
New RNA-Based Breakthroughs in Alzheimer’s Disease Diagnosis and Therapeutics


Abstract
:
1. Dementia and Alzheimer’s Disease Impact
2. Alzheimer’s Disease Characterization
2.1. Sporadic AD vs. Familiar AD
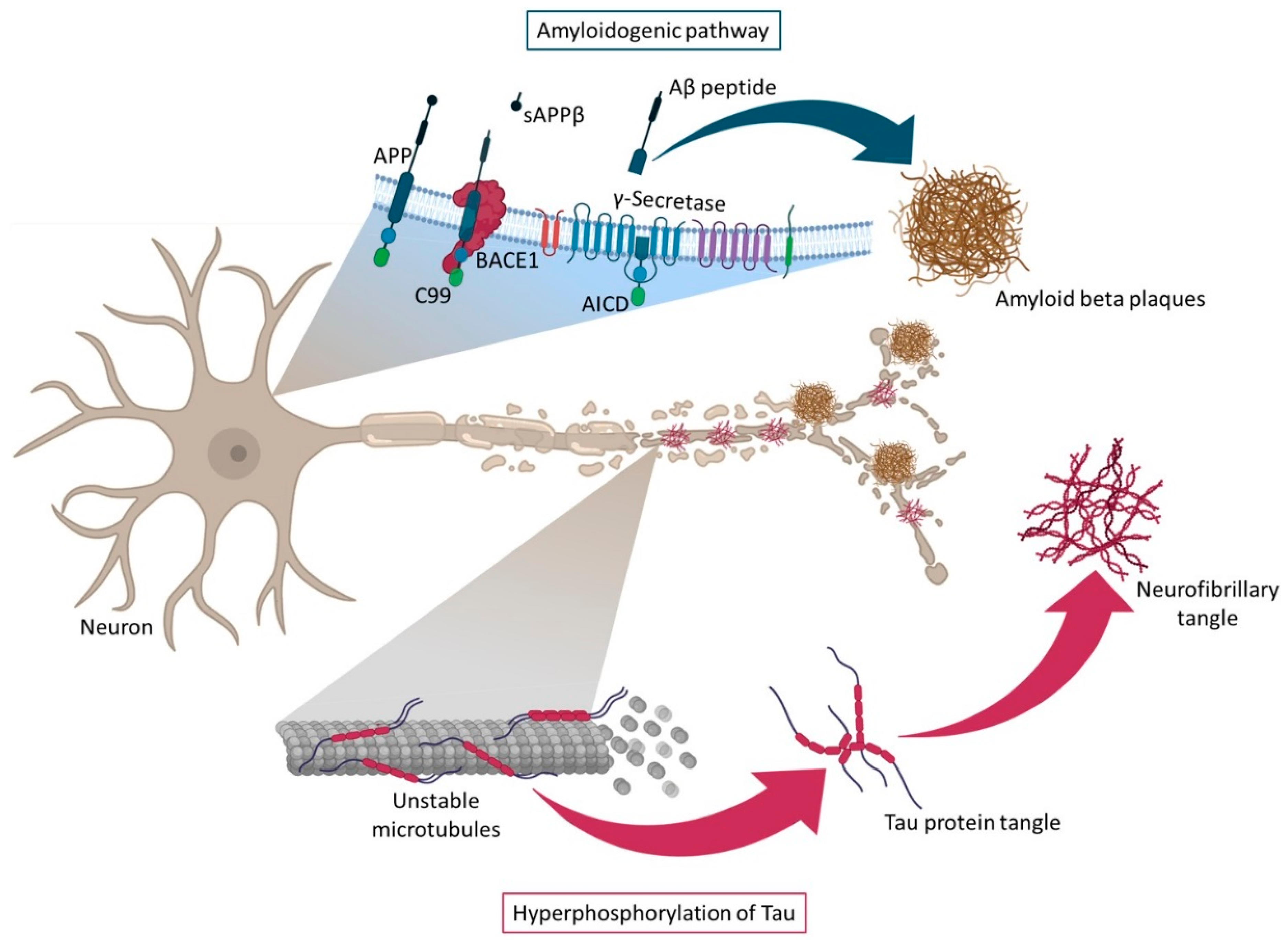
2.2. Amyloid and Tau Hypotheses
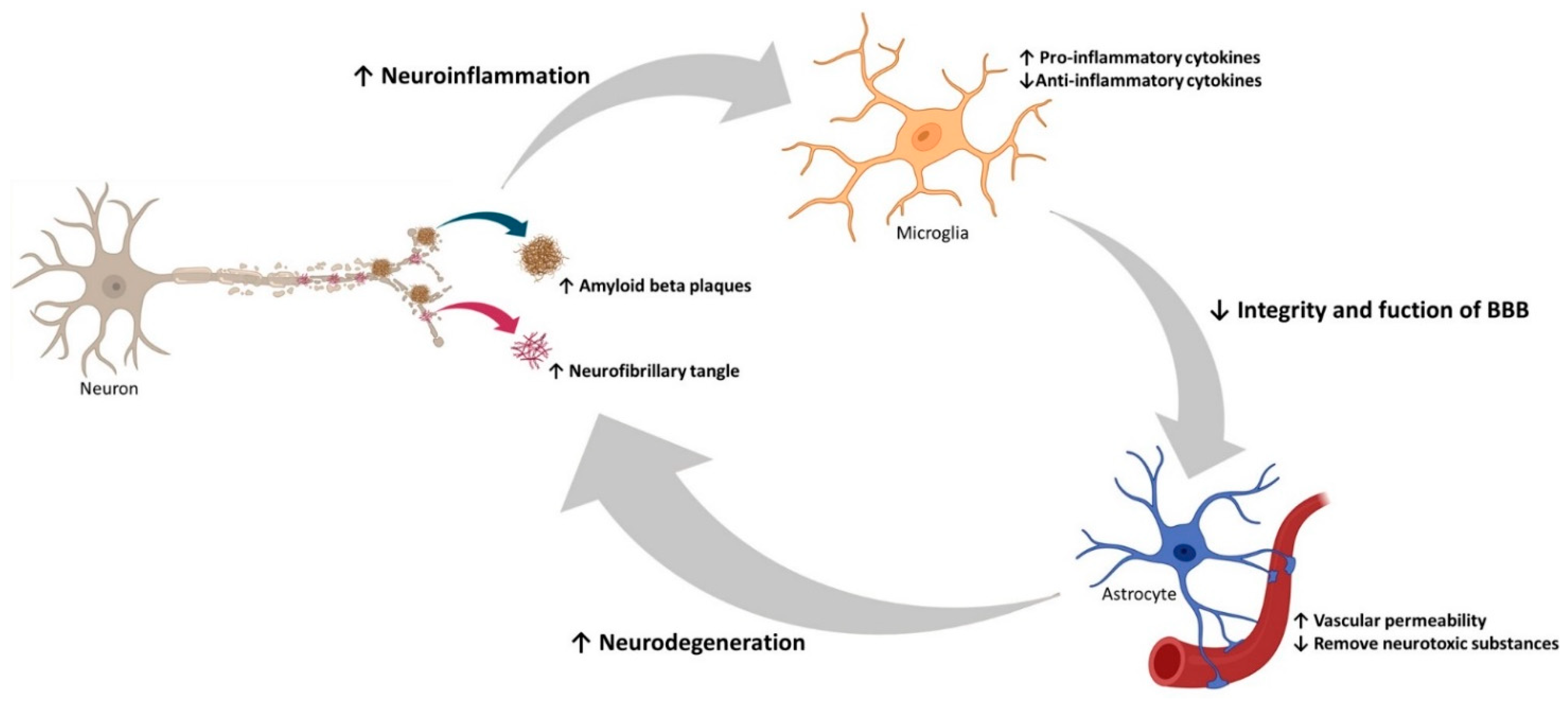
2.3. Neuroinflammation, Oxidative Stress, and Autophagy in AD
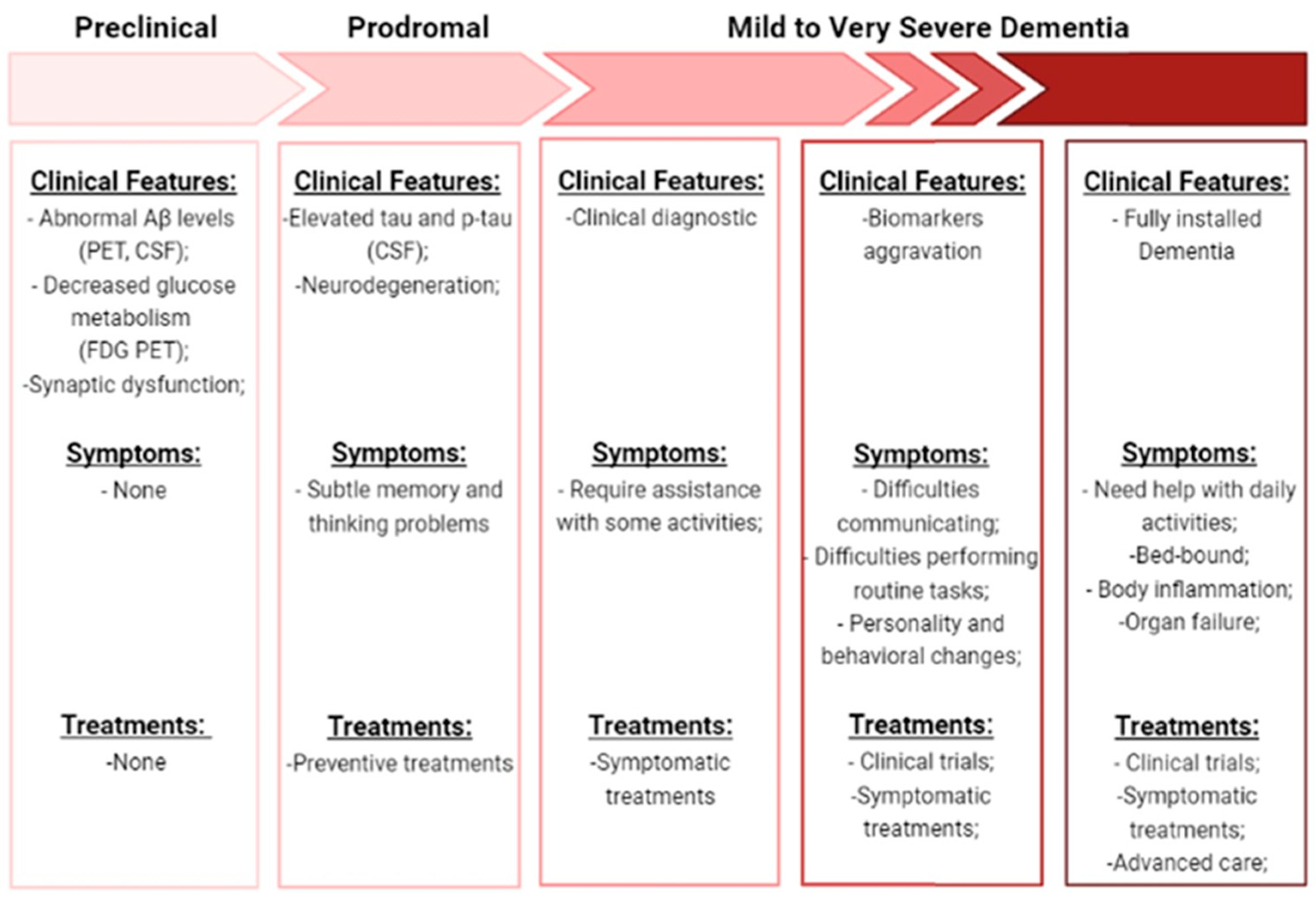
3. Diagnostic Tools for Alzheimer’s Disease
3.1. Approved Diagnostic Tools
3.2. Novel Diagnostic Approaches
3.2.1. Ribonucleic Acid
