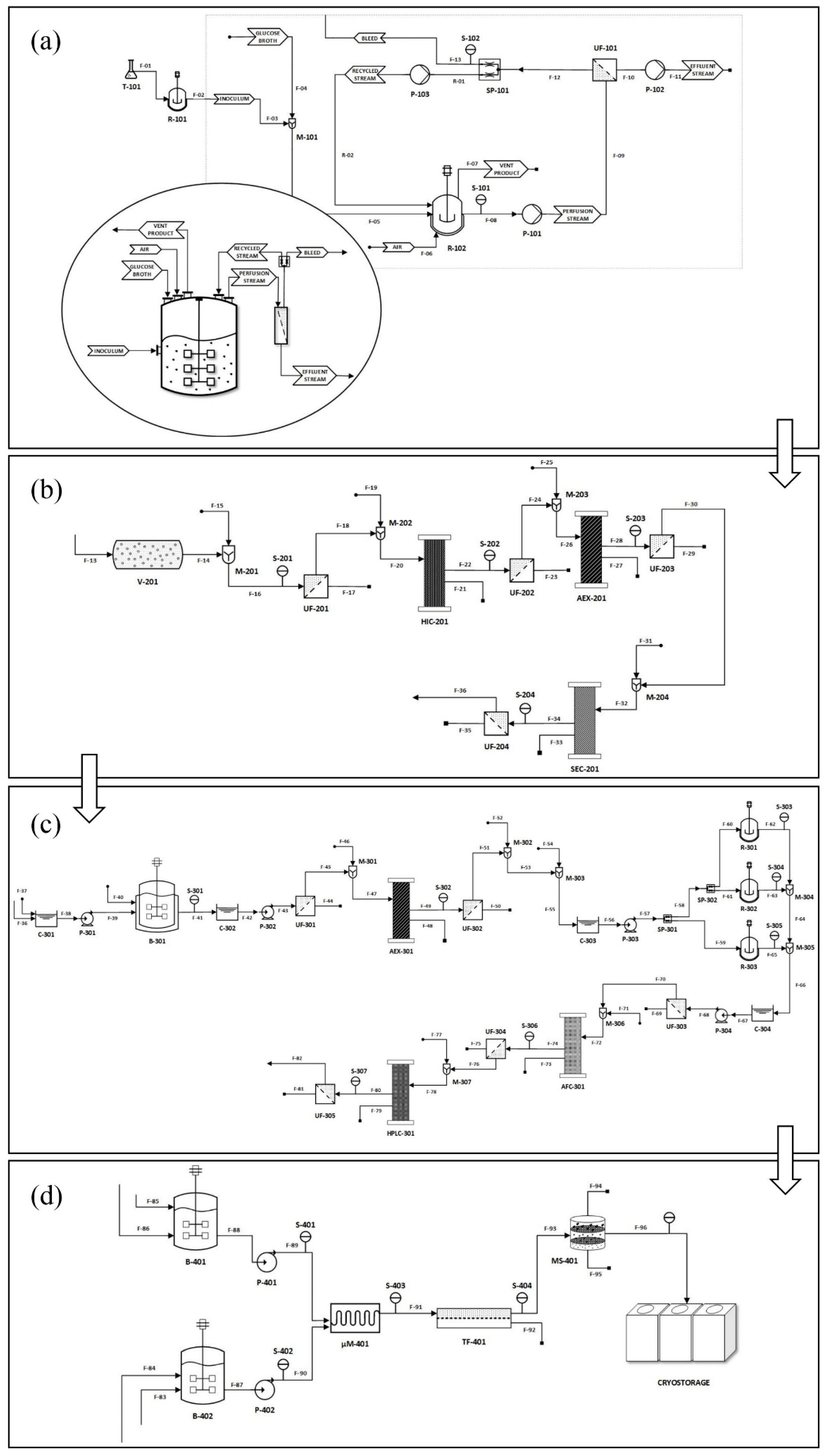
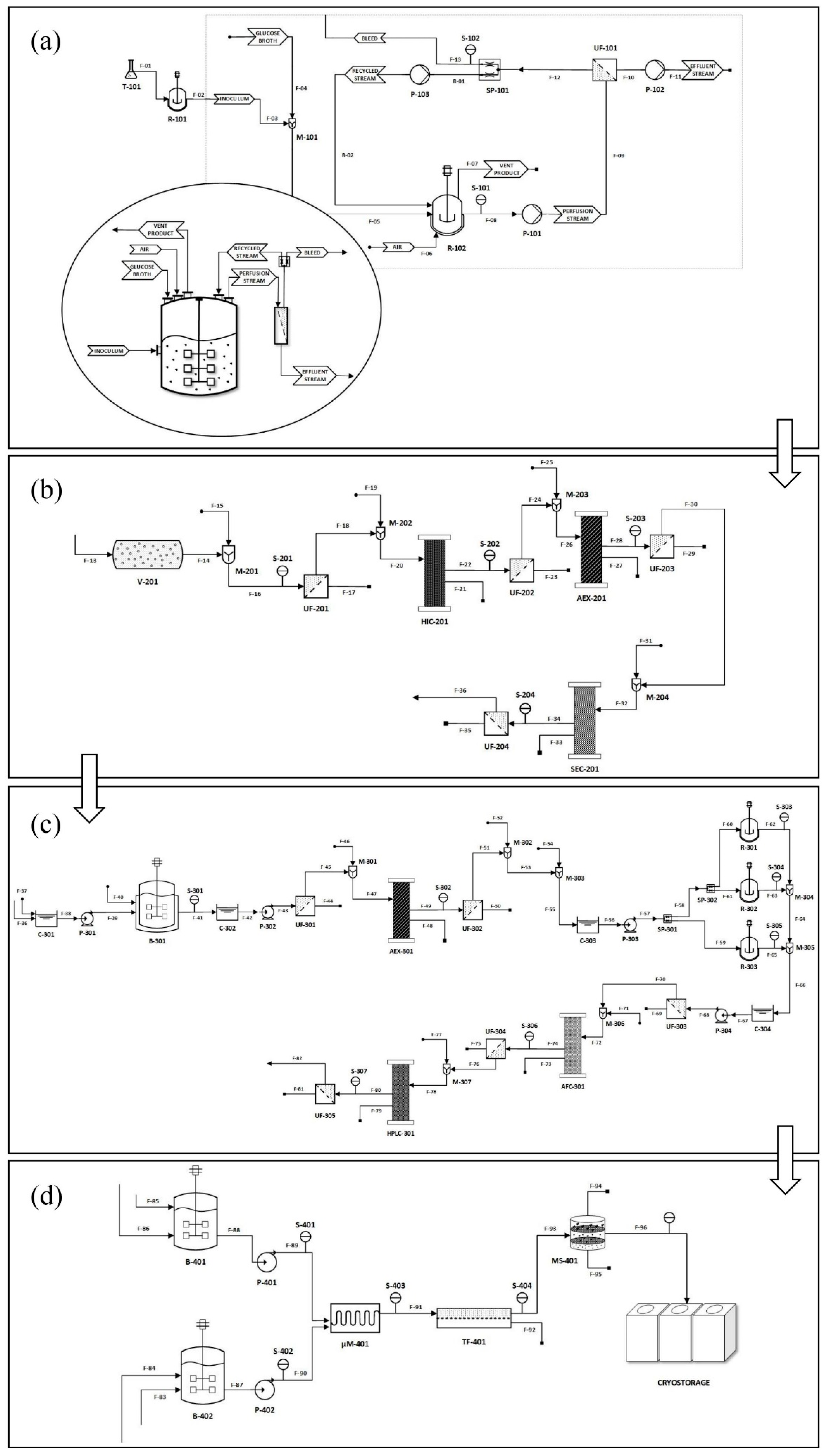
2.1. Comprehensive Overview of the Sheet Workflow and Methods Design Strategy
2.2. Inoculation, Upstream Production, and Plasmid Augmentation
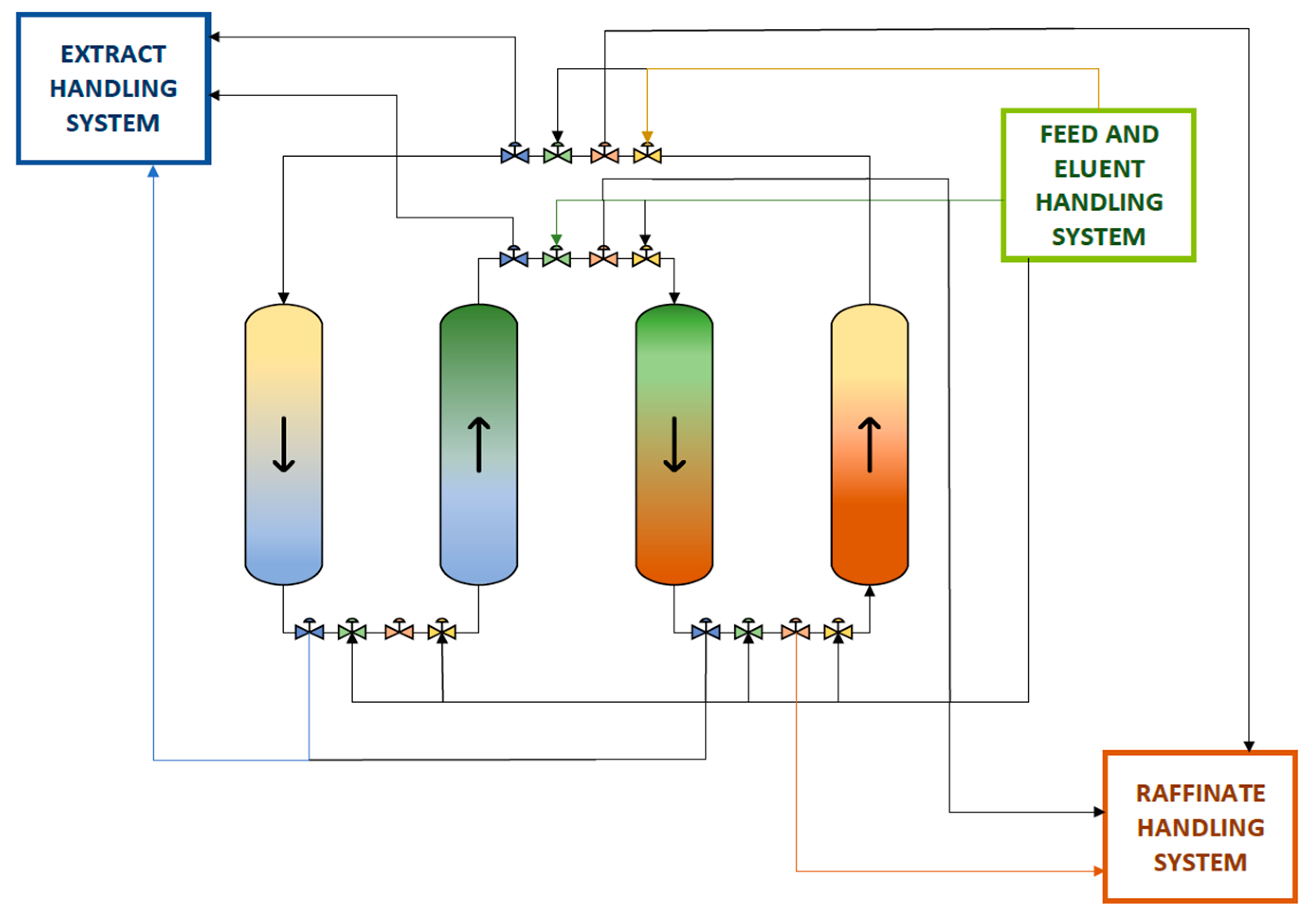
2.3. Alkaline Cell Lysis, Plasmid Purification, and DNA Linearization
2.4. Design of IVT Reactor and Purification of mRNA
2.5. mRNA Lipid Nanoparticle (LNP) Formulation Design
