
Delivery of Various Cargos into Cancer Cells and Tissues via Cell-Penetrating Peptides: A Review of the Last Decade



Abstract
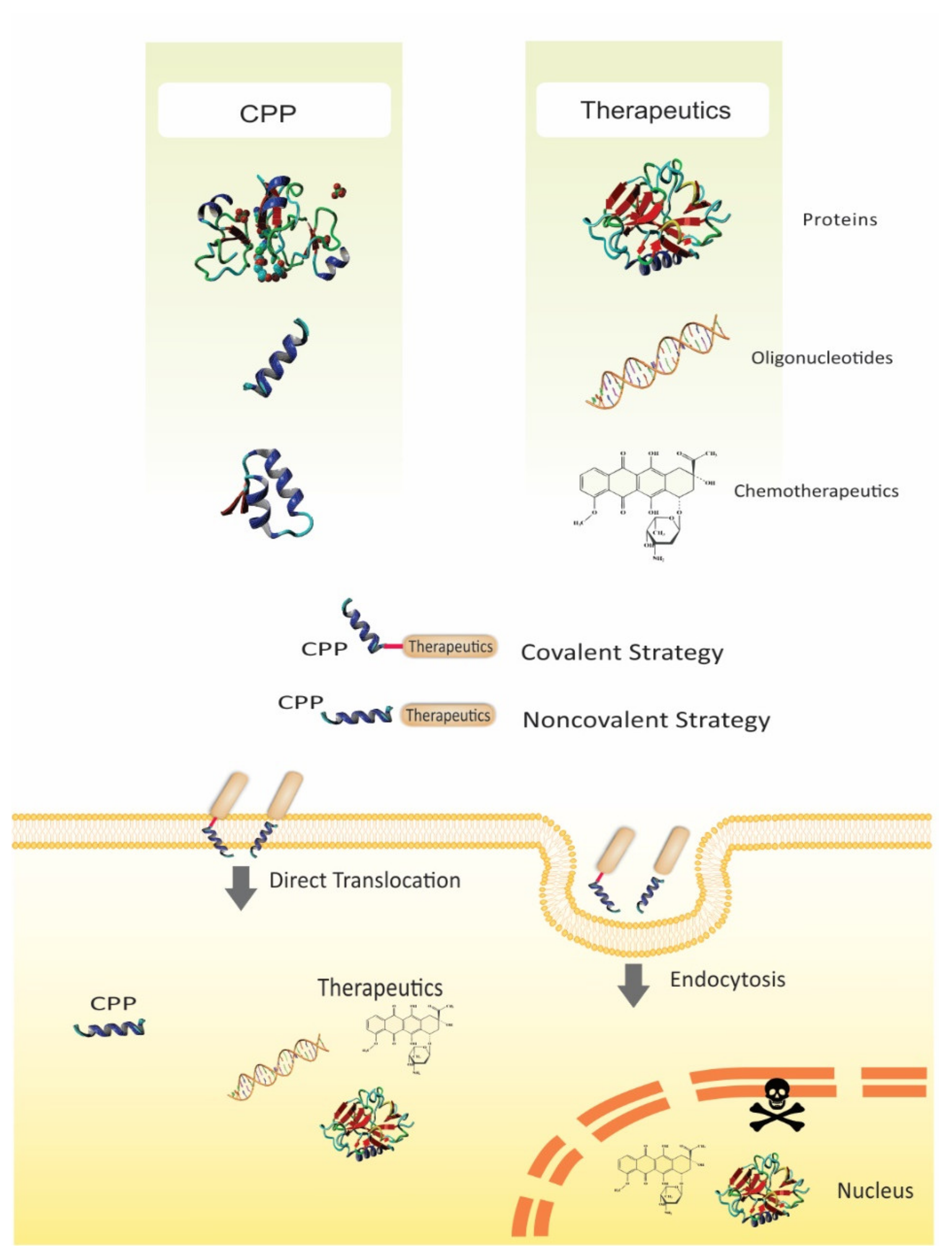
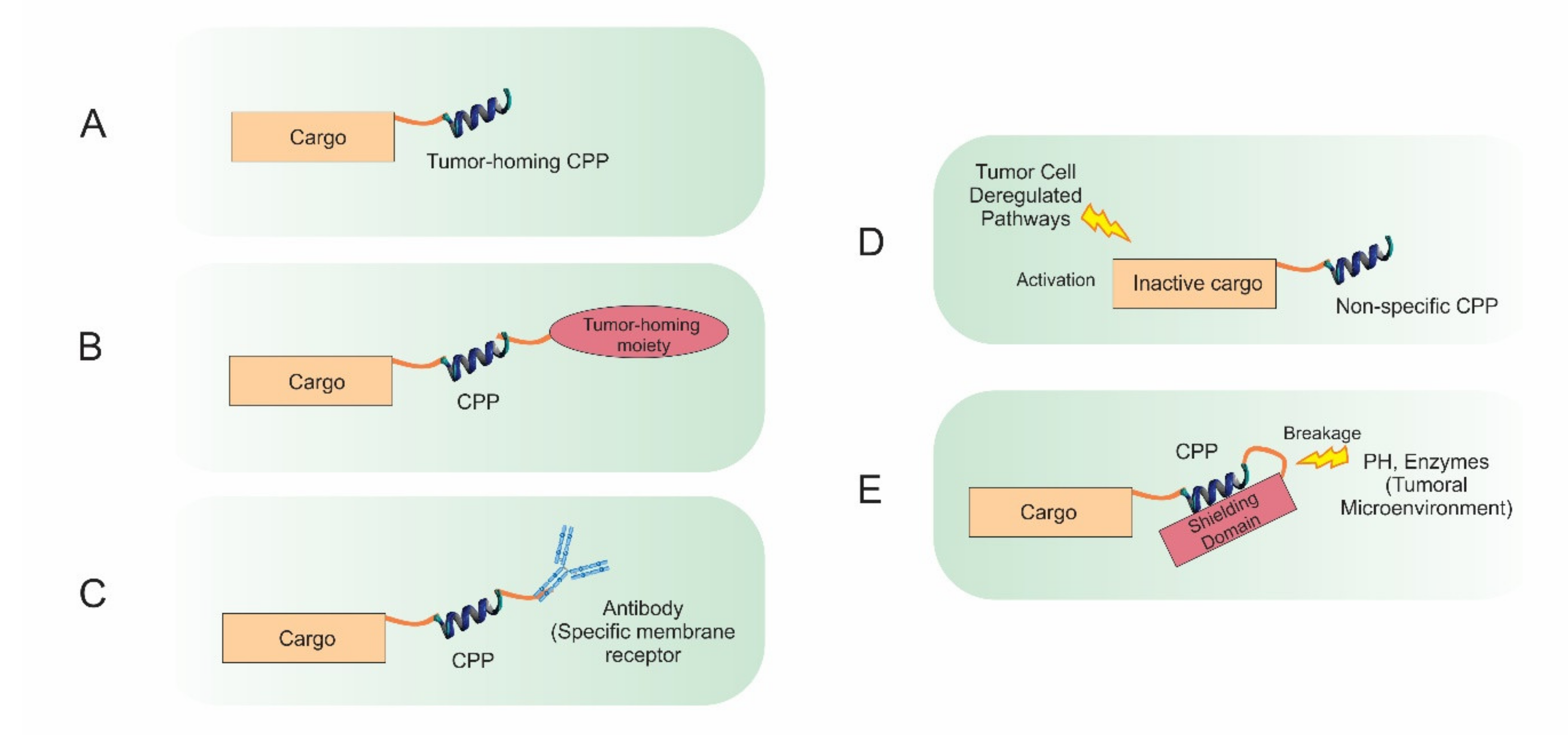
1. Introduction
2. Challenges of Biomacromolecules and Chemotherapeutics Delivery
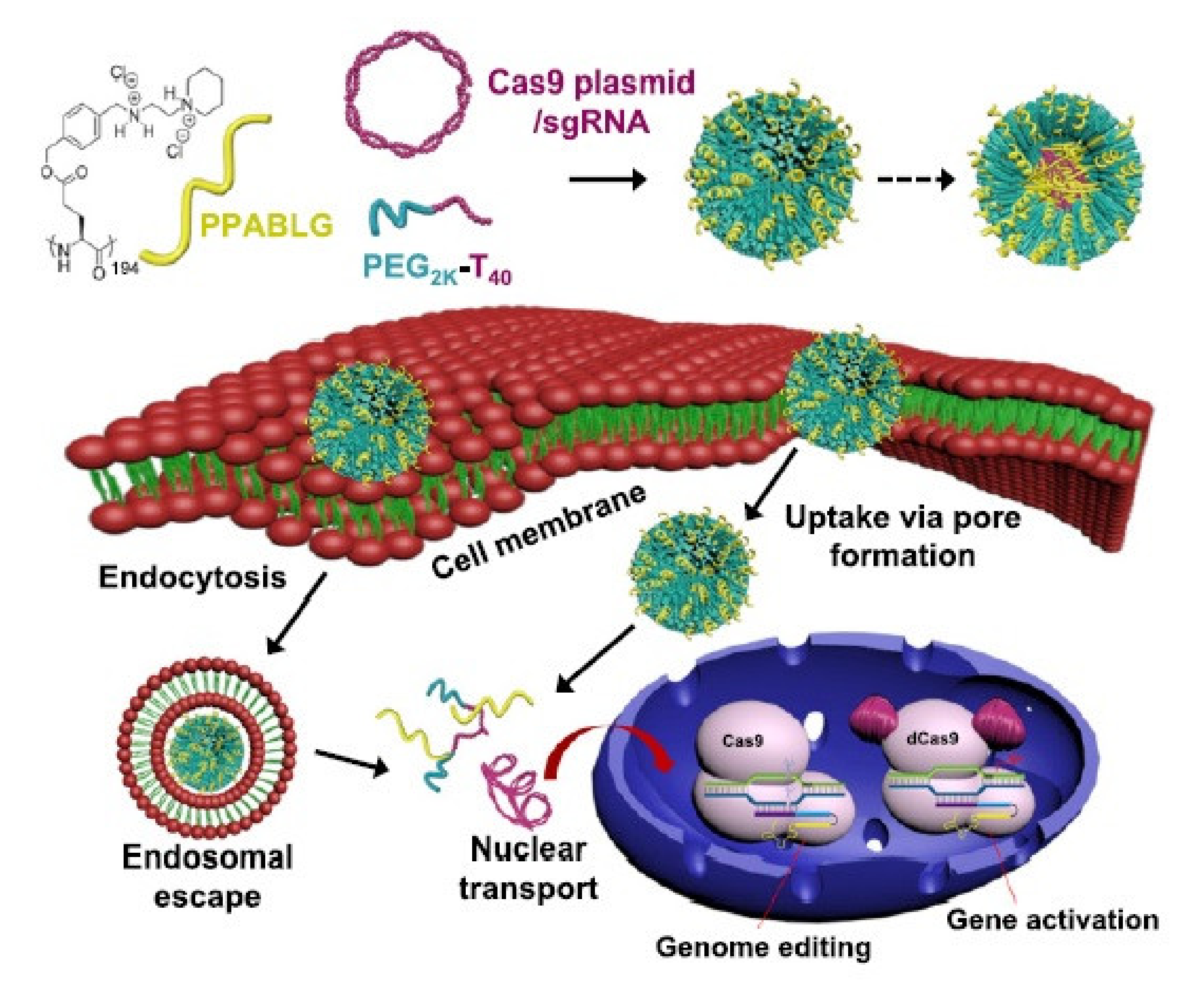
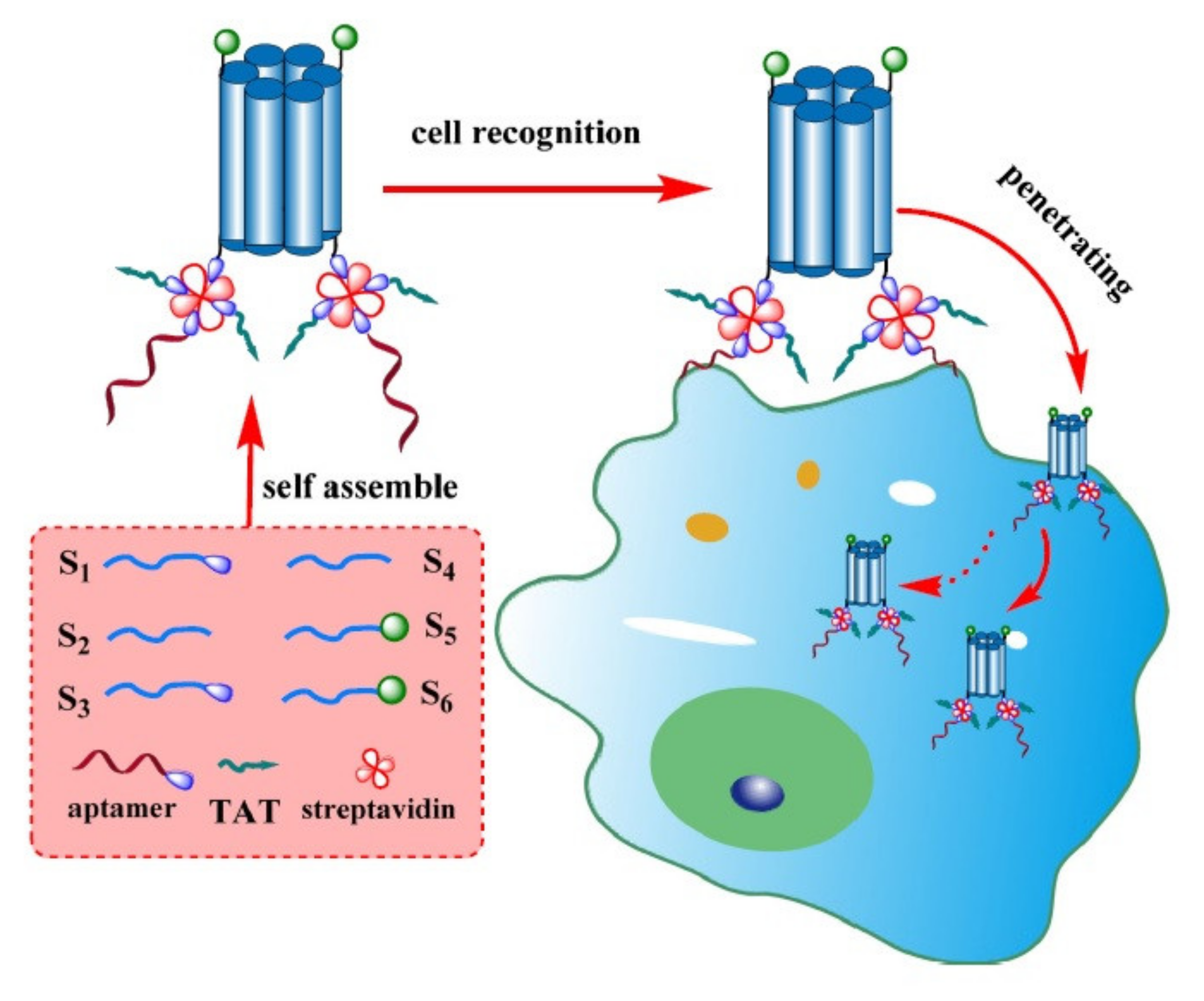
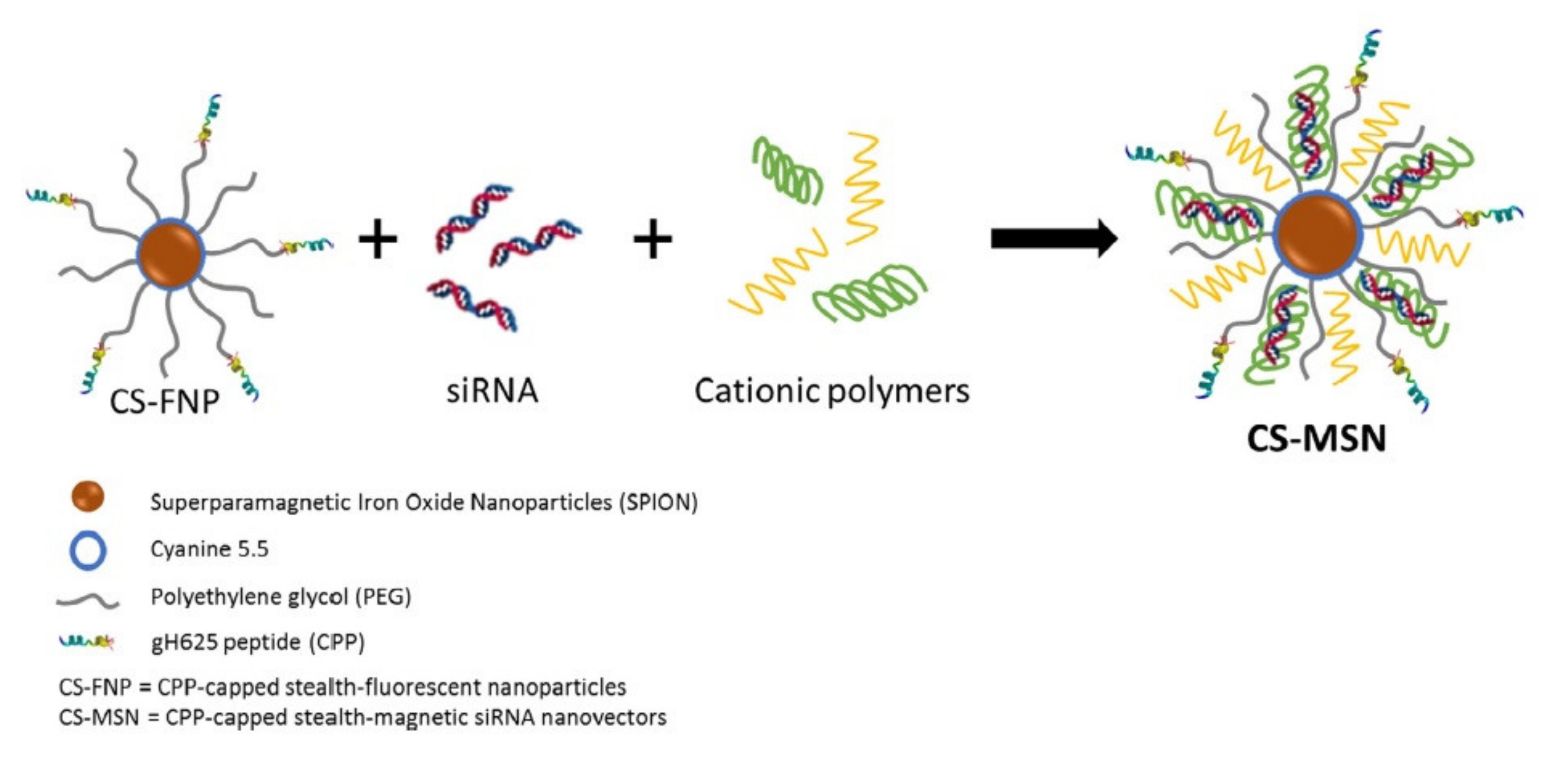
3. Delivery of Nucleic Acids
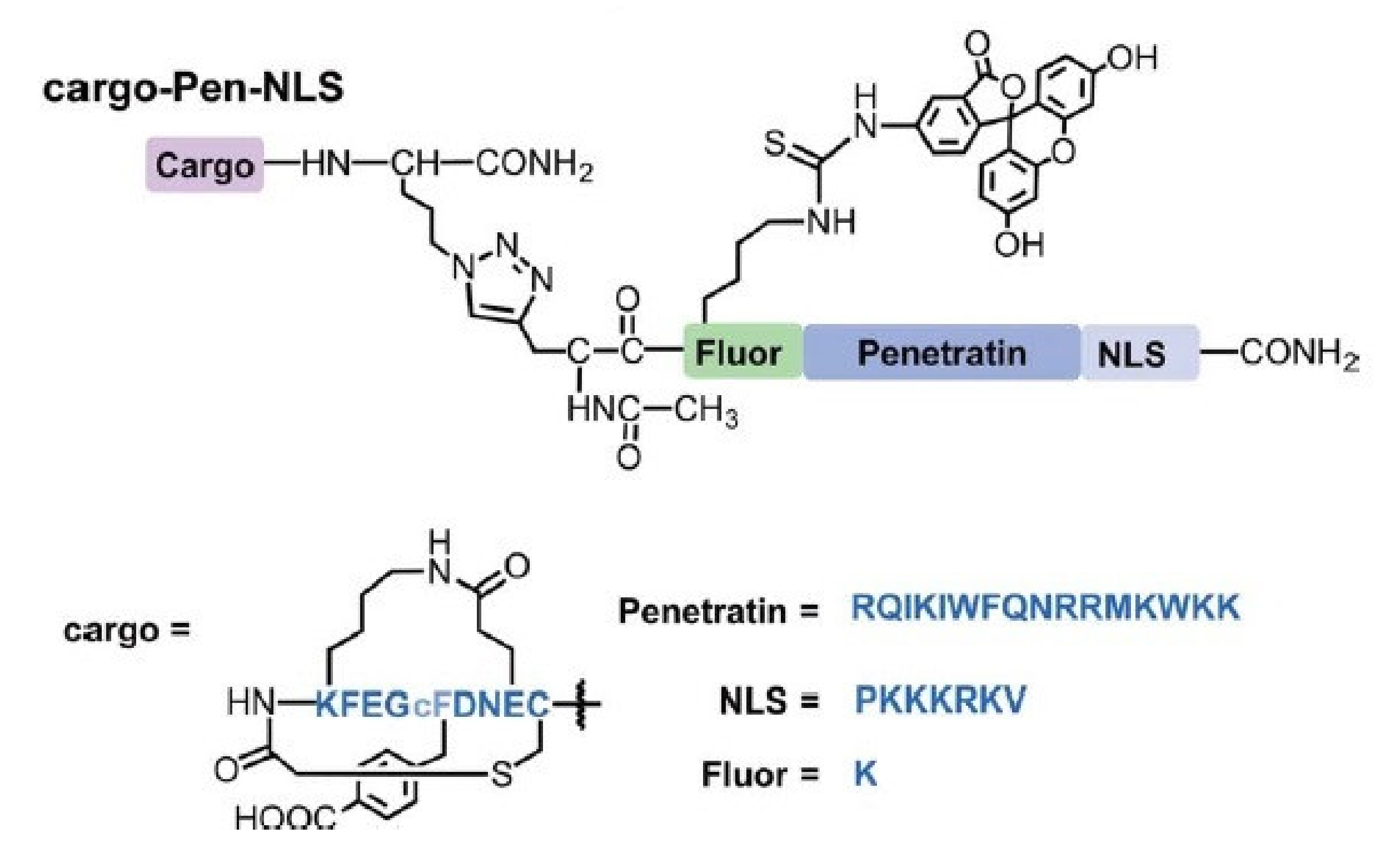
4. Delivery of Proteins/Peptides
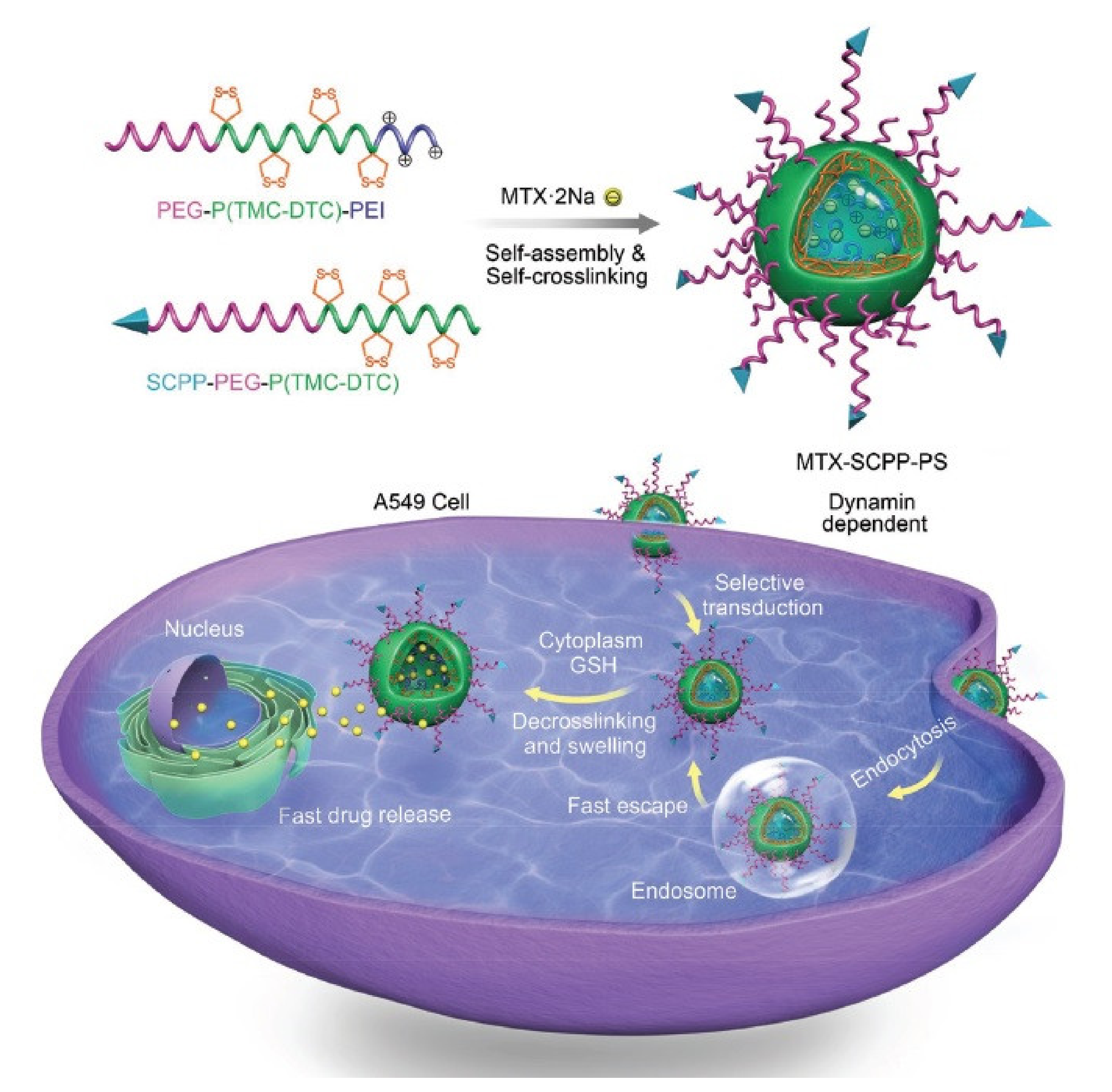
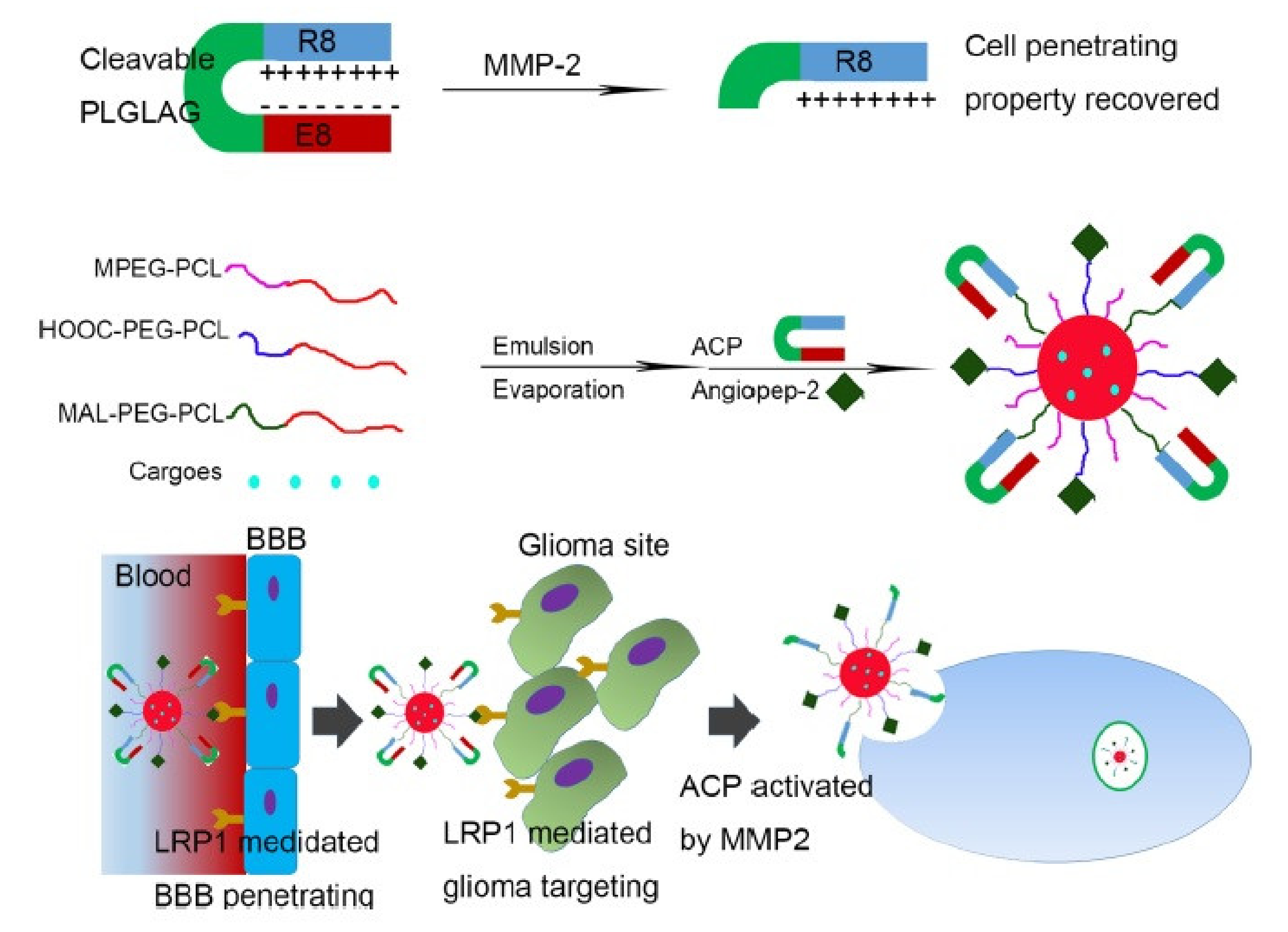
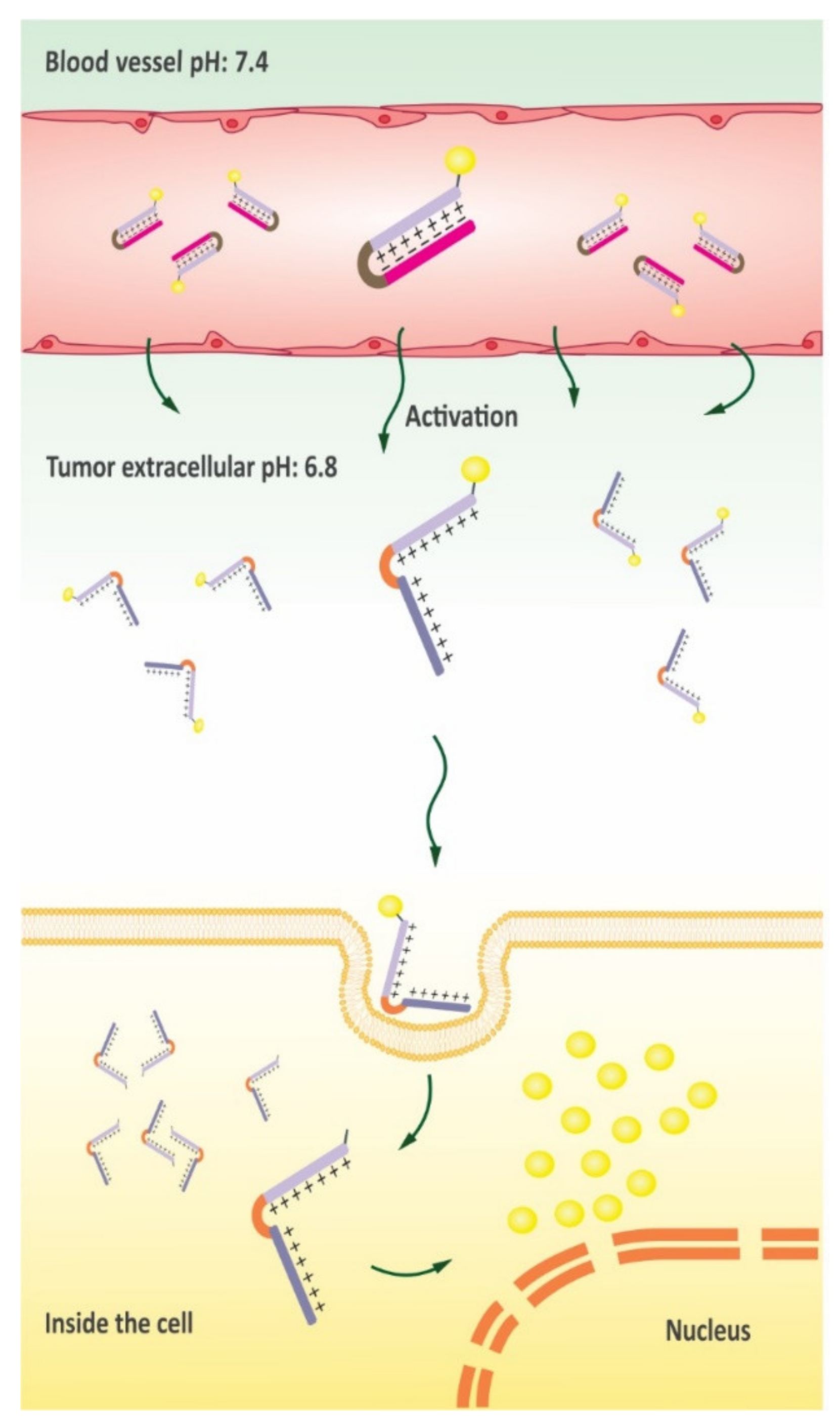
5. Delivery of Chemotherapeutics
6. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled Drug Delivery Vehicles for Cancer Treatment and Their Performance. Signal Transduct. Target. Ther. 2018, 3, 1–19. [Google Scholar] [CrossRef]
- Yang, N.J.; Hinner, M.J. Getting across the cell membrane: An overview for small molecules, peptides, and proteins. In Site-Specific Protein Labeling; Springer: New York, NY, USA, 2015; pp. 29–53. [Google Scholar]
- Tripathi, P.P.; Arami, H.; Banga, I.; Gupta, J.; Gandhi, S. Cell Penetrating Peptides in Preclinical and Clinical Cancer Diagnosis and Therapy. Oncotarget 2018, 9, 37252. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.D.; Pabo, C.O. Cellular Uptake of the Tat Protein from Human Immunodeficiency Virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Bechara, C.; Sagan, S. Cell-Penetrating Peptides: 20 Years Later, Where Do We Stand? FEBS Lett. 2013, 587, 1693–1702. [Google Scholar] [CrossRef]
- Van Nguyen, T.; Shin, M.C.; Min, K.A.; Huang, Y.; Oh, E.; Moon, C. Cell-Penetrating Peptide-Based Non-Invasive Topical Delivery Systems. J. Pharm. Investig. 2018, 48, 77–87. [Google Scholar] [CrossRef]
- Hemmati, S.; Behzadipour, Y.; Haddad, M. Decoding the Proteome of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) for Cell-Penetrating Peptides Involved in Pathogenesis or Applicable as Drug Delivery Vectors. Infect. Genet. Evol. 2020, 85, 104474. [Google Scholar] [CrossRef]
- Bolhassani, A.; Jafarzade, B.S.; Mardani, G. In Vitro and in Vivo Delivery of Therapeutic Proteins Using Cell Penetrating Peptides. Peptides 2017, 87, 50–63. [Google Scholar] [CrossRef]
- De Figueiredo, I.R.; Freire, J.M.; Flores, L.; Veiga, A.S.; Castanho, M.A.R.B. Cell-penetrating Peptides: A Tool for Effective Delivery in Gene-targeted Therapies. IUBMB Life 2014, 66, 182–194. [Google Scholar] [CrossRef]
- Xie, J.; Bi, Y.; Zhang, H.; Dong, S.; Teng, L.; Lee, R.J.; Yang, Z. Cell-Penetrating Peptides in Diagnosis and Treatment of Human Diseases: From Preclinical Research to Clinical Application. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, M.; Birch, D.; Mørck Nielsen, H. Applications and Challenges for Use of Cell-Penetrating Peptides as Delivery Vectors for Peptide and Protein Cargos. Int. J. Mol. Sci. 2016, 17, 185. [Google Scholar] [CrossRef]
- Johnson, R.M.; Harrison, S.D.; Maclean, D. Therapeutic applications of cell-penetrating peptides. In Cell-Penetrating Peptides; Springer: Totowa, NJ, USA, 2011; pp. 535–551. [Google Scholar]
- Navya, P.N.; Kaphle, A.; Srinivas, S.P.; Bhargava, S.K.; Rotello, V.M.; Daima, H.K. Current Trends and Challenges in Cancer Management and Therapy Using Designer Nanomaterials. Nano Converg. 2019, 6, 23. [Google Scholar] [CrossRef]
- Tiwari, G.; Tiwari, R.; Sriwastawa, B.; Bhati, L.; Pandey, S.; Pandey, P.; Bannerjee, S.K. Drug Delivery Systems: An Updated Review. Int. J. Pharm. Investig. 2012, 2, 2. [Google Scholar] [CrossRef]
- Mitra, A.K.; Agrahari, V.; Mandal, A.; Cholkar, K.; Natarajan, C.; Shah, S.; Joseph, M.; Trinh, H.M.; Vaishya, R.; Yang, X. Novel Delivery Approaches for Cancer Therapeutics. J. Control. release 2015, 219, 248–268. [Google Scholar] [CrossRef]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable Linkers in Antibody–Drug Conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- Thomas, A.; Teicher, B.A.; Hassan, R. Antibody–Drug Conjugates for Cancer Therapy. Lancet Oncol. 2016, 17, e254–e262. [Google Scholar] [CrossRef]
- Alas, M.; Saghaeidehkordi, A.; Kaur, K. Peptide–Drug Conjugates with Different Linkers for Cancer Therapy. J. Med. Chem. 2020, 64, 216–232. [Google Scholar] [CrossRef]
- Ritchie, M.; Tchistiakova, L.; Scott, N. Implications of Receptor-Mediated Endocytosis and Intracellular Trafficking Dynamics in the Development of Antibody Drug Conjugates. MAbs 2013, 5, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody–Drug Conjugates for Cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef]
- He, R.; Finan, B.; Mayer, J.P.; DiMarchi, R.D. Peptide Conjugates with Small Molecules Designed to Enhance Efficacy and Safety. Molecules 2019, 24, 1855. [Google Scholar] [CrossRef] [PubMed]
- Hoppenz, P.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide-Drug Conjugates and Their Targets in Advanced Cancer Therapies. Front. Chem. 2020, 8, 571. [Google Scholar] [CrossRef] [PubMed]
- Kardani, K.; Milani, A.; Shabani, S.H.; Bolhassani, A. Cell Penetrating Peptides: The Potent Multi-Cargo Intracellular Carriers. Expert Opin. Drug Deliv. 2019, 16, 1227–1258. [Google Scholar] [CrossRef]
- Böhmová, E.; Machová, D.; Pechar, M.; Pola, R.; Venclíková, K.; Janoušková, O.; Etrych, T. Cell-Penetrating Peptides: A Useful Tool for the Delivery of Various Cargoes into Cells. Physiol. Res. 2018, 67, S267–S279. [Google Scholar] [CrossRef]
- Zaro, J.L.; Shen, W.-C. Cationic and Amphipathic Cell-Penetrating Peptides (CPPs): Their Structures and in Vivo Studies in Drug Delivery. Front. Chem. Sci. Eng. 2015, 9, 407–427. [Google Scholar] [CrossRef]
- Taylor, R.E.; Zahid, M. Cell Penetrating Peptides, Novel Vectors for Gene Therapy. Pharmaceutics 2020, 12, 225. [Google Scholar] [CrossRef] [PubMed]
- Madani, F.; Lindberg, S.; Langel, Ü.; Futaki, S.; Gräslund, A. Mechanisms of Cellular Uptake of Cell-Penetrating Peptides. J. Biophys. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Sawant, R.; Torchilin, V. Intracellular Transduction Using Cell-Penetrating Peptides. Mol. Biosyst. 2010, 6, 628–640. [Google Scholar] [CrossRef]
- Milletti, F. Cell-Penetrating Peptides: Classes, Origin, and Current Landscape. Drug Discov. Today 2012, 17, 850–860. [Google Scholar] [CrossRef]
- Kamei, N.; Shingaki, T.; Kanayama, Y.; Tanaka, M.; Zochi, R.; Hasegawa, K.; Watanabe, Y.; Takeda-Morishita, M. Visualization and Quantitative Assessment of the Brain Distribution of Insulin through Nose-to-Brain Delivery Based on the Cell-Penetrating Peptide Noncovalent Strategy. Mol. Pharm. 2016, 13, 1004–1011. [Google Scholar] [CrossRef]
- Gui, L.; Zhang, X.; Qiao, Z.; Wang, H. Cell-Penetrating Peptides and Polymers for Improved Drug Delivery. ChemNanoMat 2020, 6, 1138–1148. [Google Scholar] [CrossRef]