3.2.2. Novel RNA-Based Diagnostic Tools
4. Therapeutic Applications for Alzheimer’s Disease
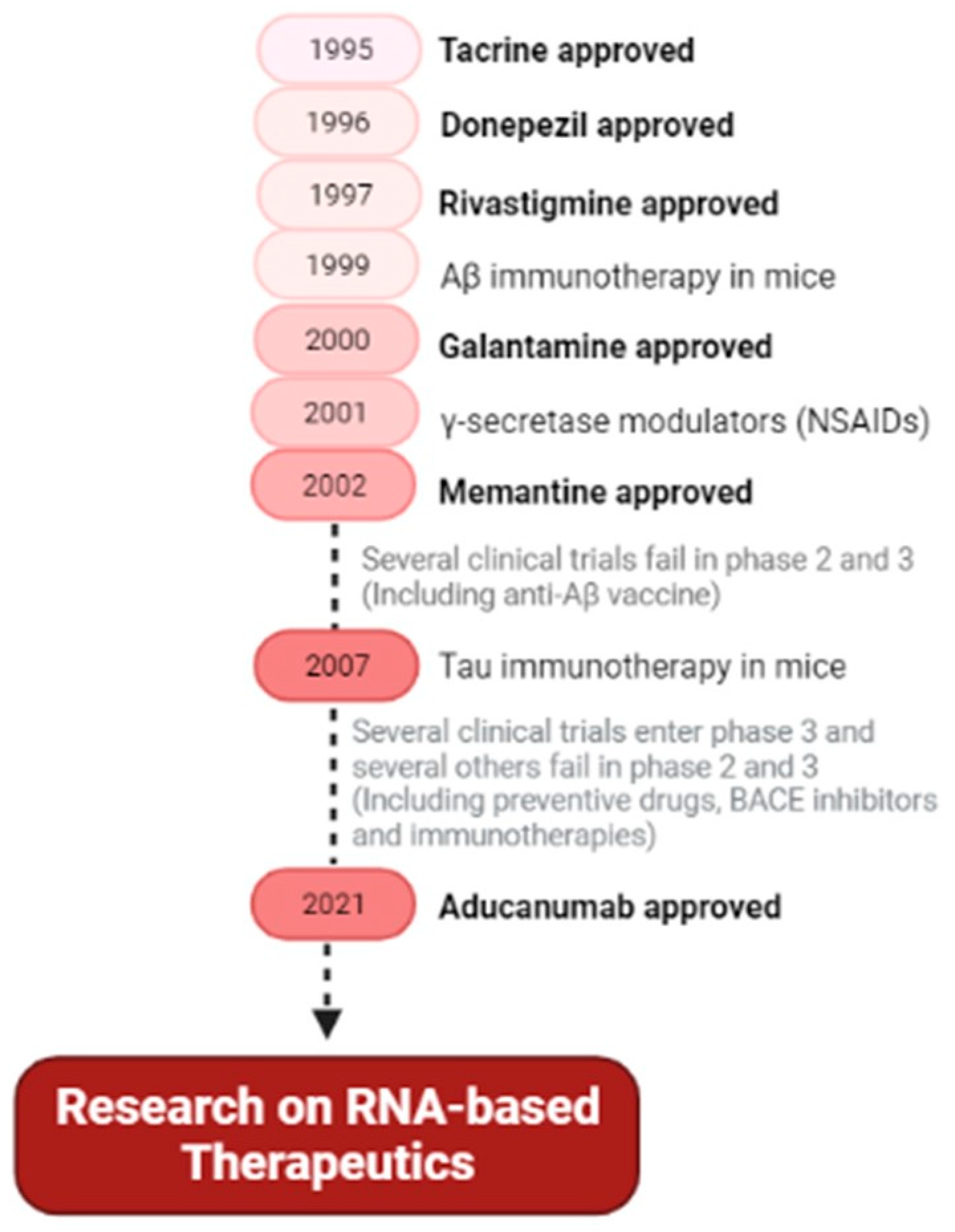
4.1. Approved Therapeutics
4.2. Novel Therapeutic Approaches
4.2.1. RNA-Based Therapeutic Approaches
Coding RNAs
Small Non-Coding RNAs
Long Non-Coding RNAs
Synthetic Oligonucleotides
4.2.2. Challenges in the RNA-Based Therapeutic Applications
5. Conclusions and Trends in AD Diagnosis and Treatment
Author Contributions
Funding
Conflicts of Interest
References
- WHO. The Top 10 Causes of Death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 26 July 2021).