2.6. Clean Room Design
2.7. Resource Analysis Method, Total Capital Investment, and Production Cost
2.7.1. Minimum Selling Price of Dose (MSPD) Estimation
2.7.2. Uncertainty Analysis
3. Results
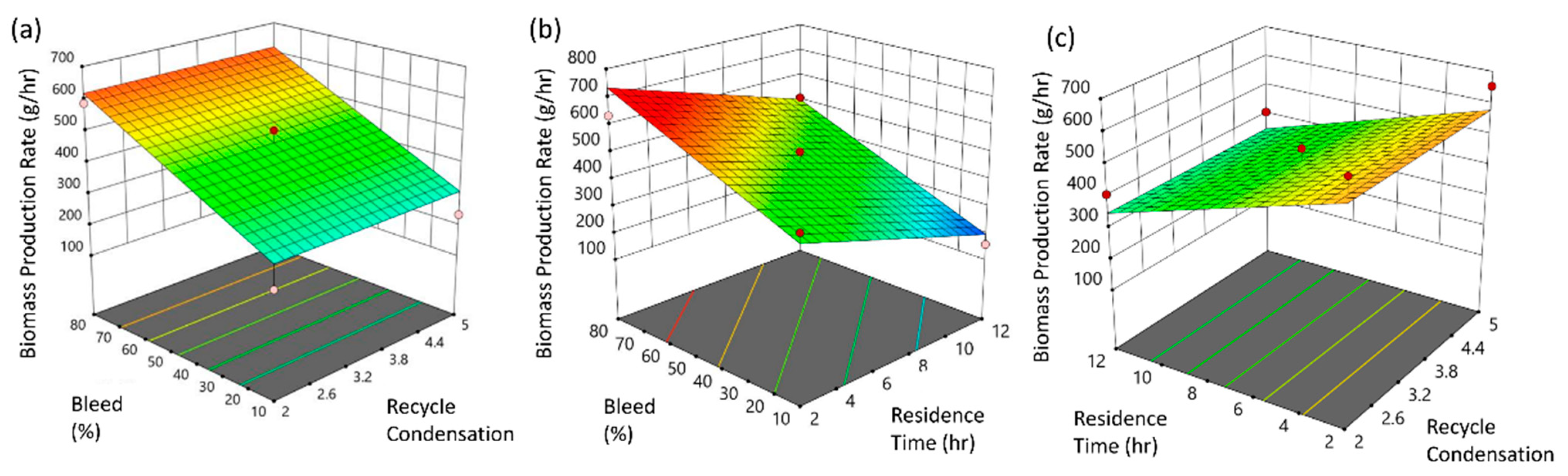
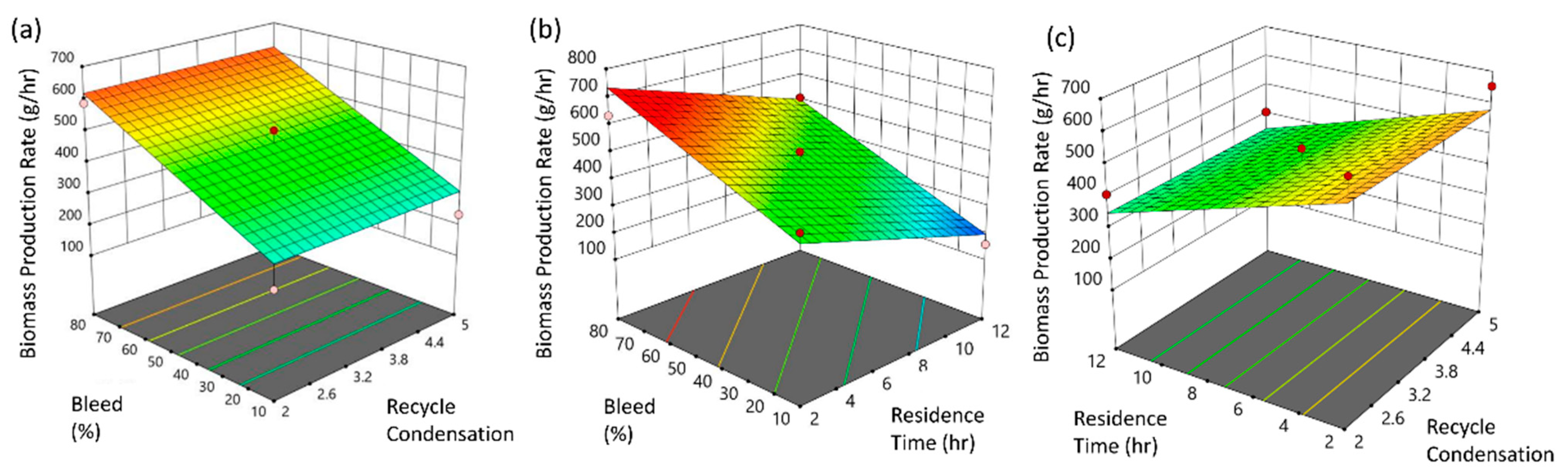
3.1. Upstream Perfusion Reactor Design
3.2. Plasmid Deliverance by Alkaline lysis, Purification, and Linearization
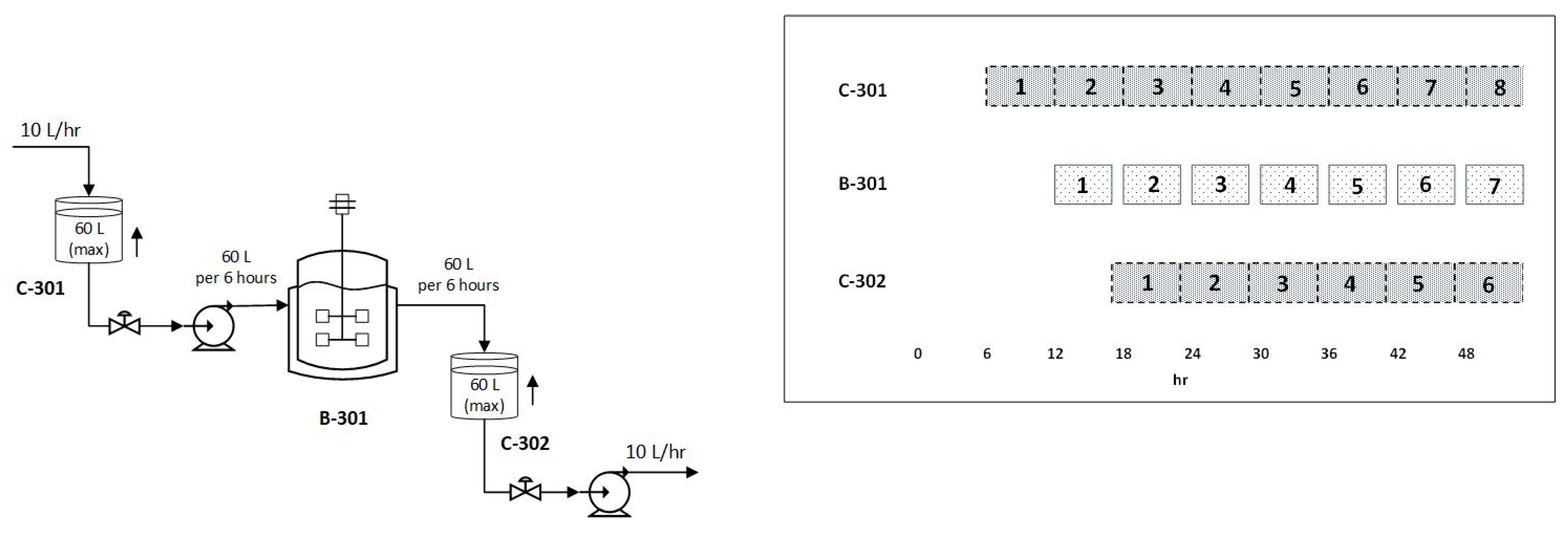