- Khan, M.M.; Filipczak, N.; Torchilin, V.P. Cell Penetrating Peptides: A Versatile Vector for Co-Delivery of Drug and Genes in Cancer. J. Control. Release 2020. [Google Scholar]
- Silva, S.; Almeida, A.J.; Vale, N. Combination of Cell-Penetrating Peptides with Nanoparticles for Therapeutic Application: A Review. Biomolecules 2019, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Vale, N.; Duarte, D.; Silva, S.; Correia, A.S.; Costa, B.; Gouveia, M.J.; Ferreira, A. Cell-Penetrating Peptides in Oncologic Pharmacotherapy: A Review. Pharmacol. Res. 2020, 105231. [Google Scholar] [CrossRef] [PubMed]
- Habault, J.; Poyet, J.-L. Recent Advances in Cell Penetrating Peptide-Based Anticancer Therapies. Molecules 2019, 24, 927. [Google Scholar] [CrossRef]
- Feni, L.; Neundorf, I. The Current Role of Cell-Penetrating Peptides in Cancer Therapy. Pept. Pept. Biomater. their Biomed. Appl. 2017, 279–295. [Google Scholar]
- Amer, M.H. Gene Therapy for Cancer: Present Status and Future Perspective. Mol. Cell. Ther. 2014, 2, 1–19. [Google Scholar] [CrossRef]
- Durymanov, M.; Reineke, J. Non-Viral Delivery of Nucleic Acids: Insight into Mechanisms of Overcoming Intracellular Barriers. Front. Pharmacol. 2018, 9, 971. [Google Scholar] [CrossRef]
- Torres-Vanegas, J.D.; Cruz, J.C.; Reyes, L.H. Delivery Systems for Nucleic Acids and Proteins: Barriers, Cell Capture Pathways and Nanocarriers. Pharmaceutics 2021, 13, 428. [Google Scholar] [CrossRef]
- Walther, W.; Stein, U. Viral Vectors for Gene Transfer. Drugs 2000, 60, 249–271. [Google Scholar] [CrossRef]
- Ramamoorth, M.; Narvekar, A. Non Viral Vectors in Gene Therapy-an Overview. J. Clin. diagnostic Res. JCDR 2015, 9, GE01. [Google Scholar] [CrossRef]
- Bolhassani, A. Potential Efficacy of Cell-Penetrating Peptides for Nucleic Acid and Drug Delivery in Cancer. Biochim. Biophys. Acta BBA-Rev. Cancer 2011, 1816, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.-L.; Harris, J.L.; Khanna, K.K.; Hong, J.-H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef] [PubMed]
- Gheorghe, D.C.; Niculescu, A.-G.; Bîrcă, A.C.; Grumezescu, A.M. Biomaterials for the Prevention of Oral Candidiasis Development. Pharmaceutics 2021, 13, 803. [Google Scholar] [CrossRef]
- Kristensen, M.; Nielsen, H.M. Cell-Penetrating Peptides as Tools to Enhance Non-Injectable Delivery of Biopharmaceuticals. Tissue Barriers 2016, 4, e1178369. [Google Scholar] [CrossRef]
- Banks, W.A. Peptides and the Blood–Brain Barrier. Peptides 2015, 72, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, M.B. Is Cancer Chemotherapy Dying? Asian J. Transfus. Sci. 2016, 10, S1. [Google Scholar] [CrossRef]
- Schirrmacher, V. From Chemotherapy to Biological Therapy: A Review of Novel Concepts to Reduce the Side Effects of Systemic Cancer Treatment. Int. J. Oncol. 2019, 54, 407–419. [Google Scholar]
- Boisguérin, P.; Deshayes, S.; Gait, M.J.; O’Donovan, L.; Godfrey, C.; Betts, C.A.; Wood, M.J.A.; Lebleu, B. Delivery of Therapeutic Oligonucleotides with Cell Penetrating Peptides. Adv. Drug Deliv. Rev. 2015, 87, 52–67. [Google Scholar] [CrossRef]
- Benizri, S.; Gissot, A.; Martin, A.; Vialet, B.; Grinstaff, M.W.; Barthélémy, P. Bioconjugated Oligonucleotides: Recent Developments and Therapeutic Applications. Bioconjug. Chem. 2019, 30, 366–383. [Google Scholar] [CrossRef]
- Li, H.; Tsui, T.Y.; Ma, W. Intracellular Delivery of Molecular Cargo Using Cell-Penetrating Peptides and the Combination Strategies. Int. J. Mol. Sci. 2015, 16, 19518–19536. [Google Scholar] [CrossRef]
- Veldhoen, S.; Laufer, S.D.; Restle, T. Recent Developments in Peptide-Based Nucleic Acid Delivery. Int. J. Mol. Sci. 2008, 9, 1276–1320. [Google Scholar] [CrossRef]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef]
- Lostalé-Seijo, I.; Louzao, I.; Juanes, M.; Montenegro, J. Peptide/Cas9 Nanostructures for Ribonucleoprotein Cell Membrane Transport and Gene Edition. Chem. Sci. 2017, 8, 7923–7931. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-X.; Song, Z.; Lao, Y.-H.; Xu, X.; Gong, J.; Cheng, D.; Chakraborty, S.; Park, J.S.; Li, M.; Huang, D. Nonviral Gene Editing via CRISPR/Cas9 Delivery by Membrane-Disruptive and Endosomolytic Helical Polypeptide. Proc. Natl. Acad. Sci. USA 2018, 115, 4903–4908. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, M.; Gao, X.; Chen, Y.; Liu, T. Nanotechnology in Cancer Diagnosis: Progress, Challenges and Opportunities. J. Hematol. Oncol. 2019, 12, 137. [Google Scholar] [CrossRef]
- Chi, Q.; Yang, Z.; Xu, K.; Wang, C.; Liang, H. DNA Nanostructure as an Efficient Drug Delivery Platform for Immunotherapy. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.-L.; Yuan, D.-D.; Song, T.; Li, X.-M. DNA Nanopore Functionalized with Aptamer and Cell-Penetrating Peptide for Tumor Cell Recognition. Anal. Bioanal. Chem. 2017, 409, 3789–3797. [Google Scholar] [CrossRef]
- Xin, Y.; Huang, M.; Guo, W.W.; Huang, Q.; zhen Zhang, L.; Jiang, G. Nano-Based Delivery of RNAi in Cancer Therapy. Mol. Cancer 2017, 16, 1–9. [Google Scholar] [CrossRef]
- Wan, Y.; Dai, W.; Nevagi, R.J.; Toth, I.; Moyle, P.M. Multifunctional Peptide-Lipid Nanocomplexes for Efficient Targeted Delivery of DNA and SiRNA into Breast Cancer Cells. Acta Biomater. 2017, 59, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Golan, M.; Feinshtein, V.; David, A. Conjugates of HA2 with Octaarginine-Grafted HPMA Copolymer Offer Effective SiRNA Delivery and Gene Silencing in Cancer Cells. Eur. J. Pharm. Biopharm. 2016, 109, 103–112. [Google Scholar] [CrossRef]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A Review of the Challenges and Approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [PubMed]
- Yen, A.; Cheng, Y.; Sylvestre, M.; Gustafson, H.H.; Puri, S.; Pun, S.H. Serum Nuclease Susceptibility of MRNA Cargo in Condensed Polyplexes. Mol. Pharm. 2018, 15, 2268–2276. [Google Scholar] [CrossRef]
- Chen, Q.; Qi, R.; Chen, X.; Yang, X.; Wu, S.; Xiao, H.; Dong, W. A Targeted and Stable Polymeric Nanoformulation Enhances Systemic Delivery of MRNA to Tumors. Mol. Ther. 2017, 25, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Chinak, O.A.; Shernyukov, A.V.; Ovcherenko, S.S.; Sviridov, E.A.; Golyshev, V.M.; Fomin, A.S.; Pyshnaya, I.A.; Kuligina, E.V.; Richter, V.A.; Bagryanskaya, E.G. Structural and Aggregation Features of a Human κ-Casein Fragment with Antitumor and Cell-Penetrating Properties. Molecules 2019, 24, 2919. [Google Scholar] [CrossRef]
- Chinak, O.; Golubitskaya, E.; Pyshnaya, I.; Stepanov, G.; Zhuravlev, E.; Richter, V.; Koval, O. Nucleic Acids Delivery into the Cells Using Pro-Apoptotic Protein Lactaptin. Front. Pharmacol. 2019, 10, 1043. [Google Scholar] [CrossRef]