- Miya Shaik, M.; Tamargo, I.A.; Abubakar, M.B.; Kamal, M.A.; Greig, N.H.; Gan, S.H. The Role of microRNAs in Alzheimer’s Disease and Their Therapeutic Potentials. Genes 2018, 9, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, R.; Kennelly, S.P.; O’Neill, D. Drug treatments in Alzheimer’s disease. Clin. Med. 2016, 16, 247–253. [Google Scholar] [CrossRef] [PubMed]
- 2020 Alzheimer’s disease facts and figures. Alzheimers Dement. 2020, 16, 391–460. [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Yang, G.; Yan, Q.Q.; Zhao, J.; Li, S. Exosome-encapsulated microRNAs as promising biomarkers for Alzheimer’s disease. Rev. Neurosci. 2019, 31, 77–87. [Google Scholar] [CrossRef]
- 2017 Alzheimer’s disease facts and figures. Alzheimers Dement. 2017, 13, 325–373. [CrossRef]
- Alagiakrishnan, K.; Gill, S.S.; Fagarasanu, A. Genetics and epigenetics of Alzheimer’s disease. Postgrad. Med. J. 2012, 88, 522–529. [Google Scholar] [CrossRef]
- Wimo, A.; Ali, C.A.; Guerchet, M.; Prince, M.; Prina, M.; Wu, Y. World Alzheimer Report 2015: The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends; Alzheimer’s Disease International: London, UK, 2015. [Google Scholar]
- Reynolds, D.S. A short perspective on the long road to effective treatments for Alzheimer’s disease. Br. J. Pharmacol. 2019, 176, 3636–3648. [Google Scholar] [CrossRef]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 2018, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Amakiri, N.; Kubosumi, A.; Tran, J.; Reddy, P.H. Amyloid Beta and MicroRNAs in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 430. [Google Scholar] [CrossRef] [Green Version]
- Surguchov, A. Caveolin: A New Link Between Diabetes and AD. Cell. Mol. Neurobiol. 2020, 40, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Shampo, M.A.; Kyle, R.A.; Steensma, D.P. Alois Alzheimer—Alzheimer disease. Mayo Clin. Proc. 2013, 88, e155. [Google Scholar] [CrossRef] [Green Version]
- Glenner, G.G.; Wong, C.W.; Quaranta, V.; Eanes, E.D. The amyloid deposits in Alzheimer’s disease: Their nature and pathogenesis. Appl. Pathol. 1984, 2, 357–369. [Google Scholar]
- Wong, K.H.; Riaz, M.K.; Xie, Y.; Zhang, X.; Liu, Q.; Chen, H.; Bian, Z.; Chen, X.; Lu, A.; Yang, Z. Review of Current Strategies for Delivering Alzheimer’s Disease Drugs across the Blood-Brain Barrier. Int. J. Mol. Sci. 2019, 20, 381. [Google Scholar] [CrossRef] [Green Version]
- Siedlecki-Wullich, D.; Miñano-Molina, A.J.; Rodríguez-Álvarez, J. microRNAs as Early Biomarkers of Alzheimer’s Disease: A Synaptic Perspective. Cells 2021, 10, 113. [Google Scholar] [CrossRef] [PubMed]
- Martinez, B.; Peplow, P. MicroRNAs as diagnostic and therapeutic tools for Alzheimer’s disease: Advances and limitations. Neural Regen. Res. 2019, 14, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet. Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Nahalka, J. The role of the protein-RNA recognition code in neurodegeneration. Cell. Mol. Life Sci. 2019, 76, 2043–2058. [Google Scholar] [CrossRef] [PubMed]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Maloney, B.; Rogers, J.T.; Lahiri, D.K. Novel upregulation of amyloid-β precursor protein (APP) by microRNA-346 via targeting of APP mRNA 5′-untranslated region: Implications in Alzheimer’s disease. Mol. Psychiatry 2019, 24, 345–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, R.C. The genetics of Alzheimer’s disease. Scientifica 2012, 2012, 246210. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Ham, S.; Kim, T.K.; Ryu, J.; Kim, Y.S.; Tang, Y.-P.; Im, H.-I. Comprehensive MicroRNAome Analysis of the Relationship Between Alzheimer Disease and Cancer in PSEN Double-Knockout Mice. Int. Neurourol. J. 2018, 22, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [Green Version]
- Slota, J.A.; Booth, S.A. MicroRNAs in Neuroinflammation: Implications in Disease Pathogenesis, Biomarker Discovery and Therapeutic Applications. Non-Coding RNA 2019, 5, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barage, S.H.; Sonawane, K.D. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 2015, 52, 1–18. [Google Scholar] [CrossRef]
- Vanden Dries, V.; Stygelbout, V.; Pierrot, N.; Yilmaz, Z.; Suain, V.; De Decker, R.; Buée, L.; Octave, J.N.; Brion, J.P.; Leroy, K. Amyloid precursor protein reduction enhances the formation of neurofibrillary tangles in a mutant tau transgenic mouse model. Neurobiol. Aging 2017, 55, 202–212. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Singh, A.; Ekavali. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Bali, J.; Halima, S.B.; Felmy, B.; Goodger, Z.; Zurbriggen, S.; Rajendran, L. Cellular basis of Alzheimer’s disease. Ann. Indian Acad. Neurol. 2010, 13, S89–S93. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Gao, Y.; Sun, J.Y.; Meng, X.L.; Yang, D.; Fan, L.H.; Xiang, L.; Wang, P. Traditional Chinese Medicine: Role in Reducing β-Amyloid, Apoptosis, Autophagy, Neuroinflammation, Oxidative Stress, and Mitochondrial Dysfunction of Alzheimer’s Disease. Front. Pharm. 2020, 11, 497. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Wong, L.W.; Su, Y.; Huang, X.; Wang, N.; Chen, H.; Yi, C. Blood-brain barrier integrity in the pathogenesis of Alzheimer’s disease. Front. Neuroendocrinol. 2020, 59, 100857. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Qiao, P.F.; Wan, C.Q.; Cai, M.; Zhou, N.K.; Li, Q. Role of Blood-Brain Barrier in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 63, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhao, Y.; Liu, Y.; Wang, M.; Yu, W.; Zhang, L. Exploring the regulatory roles of circular RNAs in Alzheimer’s disease. Transl. Neurodegener. 2020, 9, 35. [Google Scholar] [CrossRef]
- Wang, X.; Shen, C.; Zhu, J.; Shen, G.; Li, Z.; Dong, J. Long Noncoding RNAs in the Regulation of Oxidative Stress. Oxid. Med. Cell Longev. 2019, 2019, 1318795. [Google Scholar] [CrossRef]
- Huang, J.L.; Su, M.; Wu, D.P. Functional roles of circular RNAs in Alzheimer’s disease. Ageing Res. Rev. 2020, 60, 101058. [Google Scholar] [CrossRef]
- Nassif, M.; Hetz, C. Autophagy impairment: A crossroad between neurodegeneration and tauopathies. BMC Biol. 2012, 10, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeromin, A.; Bowser, R. Biomarkers in Neurodegenerative Diseases. Adv. Neurobiol. 2017, 15, 491–528. [Google Scholar] [CrossRef]
- Zafari, S.; Backes, C.; Meese, E.; Keller, A. Circulating Biomarker Panels in Alzheimer’s Disease. Gerontology 2015, 61, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Vassileff, N.; Cheng, L.; Hill, A.F. Extracellular vesicles—propagators of neuropathology and sources of potential biomarkers and therapeutics for neurodegenerative diseases. J. Cell Sci. 2020, 133, jcs243139. [Google Scholar] [CrossRef]