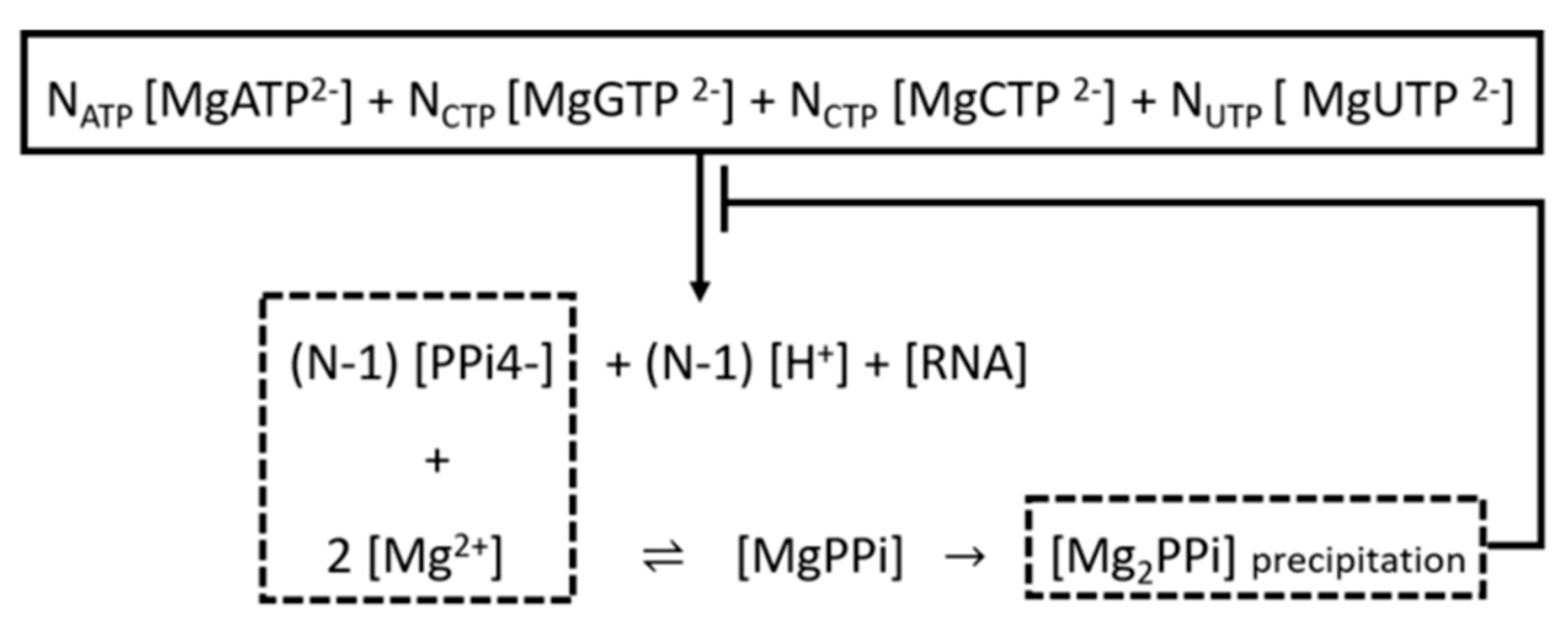
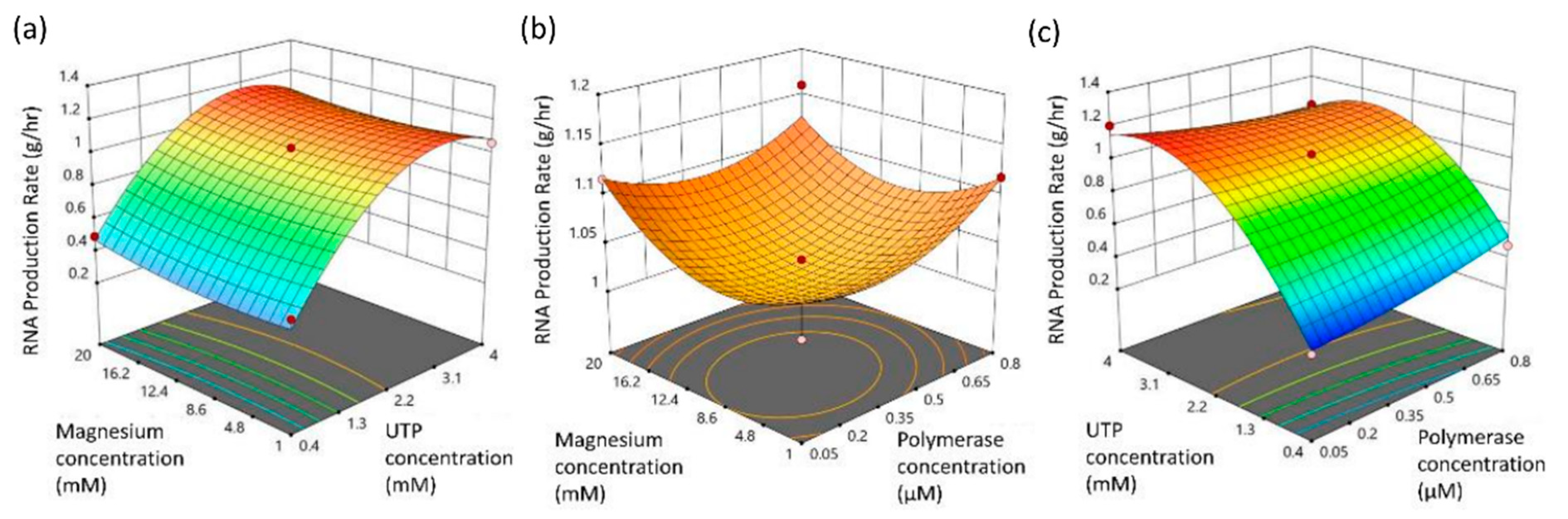
3.3. Purification of the Linearized DNA Template and IVT
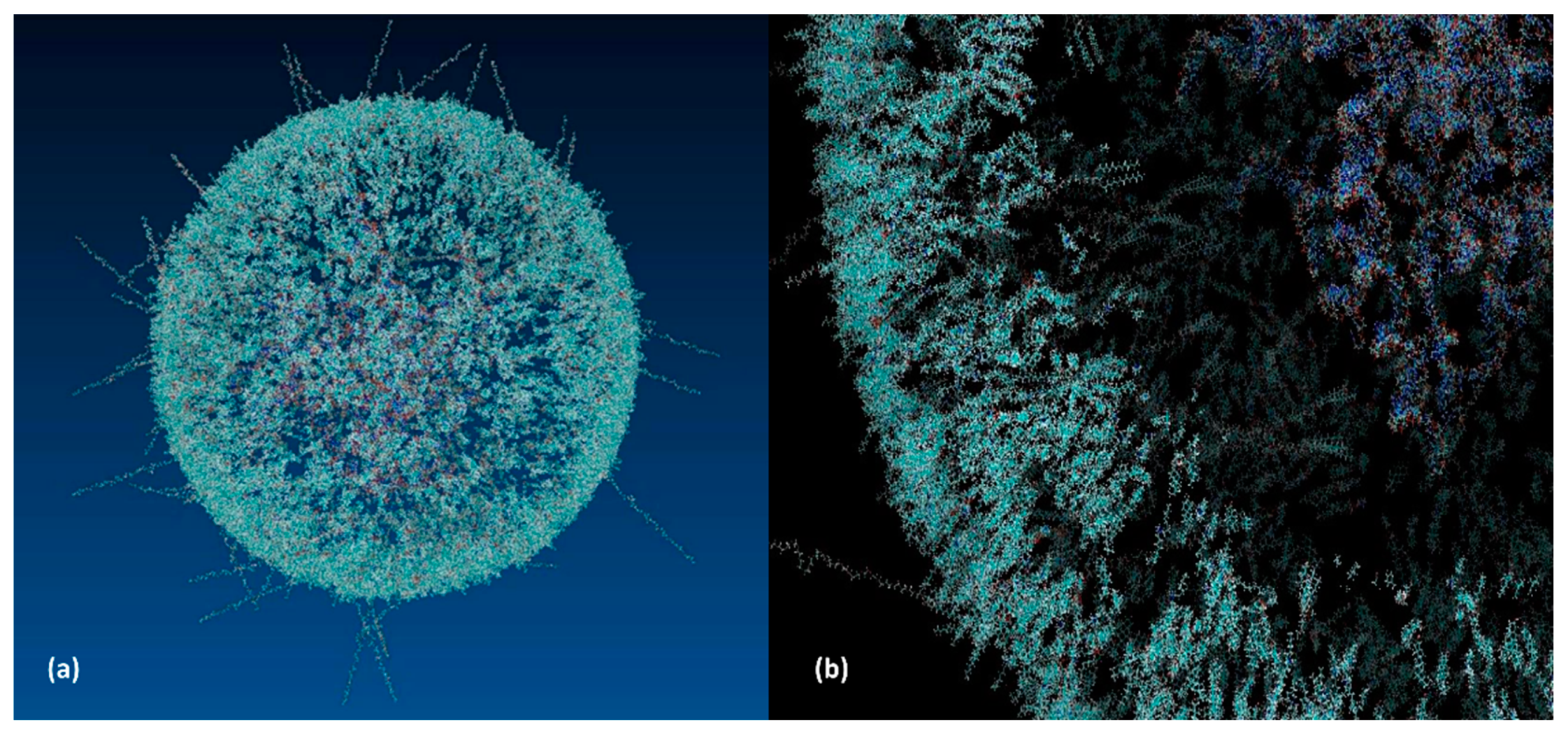
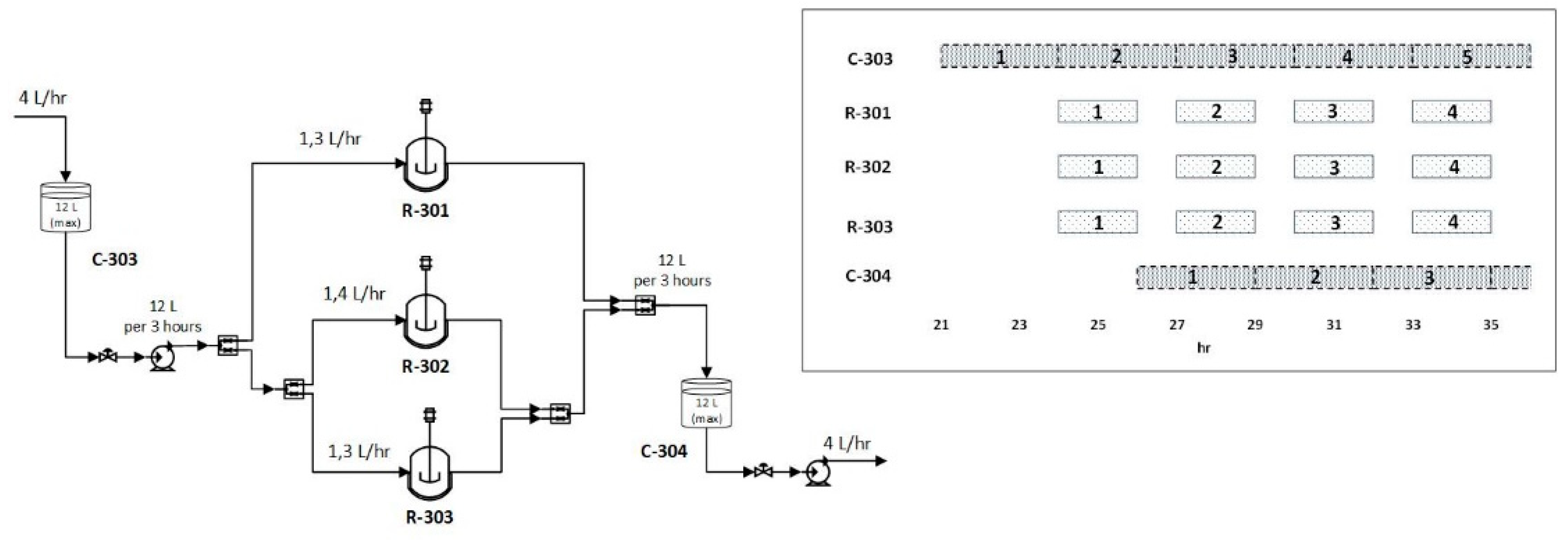
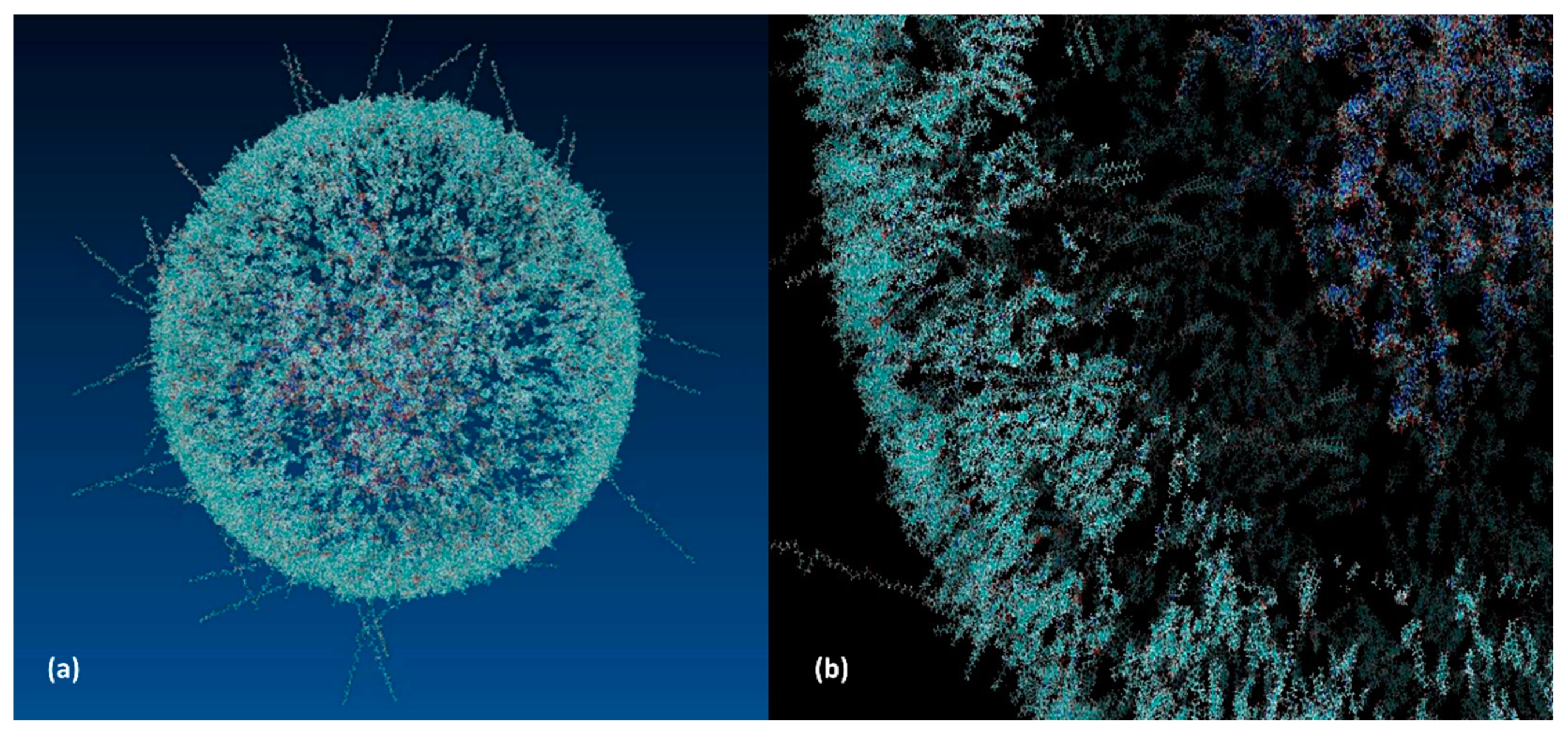
3.4. LNP Formulation and Self-Assembly
3.5. Clean Room Related Costs
3.6. Resource Commitment Analysis
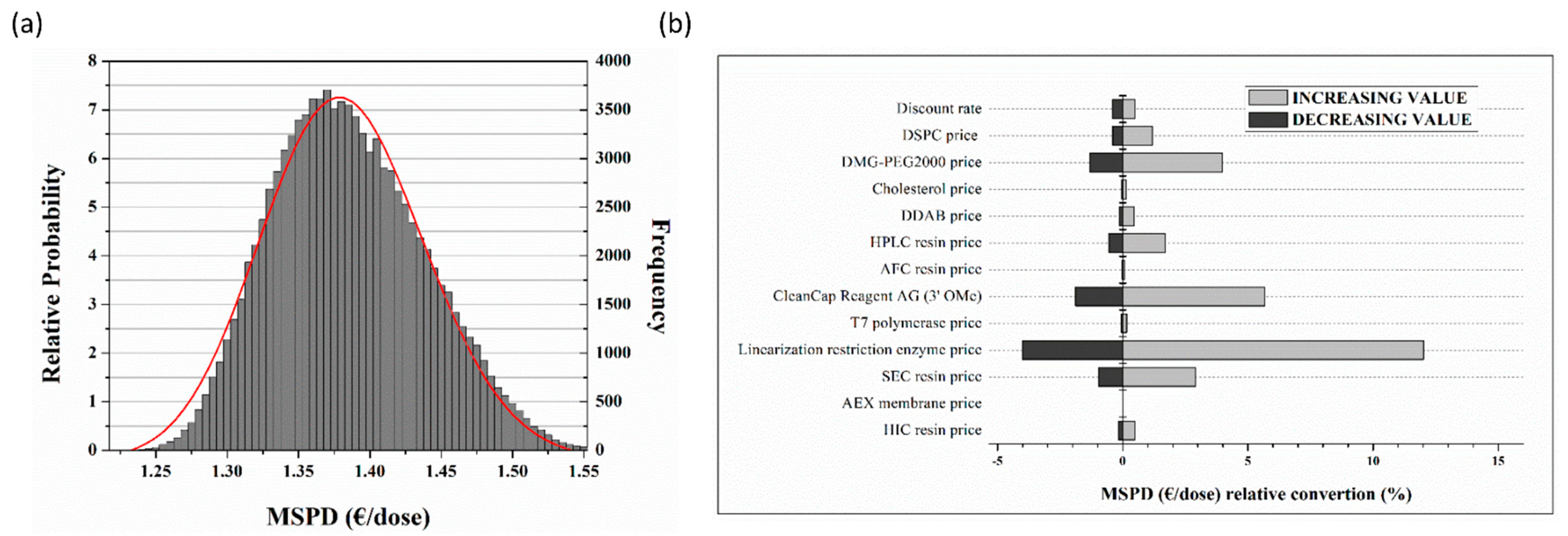
3.7. Uncertainty Analysis
4. Discussion
5. Conclusions
- (a)
- We demonstrated the feasibility of continuous end-to-end GMP compliant, mRNA LNP formulated vaccine technology adoption, launching from bacterial culture. Innovations such as perfusion reactors, SMB chromatographic steps, continuous cell lysis modules, and microfluidic formulation were introduced to approach the task. This paves the way for the future actual, modular process line development implementation, under strict quality directives.