- Ezzat, K.; EL Andaloussi, S.; Zaghloul, E.M.; Lehto, T.; Lindberg, S.; Moreno, P.M.D.; Viola, J.R.; Magdy, T.; Abdo, R.; Guterstam, P. PepFect 14, a Novel Cell-Penetrating Peptide for Oligonucleotide Delivery in Solution and as Solid Formulation. Nucleic Acids Res. 2011, 39, 5284–5298. [Google Scholar] [CrossRef]
- Margus, H.; Padari, K.; Pooga, M. Cell-Penetrating Peptides as Versatile Vehicles for Oligonucleotide Delivery. Mol. Ther. 2012, 20, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Munyendo, W.L.L.; Lv, H.; Benza-Ingoula, H.; Baraza, L.D.; Zhou, J. Cell Penetrating Peptides in the Delivery of Biopharmaceuticals. Biomolecules 2012, 2, 187–202. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, K.; Zaghloul, E.M.; Andaloussi, S.E.L.; Lehto, T.; El-Sayed, R.; Magdy, T.; Smith, C.I.E.; Langel, Ü. Solid Formulation of Cell-Penetrating Peptide Nanocomplexes with SiRNA and Their Stability in Simulated Gastric Conditions. J. Control. Release 2012, 162, 1–8. [Google Scholar] [CrossRef]
- Angeli, E.; Nguyen, T.T.; Janin, A.; Bousquet, G. How to Make Anticancer Drugs Cross the Blood–Brain Barrier to Treat Brain Metastases. Int. J. Mol. Sci. 2020, 21, 22. [Google Scholar] [CrossRef]
- Abdul Razzak, R.; Florence, G.J.; Gunn-Moore, F.J. Approaches to CNS Drug Delivery with a Focus on Transporter-Mediated Transcytosis. Int. J. Mol. Sci. 2019, 20, 3108. [Google Scholar] [CrossRef]
- Ji, X.; Wang, H.; Chen, Y.; Zhou, J.; Liu, Y. Recombinant Expressing Angiopep-2 Fused Anti-VEGF Single Chain Fab (ScFab) Could Cross Blood–Brain Barrier and Target Glioma. AMB Express 2019, 9, 165. [Google Scholar] [CrossRef]
- Srimanee, A.; Arvanitidou, M.; Kim, K.; Hällbrink, M.; Langel, Ü. Cell-Penetrating Peptides for SiRNA Delivery to Glioblastomas. Peptides 2018, 104, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Djemaa, S.B.; David, S.; Hervé-Aubert, K.; Falanga, A.; Galdiero, S.; Allard-Vannier, E.; Chourpa, I.; Munnier, E. Formulation and in Vitro Evaluation of a SiRNA Delivery Nanosystem Decorated with GH625 Peptide for Triple Negative Breast Cancer Theranosis. Eur. J. Pharm. Biopharm. 2018, 131, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Kanazawa, T.; Horiuchi, S.; Ando, T.; Sugawara, K.; Takashima, Y.; Seta, Y.; Okada, H. Cytoplasm-Responsive Nanocarriers Conjugated with a Functional Cell-Penetrating Peptide for Systemic SiRNA Delivery. Int. J. Pharm. 2013, 455, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Cheung, H.W.; von Maltzhan, G.; Agrawal, A.; Cowley, G.S.; Weir, B.A.; Boehm, J.S.; Tamayo, P.; Karst, A.M.; Liu, J.F. Targeted Tumor-Penetrating SiRNA Nanocomplexes for Credentialing the Ovarian Cancer Oncogene ID4. Sci. Transl. Med. 2012, 4, 147ra112. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Sagers, J.E.; Landegger, L.D.; Bhatia, S.N.; Stankovic, K.M. Tumor-Penetrating Delivery of SiRNA against TNFα to Human Vestibular Schwannomas. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Wang, H.; Li, Y. Stimuli-Responsive Nanomedicines for Overcoming Cancer Multidrug Resistance. Theranostics 2018, 8, 1059. [Google Scholar] [CrossRef]
- Chen, B.; Dai, W.; He, B.; Zhang, H.; Wang, X.; Wang, Y.; Zhang, Q. Current Multistage Drug Delivery Systems Based on the Tumor Microenvironment. Theranostics. 2017, 7, 538. [Google Scholar] [CrossRef]
- Zhu, L.; Torchilin, V.P. Stimulus-Responsive Nanopreparations for Tumor Targeting. Integr. Biol. 2013, 5, 96–107. [Google Scholar] [CrossRef]
- Fang, Y.; Xue, J.; Gao, S.; Lu, A.; Yang, D.; Jiang, H.; He, Y.; Shi, K. Cleavable PEGylation: A Strategy for Overcoming the “PEG Dilemma” in Efficient Drug Delivery. Drug Deliv. 2017, 24, 22–32. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef]
- Wang, H.-X.; Yang, X.-Z.; Sun, C.-Y.; Mao, C.-Q.; Zhu, Y.-H.; Wang, J. Matrix Metalloproteinase 2-Responsive Micelle for SiRNA Delivery. Biomaterials 2014, 35, 7622–7634. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, R.A.; Rouillé, Y.; Dubuisson, J. Interactions between Virus Proteins and Host Cell Membranes during the Viral Life Cycle. Int. Rev. Cytol. 2005, 245, 171–244. [Google Scholar] [PubMed]
- Lau, W.L.; Ege, D.S.; Lear, J.D.; Hammer, D.A.; Degrado, W.F. Oligomerization of Fusogenic Peptides Promotes Membrane Fusion by Enhancing Membrane Destabilization. Biophys. J. 2004, 86, 272–284. [Google Scholar] [CrossRef][Green Version]
- Cantini, L.; Attaway, C.C.; Butler, B.; Andino, L.M.; Sokolosky, M.L.; Jakymiw, A. Fusogenic-Oligoarginine Peptide-Mediated Delivery of SiRNAs Targeting the CIP2A Oncogene into Oral Cancer Cells. PLoS ONE 2013, 8, e73348. [Google Scholar] [CrossRef] [PubMed]
- Saw, P.E.; Song, E.-W. Phage Display Screening of Therapeutic Peptide for Cancer Targeting and Therapy. Protein Cell 2019, 1–21. [Google Scholar] [CrossRef]
- Ceci, C.; Atzori, M.G.; Lacal, P.M.; Graziani, G. Role of VEGFs/VEGFR-1 Signaling and Its Inhibition in Modulating Tumor Invasion: Experimental Evidence in Different Metastatic Cancer Models. Int. J. Mol. Sci. 2020, 21, 1388. [Google Scholar] [CrossRef]
- Newman, M.R.; Benoit, D.S.W. In Vivo Translation of Peptide-Targeted Drug Delivery Systems Discovered by Phage Display. Bioconjug. Chem. 2018, 29, 2161–2169. [Google Scholar] [CrossRef]
- Fang, B.; Jiang, L.; Zhang, M.; Ren, F.Z. A Novel Cell-Penetrating Peptide TAT-A1 Delivers SiRNA into Tumor Cells Selectively. Biochimie 2013, 95, 251–257. [Google Scholar] [CrossRef]
- Lee, Y.W.; Hwang, Y.E.; Lee, J.Y.; Sohn, J.-H.; Sung, B.H.; Kim, S.C. VEGF SiRNA Delivery by a Cancer-Specific Cell-Penetrating Peptide. J. Microbiol. Biotechnol. 2018, 28, 367–374. [Google Scholar] [CrossRef]
- Kulkarni, K.; Watson, G.M.; Sang, J.; Wilce, J.A. Preparation and Cellular Uptake of Bicyclic-peptide Cargo Clicked to Cell Penetrating Peptides. Pept. Sci. 2018, 110, e24037. [Google Scholar] [CrossRef]
- Bidwell, G.L. Peptides for Cancer Therapy: A Drug-Development Opportunity and a Drug-Delivery Challenge. Ther. Deliv. 2012, 3, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Rossoll, W.; Bassell, G.J. Spinal muscular atrophy and a model for survival of motor neuron protein function in axonal ribonucleoprotein complexes. In Cell Biology of the Axon; Springer: Berlin/Heidelberg, Germany, 2009; pp. 87–107. [Google Scholar]
- Chaytow, H.; Huang, Y.-T.; Gillingwater, T.H.; Faller, K.M.E. The Role of Survival Motor Neuron Protein (SMN) in Protein Homeostasis. Cell. Mol. Life Sci. 2018, 75, 3877–3894. [Google Scholar] [CrossRef]
- Brahms, H.; Meheus, L.; de Brabandere, V.; Fischer, U.; Lührmann, R. Symmetrical Dimethylation of Arginine Residues in Spliceosomal Sm Protein B/B’and the Sm-like Protein LSm4, and Their Interaction with the SMN Protein. Rna 2001, 7, 1531–1542. [Google Scholar] [CrossRef]