- Penner, G.; Lecocq, S.; Chopin, A.; Vedoya, X.; Lista, S.; Vergallo, A.; Lejeune, F.X.; Hampel, H. Blood-based diagnostics of Alzheimer’s disease. Expert Rev. Mol. Diagn. 2019, 19, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Blennow, K. Moving fluid biomarkers for Alzheimer’s disease from research tools to routine clinical diagnostics. Mol. Neurodegener. 2021, 16, 10. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Ta, Q.T.H.; Nguyen, T.K.O.; Nguyen, T.T.D.; Vo, V.G. Role of Body-Fluid Biomarkers in Alzheimer’s Disease Diagnosis. Diagnostics 2020, 10, 326. [Google Scholar] [CrossRef]
- Zetterberg, H.; Burnham, S.C. Blood-based molecular biomarkers for Alzheimer’s disease. Mol. Brain 2019, 12, 26. [Google Scholar] [CrossRef]
- Sharp, P.A. The centrality of RNA. Cell 2009, 136, 577–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.V.; Ignacimuthu, S. RNA interference—A silent but an efficient therapeutic tool. Appl. Biochem. Biotechnol. 2013, 169, 1774–1789. [Google Scholar] [CrossRef]
- Svoboda, P. Renaissance of mammalian endogenous RNAi. FEBS Lett. 2014, 588, 2550–2556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogini, D.B.; Pascoal, V.D.; Avansini, S.H.; Vieira, A.S.; Pereira, T.C.; Lopes-Cendes, I. The new world of RNAs. Genet. Mol. Biol. 2014, 37, 285–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baptista, B.; Riscado, M.; Queiroz, J.A.; Pichon, C.; Sousa, F. Non-coding RNAs: Emerging from the discovery to therapeutic applications. Biochem. Pharmacol. 2021, 189, 114469. [Google Scholar] [CrossRef]
- Li, S.; Qian, T.; Wang, X.; Liu, J.; Gu, X. Noncoding RNAs and Their Potential Therapeutic Applications in Tissue Engineering. Engineering 2017, 3, 3–15. [Google Scholar] [CrossRef]
- Piscopo, P.; Bellenghi, M.; Manzini, V.; Crestini, A.; Pontecorvi, G.; Corbo, M.; Ortona, E.; Carè, A.; Confaloni, A. A Sex Perspective in Neurodegenerative Diseases: microRNAs as Possible Peripheral Biomarkers. Int. J. Mol. Sci. 2021, 22, 4423. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Reddy, P.H. Are circulating microRNAs peripheral biomarkers for Alzheimer’s disease? Biochim. Biophys. Acta 2016, 1862, 1617–1627. [Google Scholar] [CrossRef]
- Nikolac Perkovic, M.; Videtic Paska, A.; Konjevod, M.; Kouter, K.; Svob Strac, D.; Nedic Erjavec, G.; Pivac, N. Epigenetics of Alzheimer’s Disease. Biomolecules 2021, 11, 195. [Google Scholar] [CrossRef] [PubMed]
- Leidinger, P.; Backes, C.; Deutscher, S.; Schmitt, K.; Mueller, S.C.; Frese, K.; Haas, J.; Ruprecht, K.; Paul, F.; Stähler, C.; et al. A blood based 12-miRNA signature of Alzheimer disease patients. Genome Biol. 2013, 14, R78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.G.; Song, J.; Zhang, Y.Q.; Wang, P.C. MicroRNA-193b is a regulator of amyloid precursor protein in the blood and cerebrospinal fluid derived exosomal microRNA-193b is a biomarker of Alzheimer’s disease. Mol. Med. Rep. 2014, 10, 2395–2400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Doecke, J.D.; Sharples, R.A.; Villemagne, V.L.; Fowler, C.J.; Rembach, A.; Martins, R.N.; Rowe, C.C.; Macaulay, S.L.; Masters, C.L.; et al. Prognostic serum miRNA biomarkers associated with Alzheimer’s disease shows concordance with neuropsychological and neuroimaging assessment. Mol. Psychiatry 2015, 20, 1188–1196. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer Disease and microRNA—List Results—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=Alzheimer+Disease&term=microRNA&cntry=&state=&city=&dist=&Search=Search (accessed on 26 July 2021).
- Doxtater, K.; Tripathi, M.K.; Khan, M.M. Recent advances on the role of long non-coding RNAs in Alzheimer’s disease. Neural Regen. Res. 2020, 15, 2253–2254. [Google Scholar] [CrossRef]
- Htoo, K.P.P.; Yamkamon, V.; Yainoy, S.; Suksrichavalit, T.; Viseshsindh, W.; Eiamphungporn, W. Colorimetric detection of PCA3 in urine for prostate cancer diagnosis using thiol-labeled PCR primer and unmodified gold nanoparticles. Clin. Chim. Acta Int. J. Clin. Chem. 2019, 488, 40–49. [Google Scholar] [CrossRef]
- Zhang, W.; Zhao, H.; Wu, Q.; Xu, W.; Xia, M. Knockdown of BACE1-AS by siRNA improves memory and learning behaviors in Alzheimer’s disease animal model. Exp. Ther. Med. 2018, 16, 2080–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciarlo, E.; Massone, S.; Penna, I.; Nizzari, M.; Gigoni, A.; Dieci, G.; Russo, C.; Florio, T.; Cancedda, R.; Pagano, A. An intronic ncRNA-dependent regulation of SORL1 expression affecting Aβ formation is upregulated in post-mortem Alzheimer’s disease brain samples. Dis. Models Mech. 2013, 6, 424–433. [Google Scholar] [CrossRef] [Green Version]
- Fotuhi, S.N.; Khalaj-Kondori, M.; Hoseinpour Feizi, M.A.; Talebi, M. Long Non-coding RNA BACE1-AS May Serve as an Alzheimer’s Disease Blood-Based Biomarker. J. Mol. Neurosci. 2019, 69, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Modarresi, F.; Faghihi, M.A.; Patel, N.S.; Sahagan, B.G.; Wahlestedt, C.; Lopez-Toledano, M.A. Knockdown of BACE1-AS Nonprotein-Coding Transcript Modulates Beta-Amyloid-Related Hippocampal Neurogenesis. Int. J. Alzheimers Dis. 2011, 2011, 929042. [Google Scholar] [CrossRef] [Green Version]
- Akhter, R. Circular RNA and Alzheimer’s Disease. Adv. Exp. Med. Biol. 2018, 1087, 239–243. [Google Scholar] [CrossRef]
- Lu, D.; Xu, A.D. Mini Review: Circular RNAs as Potential Clinical Biomarkers for Disorders in the Central Nervous System. Front. Genet. 2016, 7, 53. [Google Scholar] [CrossRef]
- Dube, U.; Del-Aguila, J.L.; Li, Z.; Budde, J.P.; Jiang, S.; Hsu, S.; Ibanez, L.; Fernandez, M.V.; Farias, F.; Norton, J.; et al. An atlas of cortical circular RNA expression in Alzheimer disease brains demonstrates clinical and pathological associations. Nat. Neurosci. 2019, 22, 1903–1912. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, F.; Bao, S.; Sun, J. Systematic Characterization of Circular RNA-Associated CeRNA Network Identified Novel circRNA Biomarkers in Alzheimer’s Disease. Front. Bioeng. Biotechnol. 2019, 7, 222. [Google Scholar] [CrossRef]
- Li, Y.; Fan, H.; Sun, J.; Ni, M.; Zhang, L.; Chen, C.; Hong, X.; Fang, F.; Zhang, W.; Ma, P. Circular RNA expression profile of Alzheimer’s disease and its clinical significance as biomarkers for the disease risk and progression. Int. J. Biochem. Cell Biol. 2020, 123, 105747. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Disease Research Timeline Alzforum. Available online: https://www.alzforum.org/timeline/alzheimers-disease#1906 (accessed on 25 July 2021).