- (b)
- The material-driven flowsheet converging mass and energy balances combined or leveraged by bibliographic, empirical, and experimental elements allowed us to link the operational design space framework to the critical quality attributes, hereafter offering a comprehensive mapping of the whole process, accessible for further experimental optimizations. Regarding the processes and the representative material system studied, bleed fraction, residence time, and UTP concentration were identified as the critical parameters for the component blocks of the perfusion bioreactor and the IVT reactors, respectively.
- (c)
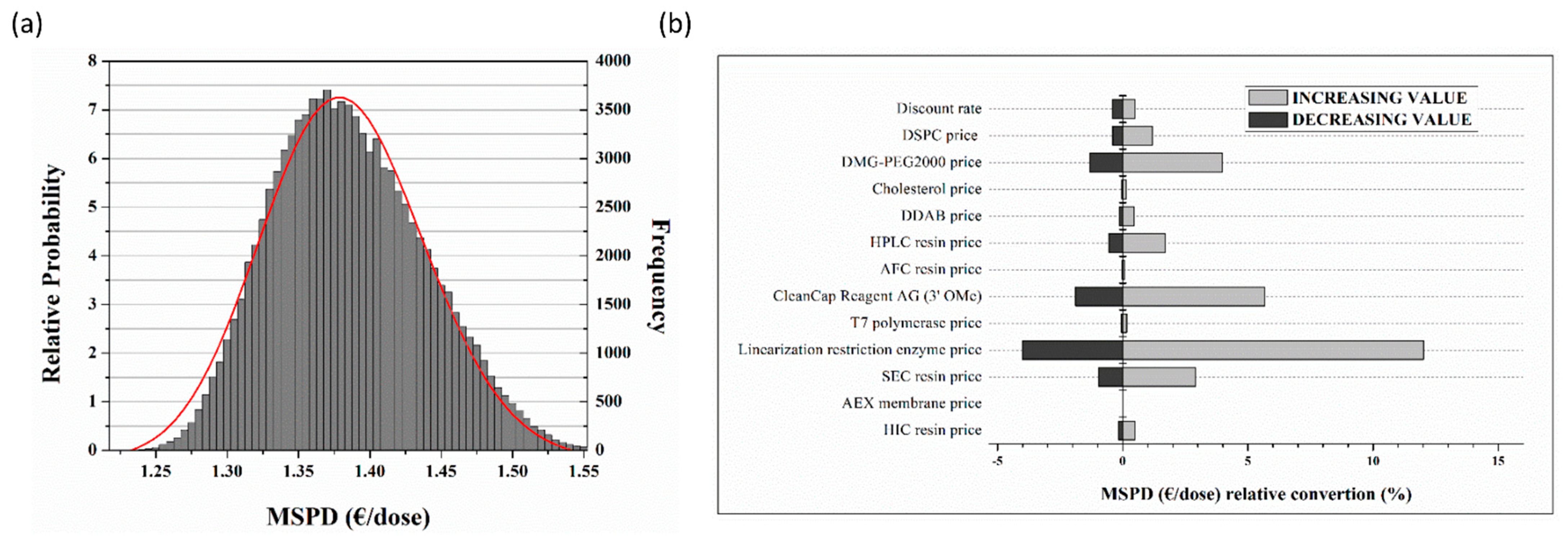
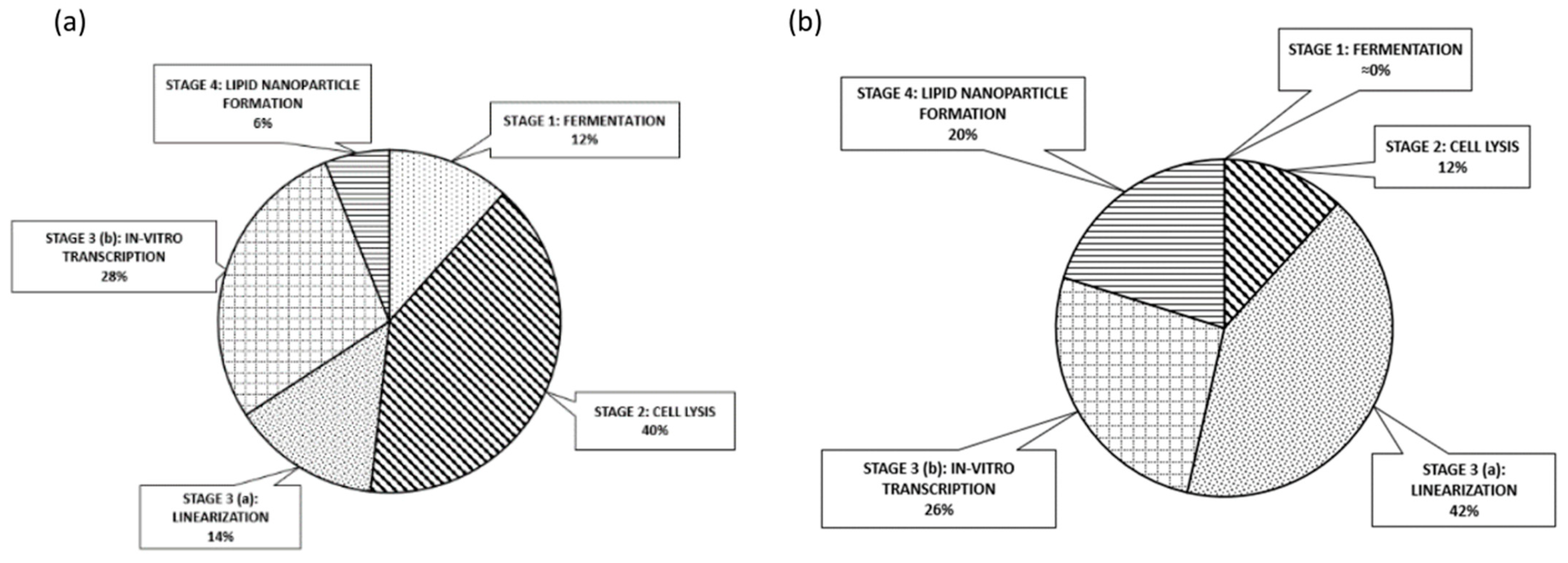
- This digital approach furthered allowed us to safely assess the economic aspects of the venture, documenting certain remarkable findings. The MSPD price correlated to single dose regime is expected to range most probably between 1.30 to 1.45 Euros. Indeed, the continuous production of mRNA vaccines overall cost conveys marginally the same price, when compared to batch, being attributed to the limited, applied scale-up grade. MSPD featured reserved dispersion and therefore limited dependence borne by fluctuations. Finally, cell lysis devices including their related purification modules and linearization enzymes ascend as the principal cost factors accounting for 40% and 42% of the equipment and raw material, respectively.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct Gene Transfer into Mouse Muscle in Vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef]
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics—Developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Rissanou, A.N.; Ouranidis, A.; Karatasos, K. Complexation of single stranded RNA with an ionizable lipid: An all-atom molecular dynamics simulation study. Soft Matter 2020, 16, 6993–7005. [Google Scholar] [CrossRef]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of Pseudouridine Into mRNA Yields Superior Nonimmunogenic Vector with Increased Translational Capacity and Biological Stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef]
- Baiersdörfer, M.; Boros, G.; Muramatsu, H.; Mahiny, A.; Vlatkovic, I.; Sahin, U.; Karikó, K. A Facile Method for the Removal of dsRNA Contaminant from In Vitro-Transcribed mRNA. Mol. Ther.—Nucleic Acids 2019, 15, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Weissman, D.; Karikó, K. mRNA: Fulfilling the Promise of Gene Therapy. Mol. Ther. 2015, 23, 1416–1417. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Udugama, I.A.; Gargalo, C.L.; Gernaey, K.V. Why Is Batch Processing Still Dominating the Biologics Landscape? Towards an Integrated Continuous Bioprocessing Alternative. Processes 2020, 8, 1641. [Google Scholar] [CrossRef]
- Crommelin, D.J.; Anchordoquy, T.J.; Volkin, D.B.; Jiskoot, W.; Mastrobattista, E. Addressing the Cold Reality of mRNA Vaccine Stability. J. Pharm. Sci. 2020, 110, 997–1001. [Google Scholar] [CrossRef]
- Nicoud, R.-M. The Amazing Ability of Continuous Chromatography to Adapt to a Moving Environment. Ind. Eng. Chem. Res. 2014, 53, 3755–3765. [Google Scholar] [CrossRef]
- Rajendran, A.; Paredes, G.; Mazzotti, M. Simulated moving bed chromatography for the separation of enantiomers. J. Chromatogr. A 2009, 1216, 709–738. [Google Scholar] [CrossRef]
- Khairkhah, N.; Aghasadeghi, M.R.; Namvar, A.; Bolhassani, A. Design of novel multiepitope constructs-based peptide vaccine against the structural S, N and M proteins of human COVID-19 using immunoinformatics analysis. PLoS ONE 2020, 15, e0240577. [Google Scholar] [CrossRef]
- Hu, W.-S.; Ozturk, S. (Eds.) Cell Culture Technology for Pharmaceutical and Cell-Based Therapies, 1st ed.; CRC Press: Boca Raton, FL, USA, 2005; Available online: https://www.taylorfrancis.com/books/edit/10.1201/9780849351068/cell-culture-technology-pharmaceutical-cell-based-therapies-sadettin-ozturk-wei-shou-hu (accessed on 27 August 2021).