- Bidwell III, G.L.; Whittom, A.A.; Thomas, E.; Lyons, D.; Hebert, M.D.; Raucher, D. A Thermally Targeted Peptide Inhibitor of Symmetrical Dimethylation Inhibits Cancer-Cell Proliferation. Peptides 2010, 31, 834–841. [Google Scholar] [CrossRef][Green Version]
- Moktan, S.; Raucher, D. Anticancer Activity of Proapoptotic Peptides Is Highly Improved by Thermal Targeting Using Elastin-like Polypeptides. Int. J. Pept. Res. Ther. 2012, 18, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Massodi, I.; Moktan, S.; Rawat, A.; Bidwell III, G.L.; Raucher, D. Inhibition of Ovarian Cancer Cell Proliferation by a Cell Cycle Inhibitory Peptide Fused to a Thermally Responsive Polypeptide Carrier. Int. J. cancer 2010, 126, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.S.; Raucher, D. Anti-Tumor Efficacy of a Therapeutic Peptide Based on Thermo-Responsive Elastin-like Polypeptide in Combination with Gemcitabine. Cancer Lett. 2014, 348, 177–184. [Google Scholar] [CrossRef]
- Wang, H.; Chen, X.; Chen, Y.; Sun, L.; Li, G.; Zhai, M.; Zhai, W.; Kang, Q.; Gao, Y.; Qi, Y. Antitumor Activity of Novel Chimeric Peptides Derived from CyclinD/CDK4 and the Protein Transduction Domain 4. Amino Acids 2013, 44, 499–510. [Google Scholar] [CrossRef]
- Garner, D.K.; Vaughan, M.D.; Hwang, H.J.; Savelieff, M.G.; Berry, S.M.; Honek, J.F.; Lu, Y. Reduction Potential Tuning of the Blue Copper Center in Pseudomonas Aeruginosa Azurin by the Axial Methionine as Probed by Unnatural Amino Acids. J. Am. Chem. Soc. 2006, 128, 15608–15617. [Google Scholar] [CrossRef]
- Yaghoubi, A.; Khazaei, M.; Avan, A.; Hasanian, S.M.; Cho, W.C.; Soleimanpour, S. P28 Bacterial Peptide, as an Anticancer Agent. Front. Oncol. 2020, 10, 1303. [Google Scholar] [CrossRef]
- Mehta, R.R.; Yamada, T.; Taylor, B.N.; Christov, K.; King, M.L.; Majumdar, D.; Lekmine, F.; Tiruppathi, C.; Shilkaitis, A.; Bratescu, L. A Cell Penetrating Peptide Derived from Azurin Inhibits Angiogenesis and Tumor Growth by Inhibiting Phosphorylation of VEGFR-2, FAK and Akt. Angiogenesis 2011, 14, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Warso, M.A.; Richards, J.M.; Mehta, D.; Christov, K.; Schaeffer, C.; Bressler, L.R.; Yamada, T.; Majumdar, D.; Kennedy, S.A.; Beattie, C.W. A First-in-Class, First-in-Human, Phase I Trial of P28, a Non-HDM2-Mediated Peptide Inhibitor of P53 Ubiquitination in Patients with Advanced Solid Tumours. Br. J. Cancer 2013, 108, 1061–1070. [Google Scholar] [CrossRef]
- De Pinto, V.; Messina, A.; Lane, D.J.R.; Lawen, A. Voltage-Dependent Anion-Selective Channel (VDAC) in the Plasma Membrane. FEBS Lett. 2010, 584, 1793–1799. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Maldonado, E.N.; Krelin, Y. VDAC1 at the Crossroads of Cell Metabolism, Apoptosis and Cell Stress. Cell Stress 2017, 1, 11. [Google Scholar] [CrossRef]
- Prezma, T.; Shteinfer, A.; Admoni, L.; Raviv, Z.; Sela, I.; Levi, I.; Shoshan-Barmatz, V. VDAC1-Based Peptides: Novel pro-Apoptotic Agents and Potential Therapeutics for B-Cell Chronic Lymphocytic Leukemia. Cell Death Dis. 2013, 4, e809. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tints, K.; Prink, M.; Neuman, T.; Palm, K. LXXLL Peptide Converts Transportan 10 to a Potent Inducer of Apoptosis in Breast Cancer Cells. Int. J. Mol. Sci. 2014, 15, 5680–5698. [Google Scholar] [CrossRef]
- Grapa, C.M.; Mocan, T.; Gonciar, D.; Zdrehus, C.; Mosteanu, O.; Pop, T.; Mocan, L. Epidermal Growth Factor Receptor and Its Role in Pancreatic Cancer Treatment Mediated by Nanoparticles. Int. J. Nanomed. 2019, 14, 9693. [Google Scholar] [CrossRef]
- Kuroda, Y.; Kato-Kogoe, N.; Tasaki, E.; Murata, E.; Ueda, K.; Abe, M.; Miyamoto, K.; Nakase, I.; Futaki, S.; Tohyama, Y. Oligopeptides Derived from Autophosphorylation Sites of EGF Receptor Suppress EGF-Stimulated Responses in Human Lung Carcinoma A549 Cells. Eur. J. Pharmacol. 2013, 698, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wang, Y.; Zhang, Y.; Li, Y.; Zhang, X.; Xu, Y.; Chen, L.; Li, C.; Ju, Y.; Meng, S. TAT-Mediated Gp96 Transduction to APCs Enhances Gp96-Induced Antiviral and Antitumor T Cell Responses. Vaccine 2013, 31, 545–552. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming Cancer Therapeutic Bottleneck by Drug Repurposing. Signal Transduct. Target. Ther. 2020, 5, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Schrot, J.; Weng, A.; Melzig, M.F. Ribosome-Inactivating and Related Proteins. Toxins 2015, 7, 1556–1615. [Google Scholar] [CrossRef]
- Shin, M.C.; Zhang, J.; David, A.E.; Trommer, W.E.; Kwon, Y.M.; Min, K.A.; Kim, J.H.; Yang, V.C. Chemically and Biologically Synthesized CPP-Modified Gelonin for Enhanced Anti-Tumor Activity. J. Control. Release 2013, 172, 169–178. [Google Scholar] [CrossRef]
- Shin, M.C.; Zhang, J.; Min, K.A.; He, H.; David, A.E.; Huang, Y.; Yang, V.C. PTD-Modified ATTEMPTS for Enhanced Toxin-Based Cancer Therapy: An in Vivo Proof-of-Concept Study. Pharm. Res. 2015, 32, 2690–2703. [Google Scholar] [CrossRef]
- Fukazawa, T.; Walter, B.; Owen-Schaub, L.B. Adenoviral Bid Overexpression Induces Caspase-Dependent Cleavage of Truncated Bid and P53-Independent Apoptosis in Human Non-Small Cell Lung Cancers. J. Biol. Chem. 2003, 278, 25428–25434. [Google Scholar] [CrossRef]
- Orzechowska, E.J.; Kozlowska, E.; Czubaty, A.; Kozlowski, P.; Staron, K.; Trzcinska-Danielewicz, J. Controlled Delivery of BID Protein Fused with TAT Peptide Sensitizes Cancer Cells to Apoptosis. BMC Cancer 2014, 14, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; Jang, C.; Lee, K.-A. Polo-like Kinases (Plks), a Key Regulator of Cell Cycle and New Potential Target for Cancer Therapy. Dev. Reprod. 2014, 18, 65. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Hou, G.; Yang, J.; Yuan, J.; Wang, C.; Chai, T.; Liu, Z. Effects of PLK1 on Proliferation, Invasion and Metastasis of Gastric Cancer Cells through Epithelial-Mesenchymal Transition. Oncol. Lett. 2018, 16, 5739–5744. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Chae, M.K.; Lee, C.; Yim, M.S.; Bang, J.K.; Ryu, E.K. Enhanced Cellular Uptake of a TAT-Conjugated Peptide Inhibitor Targeting the Polo-Box Domain of Polo-like Kinase 1. Amino Acids 2014, 46, 2595–2603. [Google Scholar] [CrossRef]
- Dupont, E.; Prochiantz, A.; Joliot, A. Penetratin story: An overview. In Cell-Penetrating Peptides; Springer: Totowa, NJ, USA, 2011; pp. 21–29. [Google Scholar]
- Alves, I.D.; Carré, M.; Montero, M.-P.; Castano, S.; Lecomte, S.; Marquant, R.; Lecorché, P.; Burlina, F.; Schatz, C.; Sagan, S. A Proapoptotic Peptide Conjugated to Penetratin Selectively Inhibits Tumor Cell Growth. Biochim. Biophys. Acta BBA-Biomembranes 2014, 1838, 2087–2098. [Google Scholar] [CrossRef]
- Koci, L.; Chlebova, K.; Hyzdalova, M.; Hofmanova, J.; Jira, M.; Kysela, P.; Kozubik, A.; Kala, Z.; Krejci, P. Apoptosis Inhibitor 5 (API-5; AAC-11; FIF) Is Upregulated in Human Carcinomas in Vivo. Oncol. Lett. 2012, 3, 913–916. [Google Scholar]