- Romero, A.; Cacabelos, R.; Oset-Gasque, M.J.; Samadi, A.; Marco-Contelles, J. Novel tacrine-related drugs as potential candidates for the treatment of Alzheimer’s disease. Bioorganic Med. Chem. Lett. 2013, 23, 1916–1922. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; Delon, M.R. Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef]
- Chu, L.W. Alzheimer’s disease: Early diagnosis and treatment. Hong Kong Med. J. Xianggang Yi Xue Za Zhi 2012, 18, 228–237. [Google Scholar]
- Muir, J.L.; Everitt, B.J.; Robbins, T.W. AMPA-induced excitotoxic lesions of the basal forebrain: A significant role for the cortical cholinergic system in attentional function. J. Neurosci. Off. J. Soc. Neurosci. 1994, 14, 2313–2326. [Google Scholar] [CrossRef]
- Sahakian, B.J.; Coull, J.T. Tetrahydroaminoacridine (THA) in Alzheimer’s disease: An assessment of attentional and mnemonic function using CANTAB. Acta Neurol. Scandinavica. Suppl. 1993, 149, 29–35. [Google Scholar] [CrossRef]
- Weller, J.; Budson, A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Research 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Lipton, S.A. Paradigm shift in neuroprotection by NMDA receptor blockade: Memantine and beyond. Nat. Rev. Drug Discov. 2006, 5, 160–170. [Google Scholar] [CrossRef]
- McShane, R.; Areosa Sastre, A.; Minakaran, N. Memantine for dementia. Cochrane Database Syst. Rev. 2006, Cd003154. [Google Scholar] [CrossRef] [PubMed]
- Nisticò, R.; Borg, J.J. Aducanumab for Alzheimer’s disease: A regulatory perspective. Pharmacol. Res. 2021, 171, 105754. [Google Scholar] [CrossRef]
- Marasco, R.A. Current and evolving treatment strategies for the Alzheimer disease continuum. Am. J. Manag. Care 2020, 26, S167–s176. [Google Scholar] [CrossRef]
- Kennedy, M.E.; Stamford, A.W.; Chen, X.; Cox, K.; Cumming, J.N.; Dockendorf, M.F.; Egan, M.; Ereshefsky, L.; Hodgson, R.A.; Hyde, L.A.; et al. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS β-amyloid in animal models and in Alzheimer’s disease patients. Sci. Transl. Med. 2016, 8, 363ra150. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Gutiérrez, L.; Bammens, L.; Benilova, I.; Vandersteen, A.; Benurwar, M.; Borgers, M.; Lismont, S.; Zhou, L.; Van Cleynenbreugel, S.; Esselmann, H.; et al. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012, 31, 2261–2274. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef]
- Bursavich, M.G.; Harrison, B.A.; Blain, J.F. Gamma Secretase Modulators: New Alzheimer’s Drugs on the Horizon? J. Med. Chem. 2016, 59, 7389–7409. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.; Patel, T.R. γ-Secretase modulators: Current status and future directions. Prog. Med. Chem. 2014, 53, 101–145. [Google Scholar] [CrossRef] [PubMed]
- Szaruga, M.; Munteanu, B.; Lismont, S.; Veugelen, S.; Horré, K.; Mercken, M.; Saido, T.C.; Ryan, N.S.; De Vos, T.; Savvides, S.N.; et al. Alzheimer’s-Causing Mutations Shift Aβ Length by Destabilizing γ-Secretase-Aβn Interactions. Cell 2017, 170, 443–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doig, A.J.; Del Castillo-Frias, M.P.; Berthoumieu, O.; Tarus, B.; Nasica-Labouze, J.; Sterpone, F.; Nguyen, P.H.; Hooper, N.M.; Faller, P.; Derreumaux, P. Why Is Research on Amyloid-β Failing to Give New Drugs for Alzheimer’s Disease? ACS Chem. Neurosci. 2017, 8, 1435–1437. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.H.; Ramamoorthy, A.; Sahoo, B.R.; Zheng, J.; Faller, P.; Straub, J.E.; Dominguez, L.; Shea, J.E.; Dokholyan, N.V.; De Simone, A.; et al. Amyloid Oligomers: A Joint Experimental/Computational Perspective on Alzheimer’s Disease, Parkinson’s Disease, Type II Diabetes, and Amyotrophic Lateral Sclerosis. Chem. Rev. 2021, 121, 2545–2647. [Google Scholar] [CrossRef]
- Alzheimer Disease—List Results—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=Alzheimer%20Disease&term=&cntry=&state=&city=&dist=&Search=Search (accessed on 26 July 2021).
- Therapeutics|ALZFORUM. Available online: https://www.alzforum.org/therapeutics (accessed on 26 July 2021).
- Chakravarthy, M.; Chen, S.; Dodd, P.R.; Veedu, R.N. Nucleic Acid-Based Theranostics for Tackling Alzheimer’s Disease. Theranostics 2017, 7, 3933–3947. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Perche, F.; Ikegami, M.; Uchida, S.; Kataoka, K.; Itaka, K. Messenger RNA-based therapeutics for brain diseases: An animal study for augmenting clearance of beta-amyloid by intracerebral administration of neprilysin mRNA loaded in polyplex nanomicelles. J. Control. Release 2016, 235, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Liu, M.; Du, K.; Zhong, X.; Gong, S.; Jiao, L.; Wei, M. Differential Expression of mRNAs in the Brain Tissues of Patients with Alzheimer’s Disease Based on GEO Expression Profile and Its Clinical Significance. Biomed. Res. Int. 2019, 2019, 8179145. [Google Scholar] [CrossRef] [Green Version]
- Lam, J.K.; Chow, M.Y.; Zhang, Y.; Leung, S.W. siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol. Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef] [Green Version]
- McSwiggen, J.; Beigelman, L. RNA Interference Mediated Treatment of Alzheimer’s Disease Using Short Interfering Nucleic Acid (SINA). US Patent US20100168208, 1 July 2010. [Google Scholar]
- Kao, S.C.; Krichevsky, A.M.; Kosik, K.S.; Tsai, L.H. BACE1 suppression by RNA interference in primary cortical neurons. J. Biol. Chem. 2004, 279, 1942–1949. [Google Scholar] [CrossRef] [Green Version]
- Qazi, T.J.; Quan, Z.; Mir, A.; Qing, H. Epigenetics in Alzheimer’s Disease: Perspective of DNA Methylation. Mol. Neurobiol. 2018, 55, 1026–1044. [Google Scholar] [CrossRef]
- Maoz, R.; Garfinkel, B.P.; Soreq, H. Alzheimer’s Disease and ncRNAs. Adv. Exp. Med. Biol. 2017, 978, 337–361. [Google Scholar] [CrossRef]
- Contiliani, D.F.; Ribeiro, Y.A.; de Moraes, V.N.; Pereira, T.C. MicroRNAs in Prion Diseases-From Molecular Mechanisms to Insights in Translational Medicine. Cells 2021, 10, 1620. [Google Scholar] [CrossRef]
- Patel, A.A.; Ganepola, G.A.P.; Rutledge, J.R.; Chang, D.H. The Potential Role of Dysregulated miRNAs in Alzheimer’s Disease Pathogenesis and Progression. J. Alzheimers Dis. 2019, 67, 1123–1145. [Google Scholar] [CrossRef]
- Liu, X.; Jiao, B.; Shen, L. The Epigenetics of Alzheimer’s Disease: Factors and Therapeutic Implications. Front. Genet. 2018, 9, 579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, V.K.; Mehta, V.; Singh, T.G. Alzheimer’s Disorder: Epigenetic Connection and Associated Risk Factors. Curr. Neuropharmacol. 2020, 18, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Liu, X.; Jiao, B. Epigenetics: Recent Advances and Its Role in the Treatment of Alzheimer’s Disease. Front. Neurol. 2020, 11, 538301. [Google Scholar] [CrossRef]
- Liu, J.; Zuo, X.; Han, J.; Dai, Q.; Xu, H.; Liu, Y.; Cui, S. MiR-9-5p inhibits mitochondrial damage and oxidative stress in AD cell models by targeting GSK-3β. Biosci. Biotechnol. Biochem. 2020, 84, 2273–2280. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, H. miR-15b reduces amyloid-β accumulation in SH-SY5Y cell line through targetting NF-κB signaling and BACE1. Biosci. Rep. 2018, 38, BSR20180051. [Google Scholar] [CrossRef] [Green Version]