- Voisard, D.; Meuwly, F.; Ruffieux, P.-A.; Baer, G.; Kadouri, A. Potential of cell retention techniques for large-scale high-density perfusion culture of suspended mammalian cells. Biotechnol. Bioeng. 2003, 82, 751–765. [Google Scholar] [CrossRef]
- Woodside, S.M.; Bowen, B.D.; Piret, J.M. Mammalian cell retention devices for stirred perfusion bioreactors. Cytotechnology 1998, 28, 163–175. [Google Scholar] [CrossRef]
- Urthaler, J.; Buchinger, W.; Necina, R. Improved downstream process for the production of plasmid DNA for gene therapy. Acta Biochim. Pol. 2005, 52, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Urthaler, J.; Ascher, C.; Wöhrer, H.; Necina, R. Automated alkaline lysis for industrial scale cGMP production of pharmaceutical grade plasmid-DNA. J. Biotechnol. 2007, 128, 132–149. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, E.; George, A.; Wolk, B. Improving affinity chromatography resin efficiency using semi-continuous chromatography. J. Chromatogr. A 2012, 1227, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Ziomek, G.; Antos, D.; Tobiska, L.; Seidel-Morgenstern, A. Comparison of possible arrangements of five identical columns in preparative chromatography. J. Chromatogr. A 2006, 1116, 179–188. [Google Scholar] [CrossRef]
- Karikó, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latulippe, D.R.; Zydney, A.L. Size exclusion chromatography of plasmid DNA isoforms. J. Chromatogr. A 2009, 1216, 6295–6302. [Google Scholar] [CrossRef]
- Chan, S.; Titchener-Hooker, N.; Sørensen, E. Optimal Economic Design and Operation of Single- and Multi-column Chromatographic Processes. Biotechnol. Prog. 2008, 24, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Diogo, M.; Ribeiro, S.; Queiroz, J.; Monteiro, G.; Perrin, P.; Tordo, N.; Prazeres, D.M. Scale-up of hydrophobic interaction chromatography for the purification of a DNA vaccine against rabies. Biotechnol. Lett. 2000, 22, 1397–1400. [Google Scholar] [CrossRef]
- Hansen, E.B. Chromatographic Scale-Up on a Volume Basis. Preparative Chromatography for Separation of Proteins; Staby, A., Rathore, A.S., Ahuja, S., Eds.; Wiley Online Library: Hoboken, NJ, USA, 2017. [Google Scholar] [CrossRef]
- Eon-Duval, A.; Burke, G. Purification of pharmaceutical-grade plasmid DNA by anion-exchange chromatography in an RNase-free process. J. Chromatogr. B 2004, 804, 327–335. [Google Scholar] [CrossRef]
- Endres, H.N.; Johnson, J.A.C.; Ross, C.A.; Welp, J.K.; Etzel, M.R. Evaluation of an ion-exchange membrane for the purification of plasmid DNA. Biotechnol. Appl. Biochem. 2003, 37, 259–266. [Google Scholar] [CrossRef]
- Stickel, J.; Fotopoulos, A. Pressure-Flow Relationships for Packed Beds of Compressible Chromatography Media at Laboratory and Production Scale. Biotechnol. Prog. 2001, 17, 744–751. [Google Scholar] [CrossRef]
- Kern, J.A.; Davis, R.H. Application of a Fed-Batch System to Produce RNA by In Vitro Transcription. Biotechnol. Prog. 1999, 15, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Gholamalipour, Y.; Mudiyanselage, A.K.; Martin, C.T. 3′ end additions by T7 RNA polymerase are RNA self-templated, distributive and diverse in character—RNA-Seq analyses. Nucleic Acids Res. 2018, 46, 9253–9263. [Google Scholar] [CrossRef] [Green Version]
- Ray, P.D.; Fry, R.C. Chapter 2—The Cell: The Fundamental Unit in Systems Biology. Syst. Biol. Toxicol. Environ. Health 2015, 11–42. Available online: https://www.sciencedirect.com/science/article/pii/B978012801564300002X?via%3Dihub (accessed on 27 August 2021). [CrossRef]
- Slater, R.J. The purification of poly(a)-containing RNA by affinity chromatography. In Methods in Molecular Biology; Walker, J.M., Ed.; Humana Press: Clifton, NJ, USA, 1984; pp. 117–120. [Google Scholar]
- Columns Life Technologies Oligo(dT) Cellulose Columns Quality Control Data 15939-010. Available online: https://tools.thermofisher.com/content/sfs/manuals/15939010.pdf (accessed on 27 August 2021).
- Roces, C.B.; Lou, G.; Jain, N.; Abraham, S.; Thomas, A.; Halbert, G.W.; Perrie, Y. Manufacturing Considerations for the Development of Lipid Nanoparticles Using Microfluidics. Pharmaceutics 2020, 12, 1095. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. Packmol: A package for building initial configurations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Kinnun, J.J.; Mallikarjunaiah, K.J.; Petrache, H.I.; Brown, M.F. Elastic deformation and area per lipid of membranes: Atomistic view from solid-state deuterium NMR spectroscopy. Biochim. Biophys. Acta 2015, 1848, 246–259. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. Sartorius products. J. Mol. Graph. 1996, 14, 33–38. Available online: https://www.sartorius-stedim-tap.com/tap/products/index.htm (accessed on 27 August 2021). [CrossRef]
- Yang, L.; Gan, C.E. Costing small cleanrooms. Build. Environ. 2007, 42, 743–751. [Google Scholar] [CrossRef]
- Koutinas, A.A.; Yepez, B.; Kopsahelis, N.; Freire, D.M.G.; Castro, A.; Papanikolaou, S.; Kookos, I.K. Techno-economic evaluation of a complete bioprocess for 2,3-butanediol production from renewable resources. Bioresour. Technol. 2015, 204, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Yang, O.; Prabhu, S.; Ierapetritou, M.G. Comparison between Batch and Continuous Monoclonal Antibody Production and Economic Analysis. Ind. Eng. Chem. Res. 2019, 58, 5851–5863. [Google Scholar] [CrossRef]
- Peters, M.S.; Timmerhaus, K.D.; West, R.E. Plant Design and Economics for Chemical Engineers, 5th ed.; McGraw-Hill Education: New York, NY, USA, 2002. [Google Scholar]
- Zobel, S.; Helling, C.; Ditz, R.; Strube, J. Design and Operation of Continuous Countercurrent Chromatography in Biotechnological Production. Ind. Eng. Chem. Res. 2014, 53, 9169–9185. [Google Scholar] [CrossRef]
- Spectrum Laboratories Inc. Conversion of Bioreactors to Continuous Perfusion Using Hollow Fiber Cell Separators. 1989. Available online: http://www.spectrumlabs.com/lit/CPAG.pdf (accessed on 27 August 2021).
- Pörtner, R.; Jandt, U.; Zeng, A.-P. Cell Culture Technology. Ind. Biotechnol. 2016, 1, 129–158. [Google Scholar] [CrossRef]
- Maiorella, B.; Dorin, G.; Carion, A.; Harano, D. Crossflow microfiltration of animal cells. Biotechnol. Bioeng. 1991, 37, 121–126. [Google Scholar] [CrossRef]
- Zydney, A.L.; Colton, C.K. A red cell deformation model for hemolysis in cross flow membrane plasmapheresis. Chem. Eng. Commun. 1984, 30, 191–207. [Google Scholar] [CrossRef]
- Zhang, S.; Handa-Corrigan, A.; Spier, R.E. A comparison of oxygenation methods fro high-density perfusion culture of animal cells. Biotechnol. Bioeng. 1993, 41, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Mercille, S.; Johnson, M.; Massie, B. Filtration-based perfusion of hybridoma cultures in protein-free medium: Reduction of membrane fouling by medium supplementation with DNase I. Biotechnol. Bioeng. 1994, 43, 833–846. [Google Scholar] [CrossRef]
- Van Reis, R.; Leonard, L.C.; Hsu, C.C.; Builder, S.E. Industrial scale harvest of proteins from mammalian cell culture by tangential flow filtration. Biotechnol. Bioeng. 1991, 38, 413–422. [Google Scholar] [CrossRef]
- Vallez-Chetreanu, F. Characterization of the mechanism of action of spin-filters for animal cell perfusion cultures florentina vallez-chetreanu. EPFL 2006, 3488, 188. Available online: https://infoscience.epfl.ch/record/78660 (accessed on 27 August 2021).
- Quinn, H.M. A Reconciliation of Packed Column Permeability Data: Column Permeability as a Function of Particle Porosity. J. Mater. 2014, 2014, 636507. [Google Scholar] [CrossRef]
- Noirclerc-Savoye, M.; Gallet, B.; Bernaudat, F.; Vernet, T. Large scale purification of linear plasmid DNA for efficient high throughput cloning. Biotechnol. J. 2010, 5, 978–985. [Google Scholar] [CrossRef] [Green Version]
- Kis, Z.; Kontoravdi, C.; Shattock, R.; Shah, N. Resources, Production Scales and Time Required for Producing RNA Vaccines for the Global Pandemic Demand. Vaccines 2020, 9, 3. [Google Scholar] [CrossRef]
- Ouranidis, A.; Gkampelis, N.; Markopoulou, C.; Nikolakakis, I.; Kachrimanis, K. Development of a Nanocrystal Formulation of a Low Melting Point API Following a Quality by Design Approach. Processes 2021, 9, 954. [Google Scholar] [CrossRef]
- Freydell, E.J.; van der Wielen, L.; Eppink, M.; Ottens, M. Techno-economic evaluation of an inclusion body solubilization and recombinant protein refolding process. Biotechnol. Prog. 2011, 27, 1315–1328. [Google Scholar] [CrossRef] [PubMed]
- Klutz, S.; Holtmann, L.; Lobedann, M.; Schembecker, G. Cost evaluation of antibody production processes in different operation modes. Chem. Eng. Sci. 2015, 141, 63–74. [Google Scholar] [CrossRef]
- York, N.; Brisbane, C.; Singapore, T.; Ulrich, G.D. A Guide to Chemical Engineering Process Design and Economics; John Wiley & Sons: Hoboken, NJ, USA, 1984; Available online: https://dl.icdst.org/pdfs/files1/09f2516ecf28dd4b294b160fb9527043.pdf (accessed on 27 August 2021).