- Rigou, P.; Piddubnyak, V.; Faye, A.; Rain, J.; Michel, L.; Calvo, F.; Poyet, J. The Antiapoptotic Protein AAC-11 Interacts with and Regulates Acinus-mediated DNA Fragmentation. EMBO J. 2009, 28, 1576–1588. [Google Scholar] [CrossRef] [PubMed]
- Song, K.-H.; Kim, S.-H.; Noh, K.H.; Bae, H.C.; Kim, J.H.; Lee, H.-J.; Song, J.; Kang, T.H.; Kim, D.-W.; Oh, S.-J. Apoptosis Inhibitor 5 Increases Metastasis via Erk-Mediated MMP Expression. BMB Rep. 2015, 48, 330. [Google Scholar] [CrossRef] [PubMed]
- Jagot-Lacoussiere, L.; Kotula, E.; Villoutreix, B.O.; Bruzzoni-Giovanelli, H.; Poyet, J.-L. A Cell-Penetrating Peptide Targeting AAC-11 Specifically Induces Cancer Cells Death. Cancer Res. 2016, 76, 5479–5490. [Google Scholar] [CrossRef] [PubMed]
- Pasquereau-Kotula, E.; Habault, J.; Kroemer, G.; Poyet, J.-L. The Anticancer Peptide RT53 Induces Immunogenic Cell Death. PLoS ONE 2018, 13, e0201220. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-Inducible Factors: Mediators of Cancer Progression and Targets for Cancer Therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef]
- Ziello, J.E.; Jovin, I.S.; Huang, Y. Hypoxia-Inducible Factor (HIF)-1 Regulatory Pathway and Its Potential for Therapeutic Intervention in Malignancy and Ischemia. Yale J. Biol. Med. 2007, 80, 51. [Google Scholar] [PubMed]
- Mylonis, I.; Kourti, M.; Samiotaki, M.; Panayotou, G.; Simos, G. Mortalin-Mediated and ERK-Controlled Targeting of HIF-1α to Mitochondria Confers Resistance to Apoptosis under Hypoxia. J. Cell Sci. 2017, 130, 466–479. [Google Scholar] [CrossRef]
- Karagiota, A.; Kourti, M.; Simos, G.; Mylonis, I. HIF-1α-Derived Cell-Penetrating Peptides Inhibit ERK-Dependent Activation of HIF-1 and Trigger Apoptosis of Cancer Cells under Hypoxia. Cell. Mol. Life Sci. 2019, 76, 809–825. [Google Scholar] [CrossRef]
- Qadir, M.I.; Parveen, A.; Ali, M. Cdc42: Role in Cancer Management. Chem. Biol. Drug Des. 2015, 86, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Tetley, G.J.N.; Murphy, N.P.; Bonetto, S.; Ivanova-Berndt, G.; Revell, J.; Mott, H.R.; Cooley, R.N.; Owen, D. The Discovery and Maturation of Peptide Biologics Targeting the Small G-Protein Cdc42: A Bioblockade for Ras-Driven Signaling. J. Biol. Chem. 2020, 295, 2866–2884. [Google Scholar] [CrossRef]
- Palsuledesai, C.C.; Distefano, M.D. Protein Prenylation: Enzymes, Therapeutics, and Biotechnology Applications. ACS Chem. Biol. 2015, 10, 51–62. [Google Scholar] [CrossRef]
- Klimpel, A.; Stillger, K.; Wiederstein, J.L.; Krüger, M.; Neundorf, I. Cell-permeable CaaX-peptides Affect K-Ras Downstream Signaling and Promote Cell Death in Cancer Cells. FEBS J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hurd, C.A.; Mott, H.R.; Owen, D. Therapeutic Peptides Targeting the Ras Superfamily. Pept. Sci. 2020, 112, e24165. [Google Scholar] [CrossRef]
- Dougherty, P.G.; Sahni, A.; Pei, D. Understanding Cell Penetration of Cyclic Peptides. Chem. Rev. 2019, 119, 10241–10287. [Google Scholar] [CrossRef]
- Dougherty, P.G.; Wen, J.; Pan, X.; Koley, A.; Ren, J.-G.; Sahni, A.; Basu, R.; Salim, H.; Appiah Kubi, G.; Qian, Z. Enhancing the Cell Permeability of Stapled Peptides with a Cyclic Cell-Penetrating Peptide. J. Med. Chem. 2019, 62, 10098–10107. [Google Scholar] [CrossRef]
- Falzone, L.; Salomone, S.; Libra, M. Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar] [CrossRef] [PubMed]
- Nurgali, K.; Jagoe, R.T.; Abalo, R. Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae? Front. Pharmacol. 2018, 9, 245. [Google Scholar] [CrossRef]
- Ud Din, F.; Aman, W.; Ullah, I.; Qureshi, O.S.; Mustapha, O.; Shafique, S.; Zeb, A. Effective Use of Nanocarriers as Drug Delivery Systems for the Treatment of Selected Tumors. Int. J. Nanomedicine 2017, 12, 7291. [Google Scholar] [CrossRef]
- Mahato, R.; Tai, W.; Cheng, K. Prodrugs for Improving Tumor Targetability and Efficiency. Adv. Drug Deliv. Rev. 2011, 63, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Zahreddine, H.; Borden, K. Mechanisms and Insights into Drug Resistance in Cancer. Front. Pharmacol. 2013, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Szabó, I.; Orbán, E.; Schlosser, G.; Hudecz, F.; Bánóczi, Z. Cell-Penetrating Conjugates of Pentaglutamylated Methotrexate as Potential Anticancer Drugs against Resistant Tumor Cells. Eur. J. Med. Chem. 2016, 115, 361–368. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Fang, Y.; Meng, F.; Zhang, J.; Cheng, R.; Deng, C.; Zhong, Z. Selective Cell Penetrating Peptide-functionalized Polymersomes Mediate Efficient and Targeted Delivery of Methotrexate Disodium to Human Lung Cancer In Vivo. Adv. Healthc. Mater. 2018, 7, 1701135. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Qian, J.; Cao, S.; Yang, Z.; Pang, Z.; Pan, S.; Fan, L.; Xi, Z.; Jiang, X.; Zhang, Q. Precise Glioma Targeting of and Penetration by Aptamer and Peptide Dual-Functioned Nanoparticles. Biomaterials 2012, 33, 5115–5123. [Google Scholar] [CrossRef]
- De Jong, H.; Bonger, K.M.; Löwik, D.W.P.M. Activatable Cell-Penetrating Peptides: 15 Years of Research. RSC Chem. Biol. 2020, 1, 192–203. [Google Scholar] [CrossRef]
- Gao, H.; Zhang, S.; Cao, S.; Yang, Z.; Pang, Z.; Jiang, X. Angiopep-2 and Activatable Cell-Penetrating Peptide Dual-Functionalized Nanoparticles for Systemic Glioma-Targeting Delivery. Mol. Pharm. 2014, 11, 2755–2763. [Google Scholar] [CrossRef]
- Kadari, A.; Pooja, D.; Gora, R.H.; Gudem, S.; Kolapalli, V.R.M.; Kulhari, H.; Sistla, R. Design of Multifunctional Peptide Collaborated and Docetaxel Loaded Lipid Nanoparticles for Antiglioma Therapy. Eur. J. Pharm. Biopharm. 2018, 132, 168–179. [Google Scholar] [CrossRef]
- Gao, W.; Xiang, B.; Meng, T.-T.; Liu, F.; Qi, X.-R. Chemotherapeutic Drug Delivery to Cancer Cells Using a Combination of Folate Targeting and Tumor Microenvironment-Sensitive Polypeptides. Biomaterials 2013, 34, 4137–4149. [Google Scholar] [CrossRef]
- Ma, P.; Mumper, R.J. Paclitaxel Nano-Delivery Systems: A Comprehensive Review. J. Nanomed. Nanotechnol. 2013, 4, 1000164. [Google Scholar] [CrossRef]
- Moktan, S.; Ryppa, C.; Kratz, F.; Raucher, D. A Thermally Responsive Biopolymer Conjugated to an Acid-Sensitive Derivative of Paclitaxel Stabilizes Microtubules, Arrests Cell Cycle, and Induces Apoptosis. Investig. New Drugs 2012, 30, 236–248. [Google Scholar] [CrossRef]
- Zhao, B.-X.; Zhao, Y.; Huang, Y.; Luo, L.-M.; Song, P.; Wang, X.; Chen, S.; Yu, K.-F.; Zhang, X.; Zhang, Q. The Efficiency of Tumor-Specific PH-Responsive Peptide-Modified Polymeric Micelles Containing Paclitaxel. Biomaterials 2012, 33, 2508–2520. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wang, T.; Perche, F.; Taigind, A.; Torchilin, V.P. Enhanced Anticancer Activity of Nanopreparation Containing an MMP2-Sensitive PEG-Drug Conjugate and Cell-Penetrating Moiety. Proc. Natl. Acad. Sci. 2013, 110, 17047–17052. [Google Scholar] [CrossRef]