- Feng, M.G.; Liu, C.F.; Chen, L.; Feng, W.B.; Liu, M.; Hai, H.; Lu, J.M. MiR-21 attenuates apoptosis-triggered by amyloid-β via modulating PDCD4/ PI3K/AKT/GSK-3β pathway in SH-SY5Y cells. Biomed. Pharm. 2018, 101, 1003–1007. [Google Scholar] [CrossRef]
- Hébert, S.S.; Horré, K.; Nicolaï, L.; Papadopoulou, A.S.; Mandemakers, W.; Silahtaroglu, A.N.; Kauppinen, S.; Delacourte, A.; De Strooper, B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6415–6420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Song, Y.; Zhou, X.; Deng, Y.; Liu, T.; Weng, G.; Yu, D.; Pan, S. MicroRNA-29c targets β-site amyloid precursor protein-cleaving enzyme 1 and has a neuroprotective role in vitro and in vivo. Mol. Med. Rep. 2015, 12, 3081–3088. [Google Scholar] [CrossRef] [Green Version]
- Lei, X.; Lei, L.; Zhang, Z.; Zhang, Z.; Cheng, Y. Downregulated miR-29c correlates with increased BACE1 expression in sporadic Alzheimer’s disease. Int. J. Clin. Exp. Pathol. 2015, 8, 1565–1574. [Google Scholar]
- Li, P.; Xu, Y.; Wang, B.; Huang, J.; Li, Q. miR-34a-5p and miR-125b-5p attenuate Aβ-induced neurotoxicity through targeting BACE1. J. Neurol. Sci. 2020, 413, 116793. [Google Scholar] [CrossRef]
- Barros-Viegas, A.T.; Carmona, V.; Ferreiro, E.; Guedes, J.; Cardoso, A.M.; Cunha, P.; Pereira de Almeida, L.; Resende de Oliveira, C.; Pedro de Magalhães, J.; Peça, J.; et al. miRNA-31 Improves Cognition and Abolishes Amyloid-β Pathology by Targeting APP and BACE1 in an Animal Model of Alzheimer’s Disease. Mol. Nucleic Acids 2020, 19, 1219–1236. [Google Scholar] [CrossRef]
- Lin, Y.; Yao, Y.; Liang, X.; Shi, Y.; Kong, L.; Xiao, H.; Wu, Y.; Ni, Y.; Yang, J. Osthole suppresses amyloid precursor protein expression by up-regulating miRNA-101a-3p in Alzheimer’s disease cell model. Zhejiang Da Xue Xue Bao Yi Xue Ban J. Zhejiang Univ. Med. Sci. 2018, 47, 473–479. [Google Scholar]
- Chen, F.Z.; Zhao, Y.; Chen, H.Z. MicroRNA-98 reduces amyloid β-protein production and improves oxidative stress and mitochondrial dysfunction through the Notch signaling pathway via HEY2 in Alzheimer’s disease mice. Int. J. Mol. Med. 2019, 43, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Vilardo, E.; Barbato, C.; Ciotti, M.; Cogoni, C.; Ruberti, F. MicroRNA-101 regulates amyloid precursor protein expression in hippocampal neurons. J. Biol. Chem. 2010, 285, 18344–18351. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Zhao, J.; Lu, G. miR-106b inhibits tau phosphorylation at Tyr18 by targeting Fyn in a model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2016, 478, 852–857. [Google Scholar] [CrossRef]
- Jiao, Y.; Kong, L.; Yao, Y.; Li, S.; Tao, Z.; Yan, Y.; Yang, J. Osthole decreases beta amyloid levels through up-regulation of miR-107 in Alzheimer’s disease. Neuropharmacology 2016, 108, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Kang, Q.; Xiang, Y.; Li, D.; Liang, J.; Zhang, X.; Zhou, F.; Qiao, M.; Nie, Y.; He, Y.; Cheng, J.; et al. MiR-124-3p attenuates hyperphosphorylation of Tau protein-induced apoptosis via caveolin-1-PI3K/Akt/GSK3β pathway in N2a/APP695swe cells. Oncotarget 2017, 8, 24314–24326. [Google Scholar] [CrossRef] [PubMed]
- Geekiyanage, H.; Chan, C. MicroRNA-137/181c regulates serine palmitoyltransferase and in turn amyloid β, novel targets in sporadic Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 14820–14830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.M.; Ray, B.; Lahiri, D.K. MicroRNA-153 physiologically inhibits expression of amyloid-β precursor protein in cultured human fetal brain cells and is dysregulated in a subset of Alzheimer disease patients. J. Biol. Chem. 2012, 287, 31298–31310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.C.; Wang, L.M.; Wang, M.; Song, B.; Tan, S.; Teng, J.F.; Duan, D.X. MicroRNA-195 downregulates Alzheimer’s disease amyloid-β production by targeting BACE1. Brain Res. Bull. 2012, 88, 596–601. [Google Scholar] [CrossRef]
- Wang, L.; Liu, J.; Wang, Q.; Jiang, H.; Zeng, L.; Li, Z.; Liu, R. MicroRNA-200a-3p Mediates Neuroprotection in Alzheimer-Related Deficits and Attenuates Amyloid-Beta Overproduction and Tau Hyperphosphorylation via Coregulating BACE1 and PRKACB. Front Pharm. 2019, 10, 806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higaki, S.; Muramatsu, M.; Matsuda, A.; Matsumoto, K.; Satoh, J.I.; Michikawa, M.; Niida, S. Defensive effect of microRNA-200b/c against amyloid-beta peptide-induced toxicity in Alzheimer’s disease models. PLoS ONE 2018, 13, e0196929. [Google Scholar] [CrossRef]
- Chopra, N.; Wang, R.; Maloney, B.; Nho, K.; Beck, J.S.; Pourshafie, N.; Niculescu, A.; Saykin, A.J.; Rinaldi, C.; Counts, S.E.; et al. MicroRNA-298 reduces levels of human amyloid-β precursor protein (APP), β-site APP-converting enzyme 1 (BACE1) and specific tau protein moieties. Mol. Psychiatry 2020. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Chen, W.; Zeng, J.; Tong, W.; Zheng, P. MicroRNA-326 decreases tau phosphorylation and neuron apoptosis through inhibition of the JNK signaling pathway by targeting VAV1 in Alzheimer’s disease. J. Cell Physiol. 2020, 235, 480–493. [Google Scholar] [CrossRef]
- Boissonneault, V.; Plante, I.; Rivest, S.; Provost, P. MicroRNA-298 and microRNA-328 regulate expression of mouse beta-amyloid precursor protein-converting enzyme 1. J. Biol. Chem. 2009, 284, 1971–1981. [Google Scholar] [CrossRef] [Green Version]
- Long, J.M.; Ray, B.; Lahiri, D.K. MicroRNA-339-5p down-regulates protein expression of β-site amyloid precursor protein-cleaving enzyme 1 (BACE1) in human primary brain cultures and is reduced in brain tissue specimens of Alzheimer disease subjects. J. Biol. Chem. 2014, 289, 5184–5198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatnagar, S.; Chertkow, H.; Schipper, H.M.; Yuan, Z.; Shetty, V.; Jenkins, S.; Jones, T.; Wang, E. Increased microRNA-34c abundance in Alzheimer’s disease circulating blood plasma. Front. Mol. Neurosci. 2014, 7, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zovoilis, A.; Agbemenyah, H.Y.; Agis-Balboa, R.C.; Stilling, R.M.; Edbauer, D.; Rao, P.; Farinelli, L.; Delalle, I.; Schmitt, A.; Falkai, P.; et al. microRNA-34c is a novel target to treat dementias. EMBO J. 2011, 30, 4299–4308. [Google Scholar] [CrossRef]
- Lee, S.T.; Chu, K.; Jung, K.H.; Kim, J.H.; Huh, J.Y.; Yoon, H.; Park, D.K.; Lim, J.Y.; Kim, J.M.; Jeon, D.; et al. miR-206 regulates brain-derived neurotrophic factor in Alzheimer disease model. Ann. Neurol. 2012, 72, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.R.; Chang, C.C.; Dogbevia, G.; Bryleva, E.Y.; Bowen, Z.; Hasan, M.T.; Chang, T.Y. Acat1 knockdown gene therapy decreases amyloid-β in a mouse model of Alzheimer’s disease. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 1497–1506. [Google Scholar] [CrossRef]