- Petrides, D.; Koulouris, A.; Petrides, D.P.; Koulouris, A.; Lagonikos, P.T. The role of process simulation in pharmaceutical process development and product commercialization. Pharm. Eng. 2002, 22, 56–65. Available online: https://ispe.org/sites/default/files/attachments/public/Jan-Feb-2002.pdf (accessed on 27 August 2021).
- Kwan, T.H.; Pleissner, D.; Lau, K.Y.; Venus, J.; Pommeret, A.; Lin, C.S.K. Techno-economic analysis of a food waste valorization process via microalgae cultivation and co-production of plasticizer, lactic acid and animal feed from algal biomass and food waste. Bioresour. Technol. 2015, 198, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Baral, N.R.; Kavvada, O.; Mendez-Perez, D.; Mukhopadhyay, A.; Lee, T.S.; Simmons, B.A.; Scown, C.D. Techno-economic analysis and life-cycle greenhouse gas mitigation cost of five routes to bio-jet fuel blendstocks. Energy Environ. Sci. 2019, 12, 807–824. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Liu, J.; Rubacha, M.; Shukla, A.A. A mechanistic study of Protein A chromatography resin lifetime. J. Chromatogr. A 2009, 1216, 5849–5855. [Google Scholar] [CrossRef] [PubMed]
- Nweke, M.C.; Rathore, A.S.; Bracewell, D.G. Lifetime and Aging of Chromatography Resins during Biopharmaceutical Manufacture. Trends Biotechnol. 2018, 36, 992–995. [Google Scholar] [CrossRef]
- Phosphatase, A.; Buffer, R.; Buffer, E.I.R.; Amplification, I.; Pack, B.; Reaction, E.; Pack, B. 2021–2022 Price List. 2021. Available online: https://www.neb.com/-/media/nebus/files/misc/neb_pricelist_2019-20.pdf?rev=59ae00c8c4b54f6984a79d0492736954&hash=AE886A67408677D8B99E4B1D1F646409 (accessed on 27 August 2021).
- Kis, Z.; Kontoravdi, C.; Shattock, R.; Shah, N. Correction: Kis, Z. et al. Resources, Production Scales and Time Required for Producing RNA Vaccines for the Global Pandemic Demand. Vaccines 2021, 9, 205. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Helgers, H.; Vetter, F.; Juckers, A.; Strube, J. Digital Twin of mRNA-Based SARS-COVID-19 Vaccine Manufacturing towards Autonomous Operation for Improvements in Speed, Scale, Robustness, Flexibility and Real-Time Release Testing. Processes 2021, 9, 748. [Google Scholar] [CrossRef]
- Ouranidis, A.; Choli-Papadopoulou, T.; Papachristou, E.T.; Papi, R.; Kostomitsopoulos, N. Biopharmaceutics 4.0, Advanced Pre-Clinical Development of mRNA-Encoded Monoclonal Antibodies to Immunosuppressed Murine Models. Vaccines 2021, 9, 890. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Volume (mL) | Diameter (cm) | Plate Area (cm2) | Length (cm) |
| 1 | 28 | 1.6 | 2 | 14 |
| 2 | 334 | 5 | 20 | 17 |
| Experiment | Volume Feed (mL) | Retention Time (min) | Flow Rate (mL/min) | Linear Velocity (cm/h) |
| 1 | 0.5 | 40 | 0.0125 | 0.4 |
| 2 | 5 | 50 | 0.1 | 0.3 |
| Experiment | Plate Height (cm) | Peak Time (min) | Peak Width (min) | Number of Plates |
| 1 | 0.08 | 7 | 5 | 157 |
| 2 | 0.1 | 7 | 5 | 157 |
| Direct Costs | |
|---|---|
| Purchased equipment | 100 |
| Purchased equipment installation | 39 |
| Instrumentation and controls (installed) | 26 |
| Piping (installed) | 31 |
| Electrical systems (installed) | 10 |
| Buildings (including services) | 29 |
| Yard improvements | 12 |
| Service facilities (installed) | 55 |
| Total direct plant cost | 302 |
| Indirect Costs | |
| Engineering and supervision | 32 |
| Construction expenses | 34 |
| Legal expenses | 4 |
| Contractor’s fee | 19 |
| Contingency | 37 |
| Total indirect plant cost | 126 |
| Total Capital Investment | |
| Fixed-capital investment | 428 |
| Working capital (15% of total capital investment) | 75 |
| Total capital investment | 503 |
| Parameter | Lowest Conversion | Lowest Value | Highest Conversion | Highest Value |
|---|---|---|---|---|
| HIC resin price (€/L) | −10% | 1857 | +30% | 2683 |
| AEX membrane price (€/m2) | −10% | 890 | +30% | 1285 |
| SEC resin price (€/L) | −10% | 9802 | +30% | 14,159 |
| Linearization restriction enzyme price (€/unit) | −10% | 0.00459 | +30% | 0.00663 |
| T7 polymerase price (€/mg) | −10% | 34,200 | +30% | 49,400 |
| CleanCap Reagent (3’ OMe) AG (€/mg) | −10% | 223 | +30% | 322 |
| AFC resin price (€/L) | −10% | 9802 | +30% | 14,159 |
| HPLC resin price (€/L) | −10% | 9802 | +30% | 14,159 |
| Dimethyldioctadecylammonium (DDAB) price (€/g) | −10% | 498,600 | +30% | 720,200 |
| Cholesterol price (€/g) | −10% | 10 | +30% | 14 |
| 1,2-Dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (DMG-PEG2000) price (€/g) | −10% | 149 | +30% | 215 |
| 1,2-dioctadecanoyl-sn-glycero-3-phosphocholine (DSPC) price (€/g) | −10% | 154 | +30% | 223 |
| Discount rate (-) | −40% | 0.06 | +40% | 0.4 |
| Income tax rate (-) | −40% | 0.126 | +40% | 0.294 |
| Source | Sum of Squares | df | Mean Square | F-Value | p-Value | Model A |
|---|---|---|---|---|---|---|
| Biomass Production | 2.95 × 105 | 3 | 98,287.81 | 19.4 | 0.0003 | significant |
| A-Recycling Condensation | 0.0005 | 1 | 0.0005 | 9.84 × 108 | 0.9998 | |
| B-Bleed | 1.89 × 105 | 1 | 1.89 × 105 | 37.38 | 0.0002 | |
| C-Residence time | 1.06 × 105 | 1 | 1.06 × 105 | 20.81 | 0.0014 | |
| Residual | 45,601.9 | 9 | 5066.88 | |||
| Cor Total | 3.41 × 105 | 12 | ||||
| Coded model equation: Biomass Production Rate = 464 − 0.0079 × A + 153.87 × B − 114.82 × C (14) | ||||||
| Chromatography Block | ΔP (bar) | u (cm/s) | L (cm) | kO (-) | η (cP) | ε (-) | dp (μm) | N (-) | Lc (cm) | D (cm) |
|---|---|---|---|---|---|---|---|---|---|---|
| HIC-201 | - | - | - | - | - | - | - | 4 | 42.8 | 21.4 |
| AEX-201 | 0.5 | 0.004 | 9 | 150 | 1 | 0.4 | 0.8 | 4 | 12.8 | 6.4 |
| SEC-201 | 0.003 | 0.007 | 30 | 180 | 1 | 0.3 | 47 | 4 | 42.8 | 21.4 |
| Source | Sum of Squares | df | Mean Square | F-Value | p-Value | Model RP |
|---|---|---|---|---|---|---|
| Model RNA Production Rate | 1.22 | 9 | 0.1355 | 23.96 | 0.0121 | significant |
| A-Polymerase concentration | 0.0017 | 1 | 0.0017 | 0.3032 | 0.6202 | |
| B-UTP concentration | 0.8282 | 1 | 0.8282 | 146.47 | 0.0012 | |
| C-Magnesium concentration | 0.0016 | 1 | 0.0016 | 0.2916 | 0.6267 | |
| AB | 0.0317 | 1 | 0.0317 | 5.6 | 0.0988 | |
| AC | 0.0004 | 1 | 0.0004 | 0.0716 | 0.8063 | |
| BC | 0.001 | 1 | 0.001 | 0.1843 | 0.6966 | |
| A2 | 0.0022 | 1 | 0.0022 | 0.3829 | 0.5799 | |
| B2 | 0.2271 | 1 | 0.2271 | 40.16 | 0.0079 | |
| C2 | 0.0042 | 1 | 0.0042 | 0.7374 | 0.4536 | |
| Residual | 0.017 | 3 | 0.0057 | |||
| Cor Total | 1.24 | 12 | ||||
| Coded model equation: RNA production rate = 1.03 + 0.0146 × A + 0.3218 × B + 0.0144 × C − 0.08890 × AB − 0.01001 × AC − 0.0161 × BC + 0.0308 × A2 − 0.3152 × B2 + 0.0427 × C2 | ||||||