- Liu, Y.; Ran, R.; Chen, J.; Kuang, Q.; Tang, J.; Mei, L.; Zhang, Q.; Gao, H.; Zhang, Z.; He, Q. Paclitaxel Loaded Liposomes Decorated with a Multifunctional Tandem Peptide for Glioma Targeting. Biomaterials 2014, 35, 4835–4847. [Google Scholar] [CrossRef]
- Duan, Z.; Chen, C.; Qin, J.; Liu, Q.; Wang, Q.; Xu, X.; Wang, J. Cell-Penetrating Peptide Conjugates to Enhance the Antitumor Effect of Paclitaxel on Drug-Resistant Lung Cancer. Drug Deliv. 2017, 24, 752–764. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Bai, L.; Lin, L.; Wang, S.; Yin, X. Paclitaxel-Loaded Nanoparticles Decorated with Bivalent Fragment HAb18 F (Ab’) 2 and Cell Penetrating Peptide for Improved Therapeutic Effect on Hepatocellular Carcinoma. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1076–1084. [Google Scholar] [CrossRef]
- Cappetta, D.; De Angelis, A.; Sapio, L.; Prezioso, L.; Illiano, M.; Quaini, F.; Rossi, F.; Berrino, L.; Naviglio, S.; Urbanek, K. Oxidative Stress and Cellular Response to Doxorubicin: A Common Factor in the Complex Milieu of Anthracycline Cardiotoxicity. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef]
- Aroui, S.; Mili, D.; Brahim, S.; De Waard, M.; Kenani, A. Doxorubicin Coupled to Penetratin Promotes Apoptosis in CHO Cells by a Mechanism Involving C-Jun NH2-Terminal Kinase. Biochem. Biophys. Res. Commun. 2010, 396, 908–914. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, X.; Cao, Z.; Xu, W.; Zhang, J.; Gong, M. Self-Assembled Peptide (CADY-1) Improved the Clinical Application of Doxorubicin. Int. J. Pharm. 2012, 434, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.; Perkins, E.; Kratz, F.; Raucher, D. Cell Penetrating Peptides Fused to a Thermally Targeted Biopolymer Drug Carrier Improve the Delivery and Antitumor Efficacy of an Acid-Sensitive Doxorubicin Derivative. Int. J. Pharm. 2012, 436, 825–832. [Google Scholar] [CrossRef]
- Shi, N.-Q.; Gao, W.; Xiang, B.; Qi, X.-R. Enhancing Cellular Uptake of Activable Cell-Penetrating Peptide–Doxorubicin Conjugate by Enzymatic Cleavage. Int. J. Nanomed. 2012, 7, 1613. [Google Scholar]
- Zhuo, S.; Zhang, F.; Yu, J.; Zhang, X.; Yang, G.; Liu, X. PH-Sensitive Biomaterials for Drug Delivery. Molecules 2020, 25, 5649. [Google Scholar] [CrossRef]
- Cheng, H.; Zhu, J.-Y.; Xu, X.-D.; Qiu, W.-X.; Lei, Q.; Han, K.; Cheng, Y.-J.; Zhang, X.-Z. Activable Cell-Penetrating Peptide Conjugated Prodrug for Tumor Targeted Drug Delivery. ACS Appl. Mater. Interfaces 2015, 7, 16061–16069. [Google Scholar] [CrossRef]
- Mei, Y.Q.; Kai, L.; Qun, S.; Qingyue, L.; Jinghui, H.; Fangxuan, H.; Ren-Wang, J. Reversal of Multidrug Resistance in Cancer by Multi-Functional Flavonoids. Front. Oncol. 2019, 9, 487. [Google Scholar]
- Pan, L.; Liu, J.; He, Q.; Wang, L.; Shi, J. Overcoming Multidrug Resistance of Cancer Cells by Direct Intranuclear Drug Delivery Using TAT-Conjugated Mesoporous Silica Nanoparticles. Biomaterials 2013, 34, 2719–2730. [Google Scholar] [CrossRef]
- Bruno, B.J.; Miller, G.D.; Lim, C.S. Basics and Recent Advances in Peptide and Protein Drug Delivery. Ther. Deliv. 2013, 4, 1443–1467. [Google Scholar] [CrossRef]
- Soudy, R.; Chen, C.; Kaur, K. Novel Peptide–Doxorubucin Conjugates for Targeting Breast Cancer Cells Including the Multidrug Resistant Cells. J. Med. Chem. 2013, 56, 7564–7573. [Google Scholar] [CrossRef]
- Biswas, S.; Deshpande, P.P.; Perche, F.; Dodwadkar, N.S.; Sane, S.D.; Torchilin, V.P. Octa-Arginine-Modified Pegylated Liposomal Doxorubicin: An Effective Treatment Strategy for Non-Small Cell Lung Cancer. Cancer Lett. 2013, 335, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Shamay, Y.; Shpirt, L.; Ashkenasy, G.; David, A. Complexation of Cell-Penetrating Peptide–Polymer Conjugates with Polyanions Controls Cells Uptake of HPMA Copolymers and Anti-Tumor Activity. Pharm. Res. 2014, 31, 768–779. [Google Scholar] [CrossRef]
- Li, J.; Liu, F.; Shao, Q.; Min, Y.; Costa, M.; Yeow, E.K.L.; Xing, B. Enzyme-responsive Cell-penetrating Peptide Conjugated Mesoporous Silica Quantum Dot Nanocarriers for Controlled Release of Nucleus-targeted Drug Molecules and Real-time Intracellular Fluorescence Imaging of Tumor Cells. Adv. Healthc. Mater. 2014, 3, 1230–1239. [Google Scholar] [CrossRef]
- Liu, Z.; Xiong, M.; Gong, J.; Zhang, Y.; Bai, N.; Luo, Y.; Li, L.; Wei, Y.; Liu, Y.; Tan, X. Legumain Protease-Activated TAT-Liposome Cargo for Targeting Tumours and Their Microenvironment. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef]
- Apte, A.; Koren, E.; Koshkaryev, A.; Torchilin, V.P. Doxorubicin in TAT Peptide-Modified Multifunctional Immunoliposomes Demonstrates Increased Activity against Both Drug-Sensitive and Drug-Resistant Ovarian Cancer Models. Cancer Biol. Ther. 2014, 15, 69–80. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, Y.; Xie, X.; Cai, X.; Zhang, H.; Gong, W.; Wang, Z.; Mei, X. PEGylated Liposomes with NGR Ligand and Heat-Activable Cell-Penetrating Peptide–Doxorubicin Conjugate for Tumor-Specific Therapy. Biomaterials 2014, 35, 4368–4381. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.K.; Cho, H.-Y.; Kim, K.-J.; Choi, J.-W. In Situ Monitoring of Doxorubicin Release from Biohybrid Nanoparticles Modified with Antibody and Cell-Penetrating Peptides in Breast Cancer Cells Using Surface-Enhanced Raman Spectroscopy. Biosens. Bioelectron. 2015, 71, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Morshed, R.A.; Muroski, M.E.; Dai, Q.; Wegscheid, M.L.; Auffinger, B.; Yu, D.; Han, Y.; Zhang, L.; Wu, M.; Cheng, Y. Cell-Penetrating Peptide-Modified Gold Nanoparticles for the Delivery of Doxorubicin to Brain Metastatic Breast Cancer. Mol. Pharm. 2016, 13, 1843–1854. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.-H.; Kopecček, J. Enhancing Accumulation and Penetration of HPMA Copolymer–Doxorubicin Conjugates in 2D and 3D Prostate Cancer Cells via IRGD Conjugation with an MMP-2 Cleavable Spacer. J. Am. Chem. Soc. 2015, 137, 6726–6729. [Google Scholar] [CrossRef]
- Darwish, S.; Sadeghiani, N.; Fong, S.; Mozaffari, S.; Hamidi, P.; Withana, T.; Yang, S.; Tiwari, R.K.; Parang, K. Synthesis and Antiproliferative Activities of Doxorubicin Thiol Conjugates and Doxorubicin-SS-Cyclic Peptide. Eur. J. Med. Chem. 2019, 161, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Movafegh, B.; Jalal, R.; Mohammadi, Z.; Aldaghi, S.A. Poly-L-Arginine: Enhancing Cytotoxicity and Cellular Uptake of Doxorubicin and Necrotic Cell Death. Anti-Cancer Agents Med. Chem. Formerly Curr. Med. Chem. Agents 2018, 18, 1448–1456. [Google Scholar] [CrossRef]
- Yu, M.; Li, X.; Huang, X.; Zhang, J.; Zhang, Y.; Wang, H. New Cell-Penetrating Peptide (KRP) with Multiple Physicochemical Properties Endows Doxorubicin with Tumor Targeting and Improves Its Therapeutic Index. ACS Appl. Mater. Interfaces 2018, 11, 2448–2458. [Google Scholar] [CrossRef]