- Liu, C.G.; Wang, J.L.; Li, L.; Wang, P.C. MicroRNA-384 regulates both amyloid precursor protein and β-secretase expression and is a potential biomarker for Alzheimer’s disease. Int. J. Mol. Med. 2014, 34, 160–166. [Google Scholar] [CrossRef] [Green Version]
- Riva, P.; Ratti, A.; Venturin, M. The Long Non-Coding RNAs in Neurodegenerative Diseases: Novel Mechanisms of Pathogenesis. Curr. Alzheimer Res. 2016, 13, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Modarresi, F.; Faghihi, M.A.; Lopez-Toledano, M.A.; Fatemi, R.P.; Magistri, M.; Brothers, S.P.; van der Brug, M.P.; Wahlestedt, C. Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat. Biotechnol. 2012, 30, 453–459. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Zhao, J.; Wang, W.; Zhou, J.; Zhang, J. Depletion of LncRNA NEAT1 Rescues Mitochondrial Dysfunction Through NEDD4L-Dependent PINK1 Degradation in Animal Models of Alzheimer’s Disease. Front. Cell. Neurosci. 2020, 14, 28. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhao, Y.; Xu, N.; Zhang, S.; Wang, S.; Mao, Y.; Zhu, Y.; Li, B.; Jiang, Y.; Tan, Y.; et al. NEAT1 regulates neuroglial cell mediating Aβ clearance via the epigenetic regulation of endocytosis-related genes expression. Cell. Mol. Life Sci. 2019, 76, 3005–3018. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Li, Y.; Niringiyumukiza, J.D.; Su, P.; Xiang, W. Circular RNA involvement in aging: An emerging player with great potential. Mech. Ageing Dev. 2019, 178, 16–24. [Google Scholar] [CrossRef]
- Lee, W.J.; Moon, J.; Jeon, D.; Shin, Y.W.; Yoo, J.S.; Park, D.K.; Lee, S.T.; Jung, K.H.; Park, K.I.; Jung, K.Y.; et al. Possible epigenetic regulatory effect of dysregulated circular RNAs in Alzheimer’s disease model. Sci. Rep. 2019, 9, 11956. [Google Scholar] [CrossRef]
- Shi, Z.; Chen, T.; Yao, Q.; Zheng, L.; Zhang, Z.; Wang, J.; Hu, Z.; Cui, H.; Han, Y.; Han, X.; et al. The circular RNA ciRS-7 promotes APP and BACE1 degradation in an NF-κB-dependent manner. FEBS J. 2017, 284, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Tan, L.; Wang, X. Circular HDAC9/microRNA-138/Sirtuin-1 Pathway Mediates Synaptic and Amyloid Precursor Protein Processing Deficits in Alzheimer’s Disease. Neurosci. Bull. 2019, 35, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Diling, C.; Yinrui, G.; Longkai, Q.; Xiaocui, T.; Yadi, L.; Xin, Y.; Guoyan, H.; Ou, S.; Tianqiao, Y.; Dongdong, W.; et al. Circular RNA NF1-419 enhances autophagy to ameliorate senile dementia by binding Dynamin-1 and Adaptor protein 2 B1 in AD-like mice. Aging 2019, 11, 12002–12031. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Krainer, A.R.; Cleveland, D.W. Antisense Oligonucleotide Therapies for Neurodegenerative Diseases. Annu. Rev. Neurosci. 2019, 42, 385–406. [Google Scholar] [CrossRef]
- Angelucci, F.; Cechova, K.; Valis, M.; Kuca, K.; Zhang, B.; Hort, J. MicroRNAs in Alzheimer’s Disease: Diagnostic Markers or Therapeutic Agents? Front. Pharm. 2019, 10, 665. [Google Scholar] [CrossRef]
- Darling, T.; Kumar, V.B.; Banks, W.A.; Farr, S. Antisense Modulation of Amyloid Beta Protein Expression. U.S. Patent US20110166197A1, 27 August 2009. [Google Scholar]
- Banks, W.A.; Farr, S.A.; Butt, W.; Kumar, V.B.; Franko, M.W.; Morley, J.E. Delivery across the blood-brain barrier of antisense directed against amyloid beta: Reversal of learning and memory deficits in mice overexpressing amyloid precursor protein. J. Pharmacol. Exp. Ther. 2001, 297, 1113–1121. [Google Scholar]
- Chauhan, N.B.; Siegel, G.J. Antisense inhibition at the beta-secretase-site of beta-amyloid precursor protein reduces cerebral amyloid and acetyl cholinesterase activity in Tg2576. Neuroscience 2007, 146, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Fiorini, A.; Sultana, R.; Förster, S.; Perluigi, M.; Cenini, G.; Cini, C.; Cai, J.; Klein, J.B.; Farr, S.A.; Niehoff, M.L.; et al. Antisense directed against PS-1 gene decreases brain oxidative markers in aged senescence accelerated mice (SAMP8) and reverses learning and memory impairment: A proteomics study. Free Radic. Biol. Med. 2013, 65, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caceres, A.; Kosik, K.S. Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature 1990, 343, 461–463. [Google Scholar] [CrossRef]
- Babu, E.; Muthu Mareeswaran, P.; Sathish, V.; Singaravadivel, S.; Rajagopal, S. Sensing and inhibition of amyloid-β based on the simple luminescent aptamer-ruthenium complex system. Talanta 2015, 134, 348–353. [Google Scholar] [CrossRef]
- Liang, H.; Shi, Y.; Kou, Z.; Peng, Y.; Chen, W.; Li, X.; Li, S.; Wang, Y.; Wang, F.; Zhang, X. Inhibition of BACE1 Activity by a DNA Aptamer in an Alzheimer’s Disease Cell Model. PLoS ONE 2015, 10, e0140733. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, E.; Choi, W.H.; Lee, J.; Lee, J.H.; Lee, H.; Kim, D.E.; Suh, Y.H.; Lee, M.J. Inhibitory RNA Aptamers of Tau Oligomerization and Their Neuroprotective Roles against Proteotoxic Stress. Mol. Pharm. 2016, 13, 2039–2048. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.C.; Rossi, J.J. RNA-based therapeutics: Current progress and future prospects. Chem. Biol. 2012, 19, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Wang, C.C.; Choy, K.W.; Du, Q.; Chen, J.; Wang, Q.; Li, L.; Chung, T.K.; Tang, T. Therapeutic potentials of gene silencing by RNA interference: Principles, challenges, and new strategies. Gene 2014, 538, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.Y.; Yu, A.M. Bioengineering of noncoding RNAs for research agents and therapeutics. Wiley Interdiscip. Rev. RNA 2016, 7, 186–197. [Google Scholar] [CrossRef] [Green Version]
- Pereira, P.; Pedro, A.Q.; Tomás, J.; Maia, C.J.; Queiroz, J.A.; Figueiras, A.; Sousa, F. Advances in time course extracellular production of human pre-miR-29b from Rhodovulum sulfidophilum. Appl. Microbiol. Biotechnol. 2016, 100, 3723–3734. [Google Scholar] [CrossRef] [PubMed]
- Beckert, B.; Masquida, B. Synthesis of RNA by in vitro transcription. Methods Mol. Biol. 2011, 703, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Sherlin, L.D.; Bullock, T.L.; Nissan, T.A.; Perona, J.J.; Lariviere, F.J.; Uhlenbeck, O.C.; Scaringe, S.A. Chemical and enzymatic synthesis of tRNAs for high-throughput crystallization. RNA 2001, 7, 1671–1678. [Google Scholar]
- Pereira, P.; Pedro, A.Q.; Queiroz, J.A.; Figueiras, A.R.; Sousa, F. New insights for therapeutic recombinant human miRNAs heterologous production: Rhodovolum sulfidophilum vs Escherichia coli. Bioengineered 2017, 8, 670–677. [Google Scholar] [CrossRef] [Green Version]
- Ponchon, L.; Dardel, F. Large scale expression and purification of recombinant RNA in Escherichia coli. Methods 2011, 54, 267–273. [Google Scholar] [CrossRef]
- Khurana, B.; Goyal, A.K.; Budhiraja, A.; Aora, D.; Vyas, S.P. Lipoplexes versus nanoparticles: pDNA/siRNA delivery. Drug Deliv. 2013, 20, 57–64. [Google Scholar] [CrossRef]