| Chromatography Block | ΔP (bar) | u (cm/s) | L (cm) | kO (-) | η (cP) | ε (-) | dp (μm) | N (-) | Lc (cm) | D (cm) |
|---|---|---|---|---|---|---|---|---|---|---|
| AEX-301 | 0.1 | 0.002 | 5 | 150 | 1 | 0.4 | 0.8 | 4 | 7.2 | 3.6 |
| AFC-301 | - | - | - | - | - | - | - | 4 | 23.4 | 11.7 |
| HPLC-301 | 0.5 | 0.003 | 25 | 150 | 1 | 0.3 | 2.1 | 4 | 35.6 | 17.8 |
| Unit Block | Description | Size | Cp,0 (€) | Year | Lang Factor | References | Simulation Size | Cp (€) | Cp,2021 (€) |
|---|---|---|---|---|---|---|---|---|---|
| R-101 | Fermentation reactor | V = 0.065 m3 | 34,013 * | 2013 | 0.6 | [38] & assumptions | V = 0.1 m3 | 44,045 | 49,519 |
| R-102 | Perfusion reactor | V = 2 m3 | 594,299 * | 2016 | 0.6 | [39] | V = 1 m3 | 392,091 | 461,650 |
| V-201 | Horizontal vessel | V = 2.5 m3 | 12,450 | 2013 | 0.2 | [38] & assumptions | V = 0.0005 m3 | 2267 | 2548 |
| UF-201 | Ultrafiltration filter | A = 1 m2 | 6686 * | 2015 | 0.58 | [39] | A = 0.023 m2 | 750 | 859 |
| HIC-201 | Chromatography | - | - | 2016 | - | [55] | - | 500,000 | 588,702 |
| UF-202 | Ultrafiltration filter | A = 1 m2 | 6686 * | 2015 | 0.58 | [39] | A = 0.0016 m2 | 160 | 183 |
| AEX-201 | Chromatography | - | - | 2016 | - | [55] | - | 500,000 | 588,702 |
| UF-203 | Ultrafiltration filter | A = 1 m2 | 6686 * | 2015 | 0.58 | [39] | A = 0.0016 m2 | 160 | 183 |
| SEC-201 | Chromatography | - | - | 2016 | - | [55] | - | 500,000 | 588,702 |
| UF-204 | Ultrafiltration filter | A = 1 m2 | 6686 * | 2015 | 0.58 | [39] | A = 0.0016 m2 | 160 | 183 |
| C-301 | Container | V = 0.21 m3 | 271 * | 2002 | 0.2 | [40] | V = 0.075 m3 | 221 | 356 |
| P-301 | Pump | - | - | 2021 | - | Assumption | - | 500 | 500 |
| B-301 | Jacketed mixer | V = 0.2 m3 | 29,931 | 2020 | 0.16 | [55], Pall Corporation | V = 0.075 m3 | 25,584 | 27,369 |
| C-302 | Container | V = 0.21 m3 | 271 * | 2002 | 0.2 | [40] | V = 0.075 m3 | 221 | 356 |
| P-302 | Pump | - | - | 2021 | - | Assumption | - | 500 | 500 |
| UF-301 | Ultrafiltration filter | A = 1 m2 | 6686 * | 2015 | 0.58 | [39] | A = 0.0028 m2 | 221 | 253 |
| AEX-301 | Chromatography | - | - | 2016 | - | [55] | - | 500,000 | 588,702 |
| UF-302 | Ultrafiltration filter | A = 1 m2 | 6686 * | 2015 | 0.58 | [39] | A = 0.00047 m2 | 79 | 90 |
| C-303 | Container | V = 0.21 m3 | 271 * | 2002 | 0.2 | [40] | V = 0,05 m3 | 203 | 328 |
| P-303 | Pump | - | - | 2021 | - | Assumption | - | 500 | 500 |
| R-301 | Reactor | V = 0.4 m3 | 74,699 * | 2002 | 0.5 | [40] | V = 0.01 m3 | 11,811 | 19,042 |
| R-302 | Reactor | V = 0.4 m3 | 74,699 * | 2002 | 0.5 | [40] | V = 0.01 m3 | 11,811 | 19,042 |
| R-303 | Reactor | V = 0.4 m3 | 74,699 * | 2002 | 0.5 | [40] | V = 0.01 m3 | 11,811 | 19,042 |
| C-304 | Container | V = 0.21 m3 | 271 * | 2002 | 0.2 | [40] | V = 0.05 m3 | 203 | 328 |
| P-304 | Pump | - | - | 2021 | - | Assumption | - | 500 | 500 |
| UF-303 | Ultrafiltration filter | A = 1 m2 | 6686 * | 2015 | 0.58 | [39] | A = 0.006 m2 | 344 | 394 |
| AFC-301 | Chromatography | - | - | 2016 | - | [55] | - | 500,000 | 588,702 |
| UF-304 | Ultrafiltration filter | A = 1 m2 | 6686 * | 2015 | 0.58 | [39] | A = 0.005 m2 | 309 | 354 |
| HPLC-301 | Chromatography | - | - | 2016 | - | [55] | - | 500,000 | 588,702 |
| UF-305 | Ultrafiltration filter | A = 1 m2 | 6686 * | 2015 | 0.58 | [39] | A = 0.005 m2 | 309 | 354 |
| B-401 | Non-jacketed mixer | V = 0.2 m3 | 24,994 | 2020 | 0.16 | [55], Pall Corporation | V = 0.0025 m3 | 12,398 | 13,263 |
| B-402 | Non-jacketed mixer | V = 0.2 m3 | 24,994 | 2020 | 0.16 | [55], Pall Corporation | V = 0.0075 m3 | 14,780 | 15,812 |
| P-401 | Pump | - | - | 2021 | - | Assumption | - | 500 | 500 |
| P-402 | Pump | - | - | 2021 | - | Assumption | - | 500 | 500 |
| μM-401 | Microfluidic mixer | - | 176,671 * | 2021 | - | Precision Nano-systems Corporation | - | 176,671 | 176,671 |
| TF-401 | Tangential flow filtration filter | A = 0.12 m2 | 4504 * | 2018 | 0.8 | [39] | A = 3 m2 | 59,149 | 62,553 |
| MS-401 | Molecular sieve | - | - | 2021 | - | Assumption | - | 1800 | 1800 |
| Raw Material | Price | References | Total Quantity | Total Price |
|---|---|---|---|---|
| Glucose | 0.24 * €/kg | [58] | 18,454 kg/year | 4429 €/year |
| Diammonium phosphate | 0.83 * €/kg | [59] | 6336 kg/year | 5259 €/year |
| Ammonium sulfate | 0.124 * €/kg | [38] | 21,384 kg/year | 2652 €/year |
| HIC resin | 2064 €/L (replacement every 100 cycles) | [55,60,61] | 2206 L/year | 4,553,070 €/year |
| AEX membrane | 989 €/m2 (replacement every 100 cycles) | [55,60,61] | 6 m2/year | 5505 €/year |
| HPLC resin | 10,892 €/L (replacement every 100 cycles) | [55,60,61] | 1476 L/year | 16,076,592 €/year |
| SEC resin | 10,892 €/L (replacement every 150 cycles) | [60,61] | 2546 L/year | 27,736,042 €/year |
| Linearization restriction enzyme | 0.0051 * €/unit | [62] | 22,464,000,000 units/year | 114,566,400 €/year |
| T7 RNA polymerase | 38 €/mg | Merc i | 39 g/year | 1,495,680 €/year |
| CleanCap Reagent (3’ OMe) AG | 248 €/mg | Trilink Biotechnologies ii | 216 g/year | CleanCap Reagent (3’ OMe) AG |
| AFC resin | 10,892 €/L (replacement every 100 cycles) | [60,61] | 51 L/year | 553,035 €/year |
| Ethanol | 6.8 * €/kg | [58] | 41,722 kg/year | 283,707 €/year |
| Sodium citrate | 0.7 * €/kg | [38] | 102 kg/year | 72 €/year |
| Dimethyldioctadecylammonium (DDAB) | 554 €/g | Merc iii | 8 kg/year | 4,275,947 €/year |
| Cholesterol | 11 €/g | Merc iv | 129,015 g/year | 1,419,162 €/year |
| 1,2-Dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (DMG-PEG2000) | 166 €/g | Merc v | 228,584 g/year | 37,944,885 €/year |
| 1,2-dioctadecanoyl-sn-glycero-3-phosphocholine (DSPC) | 172 €/g | Merc vi | 65,996 g/year | 11,351,268 €/year |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ouranidis, A.; Davidopoulou, C.; Tashi, R.-K.; Kachrimanis, K. Pharma 4.0 Continuous mRNA Drug Products Manufacturing. Pharmaceutics 2021, 13, 1371. https://doi.org/10.3390/pharmaceutics13091371
Ouranidis A, Davidopoulou C, Tashi R-K, Kachrimanis K. Pharma 4.0 Continuous mRNA Drug Products Manufacturing. Pharmaceutics. 2021; 13(9):1371. https://doi.org/10.3390/pharmaceutics13091371
Chicago/Turabian StyleOuranidis, Andreas, Christina Davidopoulou, Reald-Konstantinos Tashi, and Kyriakos Kachrimanis. 2021. "Pharma 4.0 Continuous mRNA Drug Products Manufacturing" Pharmaceutics 13, no. 9: 1371. https://doi.org/10.3390/pharmaceutics13091371
APA StyleOuranidis, A., Davidopoulou, C., Tashi, R.-K., & Kachrimanis, K. (2021). Pharma 4.0 Continuous mRNA Drug Products Manufacturing. Pharmaceutics, 13(9), 1371. https://doi.org/10.3390/pharmaceutics13091371