- Hagner, N.; Joerger, M. Cancer Chemotherapy: Targeting Folic Acid Synthesis. Cancer Manag. Res. 2010, 2, 293. [Google Scholar] [PubMed]
- Miklán, Z.; Orbán, E.; Bánóczi, Z.; Hudecz, F. New Pemetrexed-peptide Conjugates: Synthesis, Characterization and in Vitro Cytostatic Effect on Non-small Cell Lung Carcinoma (NCI-H358) and Human Leukemia (HL-60) Cells. J. Pept. Sci. 2011, 17, 805–811. [Google Scholar] [CrossRef]
- Koshkaryev, A.; Piroyan, A.; Torchilin, V.P. Bleomycin in Octaarginine-Modified Fusogenic Liposomes Results in Improved Tumor Growth Inhibition. Cancer Lett. 2013, 334, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Wang, H.; Niu, R.; Ding, D. Drug Delivery with Nanospherical Supramolecular Cell Penetrating Peptide–Taxol Conjugates Containing a High Drug Loading. J. Colloid Interface Sci. 2015, 453, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, F.; Li, G.; Zhou, Y.; Yang, Y. Twin-Arginine Translocation Peptide Conjugated Epirubicin-Loaded Nanoparticles for Enhanced Tumor Penetrating and Targeting. J. Pharm. Sci. 2015, 104, 4185–4196. [Google Scholar] [CrossRef]
- Soler, M.; González-Bártulos, M.; Figueras, E.; Ribas, X.; Costas, M.; Massaguer, A.; Planas, M.; Feliu, L. Enzyme-Triggered Delivery of Chlorambucil from Conjugates Based on the Cell-Penetrating Peptide BP16. Org. Biomol. Chem. 2015, 13, 1470–1480. [Google Scholar] [CrossRef]
- Florea, A.-M.; Büsselberg, D. Cisplatin as an Anti-Tumor Drug: Cellular Mechanisms of Activity, Drug Resistance and Induced Side Effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef] [PubMed]
- Aroui, S.; Dardevet, L.; Ajmia, W.B.; de Boisvilliers, M.; Perrin, F.; Laajimi, A.; Boumendjel, A.; Kenani, A.; Muller, J.M.; De Waard, M. A Novel Platinum–Maurocalcine Conjugate Induces Apoptosis of Human Glioblastoma Cells by Acting through the ROS-ERK/AKT-P53 Pathway. Mol. Pharm. 2015, 12, 4336–4348. [Google Scholar] [CrossRef] [PubMed]
- Izabela, R.; Jarosław, R.; Magdalena, A.; Piotr, R.; Ivan, K. Transportan 10 Improves the Anticancer Activity of Cisplatin. Naunyn. Schmiedebergs. Arch. Pharmacol. 2016, 389, 485–497. [Google Scholar] [CrossRef]
- Gronewold, A.; Horn, M.; Ranđelović, I.; Tóvári, J.; Vázquez, S.M.; Schomäcker, K.; Neundorf, I. Characterization of a Cell-penetrating Peptide with Potential Anticancer Activity. ChemMedChem 2017, 12, 42. [Google Scholar] [CrossRef]
- Jia, L.; Gorman, G.S.; Coward, L.U.; Noker, P.E.; McCormick, D.; Horn, T.L.; Harder, J.B.; Muzzio, M.; Prabhakar, B.; Ganesh, B. Preclinical Pharmacokinetics, Metabolism, and Toxicity of Azurin-P28 (NSC745104) a Peptide Inhibitor of P53 Ubiquitination. Cancer Chemother. Pharmacol. 2011, 68, 513–524. [Google Scholar] [CrossRef]
- Gaston, J.; Maestrali, N.; Lalle, G.; Gagnaire, M.; Masiero, A.; Dumas, B.; Dabdoubi, T.; Radošević, K.; Berne, P.-F. Intracellular Delivery of Therapeutic Antibodies into Specific Cells Using Antibody-Peptide Fusions. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Reissmann, S. Cell Penetration: Scope and Limitations by the Application of Cell-penetrating Peptides. J. Pept. Sci. 2014, 20, 760–784. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Sun, L.; Ye, J.; Liu, E.; Chen, S.; Liang, Q.; Shin, M.C.; Yang, V.C. Enzyme-Triggered, Cell Penetrating Peptide-Mediated Delivery of Anti-Tumor Agents. J. Control. Release 2016, 240, 67–76. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CPP | Cargo | Targeted Tumor | Function | Ref. |
|---|---|---|---|---|
| PT24 | Cas9 | HeLa and the human lung cancer | Delivery of Cas9 in a single incubation step/high efficiency/low toxicity | [43] |
| PPABLG | Cas9 | HeLa tumor tissue | Suppressing tumor growth/prolonging animal survival rates | [44] |
| TAT | DNA nanopore | Human Burkitt’s lymphoma | Tumor cell detection with low cytotoxicity | [45] |
| TAT | BCL-2 siRNA | Human breast cancer | Knockdown of the Bcl-2 protein/Inhibition of cancer cell migration | [46] |
| R8 | RAC1 siRNA | Human lung carcinoma and ovarian adenocarcinoma | Significantly decreasing the oncogenic RAC1 mRNA levels | [47] |
| cRGD | GFP and Luc mRNA | Human primary glioblastoma | Improving tumor accumulation and potent gene expression | [48] |
| RL2 | EGFP siRNA | Human lung adenocarcinoma and epidermoid carcinoma | Substantially decreasing cancer cell viability | [49] |
| PepFect 6/TP10 | HPRT1 siRNA | Human hepatocellular carcinoma | Significantly reducing the expression of HPRT1 without acute toxicity | [50] |
| PepFect 14 | SCOs | HeLa cell line | Significant SCO-mediated splice-correction | [51] |
| PepFect 14 | HPRT1 siRNA | Human hepatocellular carcinoma | Induction of the knockdown of endogenous genes | [52] |
| PepFect 14/28 | Firefly luciferase siRNA | Glioblastoma | Increasing gene-silencing efficiency | [53] |
| gH625 | anti-GFP siRNA | Human triple negative breast cancer | Downregulation of GFP expression | [54] |
| CH2R4H2C | VEGF siRNA | Murine sarcoma | Sufficiently suppressing neovascularization on the tumor surface | [55] |
| TP–LyP-1 | ID4-specific siRNA | Human ovarian cancer | Suppressing the growth of established tumors and significantly improving survival | [56] |
| TP-iRGD | TNFα siRNA | Human vestibular schwannomas | Silencing genes and protein secretion | [57] |
| R9 | Plk1 siRNA | Human breast cancer | Inhibition of breast tumor growth | [58] |
| 599 Peptide | CIP2A siRNA | Oral cancer cells | Significant CIP2A mRNA and protein silencing resulting in the decreasing of oral cancer cell invasiveness | [59] |
| TAT-A1 | GAPDH siRNA | Human hepatocellular carcinoma | Decreasing mRNA levels | [60] |
| BR2/R9 | VEGF siRNA | Human colon cancer cells/HeLa cells | VEGF silencing/Improving antitumor efficacy without toxicity | [61] |
| CPP | Cargo | Targeted Tumor | Function | Ref. |
|---|---|---|---|---|
| SynB1 | ELP1-GRG | Human breast cancer | Disruption of SMN function | [88] |
| SynB1 | ELP1-KLAK | Human breast cancer | Mitochondria disruption/Apoptosis induction | [89] |
| Bac | p21-ELP | Pancreatic tumor cells/Human ovarian cancer | Cell cycle inhibition | [90,91] |
| PTD4 | cyclin/CDK4 analogs | Murine sarcoma/Hepatocellular carcinoma | Induction of cell cycle arrest and apoptosis | [92] |
| p28 | - | Human melanoma cancer/Human colon carcinoma | Inhibition of angiogenesis and tumor growth by inhibiting the phosphorylation of VEGFR-2 and/or p53 ubiquitination | [93,94] |
| Antp-LP4/N-Terminal-Antp | - | B-cell chronic lymphocytic leukemia | Induction of cell death | [95] |
| TP10 | LXXLL-motif of the human SRC-1 | Breast cancer cells | Induction of dose-dependent cell death | [96] |
| LMWP | Gelonin | Murine adenocarcinoma xenograft tumor | Inhibition of protein translation | [97] |
| TAT | Gelonin/anti-CEA monoclonal antibody | Human colorectal adenocarcinoma | Inhibition of tumor growth | [98] |
| TAT | BID protein | Prostate and non-small human lung cancer | Induction of apoptosis through the TRAIL pathway | [99] |
| TAT | PLHSpT | Human colon adenocarcinoma/Human epidermoid carcinoma | Targeting of Plk1 and the induction of apoptotic cell death | [100] |
| TAT | ETD | Human hepatocellular carcinoma | Inhibition of ERK-dependent activation of HIF-1 and apoptosis triggering | [101] |
| Pen | KLA | Seven human tumor cell lines | Impacting on mitochondria tubular organization/Apoptosis induction | [102] |
| Pen | G7-B7M2 | Human breast cancer | Blockade of the interactions of Grb7 | [85] |
| RT53 | - | Melanoma xenograft tumors/Mouse Fibrosarcoma | Targeting of AAC-11 and the induction of cancer cell death/Induction of immunogenic cell death | [103,104] |
| R9 | C1 | Human lung cancer | Inhibiting Cdc42, decreasing proliferation, preventing motility and invasion | [105] |
| sC18* | CaaX motif | Human pancreatic cancer | Affect K-Ras downstream signaling and promote cell death | [106] |
| CCP9 | PDI | Human osteosarcoma SJSA-1 cells | Inhibitor against the MDM2-p53 interaction/Induction of p53-dependent apoptosis | [107] |
| Peptide 38 | Raf dimers | Malignant melanoma cells | Exhibit anti-proliferative activity and inhibit paradoxical signaling | [108] |
| Chemotherapeutic | CPP | Targeted Tumor | Function | Ref. |
|---|---|---|---|---|
| DTX | Angiopep-2/R8 | Orthotopic glioma | Higher glioma localization | [140] |
| DTX | Angiopep-2 | Glioblastoma | Selective targeting with higher accumulation | [141] |
| DTX | R9 | Human mouth epidermoid carcinoma | Higher antitumor efficacy and lower systemic toxicity | [142] |
| MTX | Pen/R8 | Breast cancer cell | Not mediating in vitro cytotoxic effects | [143] |
| MTX | RLWMRWYSPRTRAYGC | Human lung cancer | Inhibition of tumor progression/Improvement of survival rates | [144] |
| Pemetrexed | R8 | Non-small cell lung carcinoma/Human leukemia cells | Higher selective cytostatic effects | [145] |
| Bleomycin | R8 | Murine mammary carcinoma | Facilitating BLM interaction with nuclear material | [146] |
| Taxol | EEGRLYMRYYSPTTRRYG | Human hepatocellular carcinoma cells | Efficient cytotoxicity comparable to that of free Taxol | [147] |
| Epirubicin | TAT | Murine hepatic cancer | Improvement of antitumor activity and biodistribution | [148] |
| Chlorambucil | BP16 | Breast cancer cell/HeLa cells | Selective release of CLB in lysosomal compartments | [149] |
| Cisplatin | D-MCa | Human glioblastoma cells | Induction of apoptosis by triggering the ROS-ERK/AKT-p53 pathway | [150] |
| Cisplatin | TP10/PTD4 | Human osteosarcoma | Nontoxic anticancer activity | [151] |
| Actinomycin D | (sC18)2 | Breast cancer cells | Decreasing cancer cell viability | [152] |
| CPP | Target cell and tissue | Function | Ref. |
|---|---|---|---|
| SynB1 | Breast cancer cells | Induction of cell cycle arrest and apoptosis | [153] |
| H7K(R2)2 | Breast tumor-bearing nude mice | Inhibition of tumor growth | [154] |
| TAT | Non-small cell lung cancer xenograft mouse models | Tumor growth inhibition/Induction of apoptosis in tumor tissues | [155] |
| R8-RGD | Glioma-bearing mice | Prolonging survival in intracranial C6 glioma-bearing mice | [156] |
| TAT and LMWP | Drug-resistant lung cancer | Influencing mitosis/Inhibition of tumor growth | [157] |
| R9 | Hepatocellular carcinoma | Maximization of the therapeutic efficacy for targeting and effective endocytosis | [158] |
| CPP | Target Cell and Tissue | Function | Ref. |
|---|---|---|---|
| Pen | CHO cells | Initiation of apoptosis through involving c-Jun NH2-terminal kinase | [162] |
| CADY-1 | Mouse lymphocytic leukemia | Increasing the blood residence time and/or therapeutic index of the drug | [163] |
| SynB1 | Murine breast tumors | Tumor inhibition 2-fold higher than that of free doxorubicin | [164] |
| DGGDGGDGGDGPLGLAGrrrrrrrrrC | Breast cancer/Fibrosarcoma | Antiproliferative effect with less toxicity | [165] |
| CR8G3PK6 | Hepatic tumor xenograft mouse models | Significant tumor growth inhibition | [166] |
| TAT | Breast cancer cells | Decreasing cancer cell viability | [167] |
| Decapeptide 18-4 | Breast cancer cells | 4-fold more antitumor effects towards cancer cells | [168] |
| R8 | Non-small cell lung cancer | Induction of higher levels of apoptosis /inhibition of tumor growth/tumor weight reduction | [169] |
| P-R8 | Murine model of B16-F10 lung metastasis | Significantly prolonging survival rates in mice | [170] |
| TAT | Lung adenocarcinoma | Increasing Doxorubicin accumulation and inducing higher tumor elimination | [171] |
| AAN-TAT | Breast cancer cells | Limited toxicity/Increasing the tumoricidal effects of doxorubicin | [172] |
| TAT | Drug-resistant ovarian cancer models | Enhancing cytotoxicity in drug-resistant cells | [173] |
| NGR peptide | Fibrosarcoma xenograft mouse models | Inhibition of tumor growth | [174] |
| TAT | Brain metastatic breast cancer | Improving survival rate in xenograft mouse models | [175] |
| iRGD | Prostate cancer cells | Enhancing the accumulation and penetration of doxorubicin | [176] |
| C(WR)4K | Human ovarian cancer/Fibrosarcoma | Localization in the nucleus/reduction of toxicity | [177] |
| Poly-L-arginine | Human prostate cancer | Facilitating doxorubicin uptake and increasing its intracellular concentration | [178] |
| KRP | Human osteosarcoma cell | Pronounced biodistribution/Selective accumulation in tumor tissues | [179] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shoari, A.; Tooyserkani, R.; Tahmasebi, M.; Löwik, D.W.P.M. Delivery of Various Cargos into Cancer Cells and Tissues via Cell-Penetrating Peptides: A Review of the Last Decade. Pharmaceutics 2021, 13, 1391. https://doi.org/10.3390/pharmaceutics13091391
Shoari A, Tooyserkani R, Tahmasebi M, Löwik DWPM. Delivery of Various Cargos into Cancer Cells and Tissues via Cell-Penetrating Peptides: A Review of the Last Decade. Pharmaceutics. 2021; 13(9):1391. https://doi.org/10.3390/pharmaceutics13091391
Chicago/Turabian StyleShoari, Alireza, Raheleh Tooyserkani, Mehdi Tahmasebi, and Dennis W. P. M. Löwik. 2021. "Delivery of Various Cargos into Cancer Cells and Tissues via Cell-Penetrating Peptides: A Review of the Last Decade" Pharmaceutics 13, no. 9: 1391. https://doi.org/10.3390/pharmaceutics13091391
APA StyleShoari, A., Tooyserkani, R., Tahmasebi, M., & Löwik, D. W. P. M. (2021). Delivery of Various Cargos into Cancer Cells and Tissues via Cell-Penetrating Peptides: A Review of the Last Decade. Pharmaceutics, 13(9), 1391. https://doi.org/10.3390/pharmaceutics13091391