- Pereira, P.; Queiroz, J.A.; Figueiras, A.; Sousa, F. Current progress on microRNAs-based therapeutics in neurodegenerative diseases. Wiley Interdiscip. Rev. RNA 2017, 8, e1409. [Google Scholar] [CrossRef]
- Shende, P.; Ture, N.; Gaud, R.S.; Trotta, F. Lipid- and polymer-based plexes as therapeutic carriers for bioactive molecules. Int. J. Pharm. 2019, 558, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Rhea, E.M.; Banks, W.A. Role of the Blood-Brain Barrier in Central Nervous System Insulin Resistance. Front. Neurosci. 2019, 13, 521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.; Xu, B.; Yang, B.; Fu, J.; Chen, H.; Wang, X. Non-coding RNAs: The extensive and interactive regulators of the blood-brain barrier permeability. RNA Biol. 2021, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Blood-Brain Barrier and Delivery of Protein and Gene Therapeutics to Brain. Front. Aging Neurosci. 2019, 11, 373. [Google Scholar] [CrossRef]
- Pardridge, W.M. Treatment of Alzheimer’s Disease and Blood-Brain Barrier Drug Delivery. Pharmaceuticals 2020, 13, 394. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Lowes, L.P.; Alfano, L.N.; Arnold, W.D.; Shell, R.; Prior, T.W.; McColly, M.; Lehman, K.J.; Church, K.; Sproule, D.M.; Nagendran, S.; et al. Impact of Age and Motor Function in a Phase 1/2A Study of Infants with SMA Type 1 Receiving Single-Dose Gene Replacement Therapy. Pediatric Neurol. 2019, 98, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Zheng, X.; Yang, P.; Pang, X.; Qian, K.; Wang, P.; Xu, S.; Sheng, D.; Wang, L.; Cao, J.; et al. Small interfering RNA delivery to the neurons near the amyloid plaques for improved treatment of Alzheimer’s disease. Acta Pharm. Sin. B 2019, 9, 590–603. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhu, F.; Liu, Y.; Zheng, M.; Wang, Y.; Zhang, D.; Anraku, Y.; Zou, Y.; Li, J.; Wu, H.; et al. Blood-brain barrier-penetrating siRNA nanomedicine for Alzheimer’s disease therapy. Sci. Adv. 2020, 6, eabc7031. [Google Scholar] [CrossRef]
- Rezai, A.R.; Ranjan, M.; D’Haese, P.F.; Haut, M.W.; Carpenter, J.; Najib, U.; Mehta, R.I.; Chazen, J.L.; Zibly, Z.; Yates, J.R.; et al. Noninvasive hippocampal blood-brain barrier opening in Alzheimer’s disease with focused ultrasound. Proc. Natl. Acad. Sci. USA 2020, 117, 9180–9182. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Expression in AD |
|---|---|
| brain-miR-112 (Unknown) | Upregulated |
| brain-miR-161 (Unknown) | Upregulated |
| hsa-let-7d-3p | Upregulated |
| hsa-miR-5010-3p | Upregulated |
| hsa-miR-26a-5p | Upregulated |
| hsa-miR-1285-5p | Upregulated |
| hsa-miR-151a-3p | Upregulated |
| hsa-miR-103a-3p | Downregulated |
| hsa-miR-107 | Downregulated |
| hsa-miR-532-5p | Downregulated |
| hsa-miR-26b-5p | Downregulated |
| hsa-let-7f-5p | Downregulated |
| Institutions | miRNA | Sample Type | Disease | Status |
|---|---|---|---|---|
| Shanghai Mental Health Center | miRNA 107 | Plasma | MCI 1 | Unknown |
| CSF 1 | AD 1 | |||
| Sun Yat-sen University | miRNAs | Blood | MCI 1 | Unknown |
| AD 1 | ||||
| Seoul National University Hospital | miRNA 206 | Olfactory neuroepithelium tissue | AD 1 | Completed |
| Shanghai Mental Health Center | miRNAs | Plasma | MCI 1 due to AD 1 | Recruiting |
| AD 1 | ||||
| Shanghai Mental Health Center | miRNAs | Plasma | MCI 1 due to AD 1 | Not yet recruiting |
| Mild AD 1 | ||||
| Moderate AD 1 | ||||
| Severe AD 1 | ||||
| LBD 1 | ||||
| FTD 1 | ||||
| Neuromed IRCCS | miRNAs | Blood | MS 1 | Unknown |
| CSF1 | PD 1 | |||
| ALS 1 | ||||
| AD 1 | ||||
| University of Pisa | miRNA-30 | Blood | PD 1 | Completed |
| miRNA-7 | AD 1 |
| Drug | Definition | Expected Results | Phase |
|---|---|---|---|
| BAN2401 | Human monoclonal antibody Affinity for soluble Aβ 1 protofibrils | Human monoclonal antibody Reduction in Aβ 1 levels and cognitive decline | Phase 3 |
| Gantenerumab | Human monoclonal IgG1 antibody | Reduction in the Aβ 1 plaques | Phase 3 |
| Affinity for Aβ 1 aggregated forms | |||
| TRx0237 | Second generation Tau aggregation inhibitor | Prevention of Tau aggregation | Phase 3 |
| Dissolution of existing Tau aggregates | |||
| ALZT-OP1 | Cromolyn and ibuprofen (anti-inflammatory compounds) | Reduction in neuroinflammation | Phase 3 |
| Clearance of Aβ 1 | |||
| COR388 | Gingipains inhibitor (Virulence proteases from Porphyromonas gingivalis, common in AD 1 brains) | Reduction in Aβ 1 42 production, neuroinflammation, and hippocampal degeneration | Phase 2/3 |
| Masitinib | Selective tyrosine kinase inhibitor | Modulation of neuroinflammation | Phase 3 |
| AGB101 (Levetiracetam) | SV2A 1 modulator (anti-convulsant medication) | Reduction in Aβ 1-induced cognitive and functional impairment | Phase 3 |
| Blarcamesine | Sigma-1 chaperone receptor agonist | Prevention of memory loss | Phase 2/3 |
| Neuroprotective effects | |||
| Blockage of Tau hyperphosphorylation | |||
| Troriluzole | Prodrug conjugate of riluzole (mechanism of action is not fully understood) | Inhibition of glutamate release | Phase 2/3 |
| miRNA | Target Proteins | Therapeutic Potential | References |
|---|---|---|---|
| miRNA-9-5p | GSK-3β 1 | Inhibition of mitochondrial damage and oxidative stress | [115] |
| miRNA-15b | NF-κB 1 signaling | Inhibition of BACE1 1, APP 1 and Aβ 1 levels | [116] |
| BACE1 1 | |||
| miRNA-21 | PDCD4 1/ PI3K 1/AKT 1/GSK-3β 1 pathway | Inhibition of Aβ 1-apoptosis induced | [117] |
| miRNA-29a/b-1 | BACE1 1 | Regulation of BACE1 1 and Aβ 1 levels | [118] |
| miRNA-29c | BACE1 1 | Reduction in BACE1 1 and Aβ 1 levels | [119,120] |
| PKA 1/CREB 1 | Neuroprotection | ||
| miRNA-34a-5p | BACE1 1 | Inhibition of Aβ 1-induced apoptosis and oxidative stress | [121] |
| miRNA-31 | APP 1 | Improvement of cognition and memory deficits | [122] |
| BACE1 1 | Reduction in glutamate vesicles accumulation | ||
| Reduction in APP 1, BACE1 1 and Aβ 1 | |||
| miRNA-101a-3p | APP 1 | Regulation in APP 1 and Aβ 1 levels | [123] |
| miRNA-98 | HEY2 1 | Inactivation of Notch signaling pathway | [124] |
| miRNA-101 | APP 1 | Reduction in APP 1 and Aβ 1 levels | [125] |
| miRNA-106b | Fyn 1 | Inhibition of Aβ 11-42-induced Tau phosphorylation at Tyr18 1 | [126] |
| miRNA-107 | BACE1 1 | Inhibition of BACE1 1 | [127] |
| miRNA-124-3p | CAV1-PI3K/Akt/GSK3β 1 pathway | Attenuation of cell and abnormal Tau hyperphosphorylation | [128] |
| miRNA-125b-5p | BACE1 1 | Inhibition of Aβ 1-induced apoptosis and oxidative stress | [121] |
| miRNA-137 | SPT 1 | Inhibition of Aβ 1 levels | [129] |
| miRNA-153 | APP 1 | Reduction in APP 1 levels | [130] |
| miRNA-181c | SPT 1 | Inhibition of Aβ 1 levels | [129] |
| miRNA-195 | BACE1 1 | Inhibition of BACE1 1 and Aβ 1 levels | [131] |
| miRNA-200a-3p | Bax 1/CASP3 1 axis | Inhibition of apoptosis, Aβ 1 and p-Tau levels | [132] |
| BACE1 1 | |||
| PKA 1 | |||
| miRNA-200b/c | PS6KB1 1 (Insulin signaling) | Reduction in Aβ 1 secretion relieved and memory impairments | [133] |
| miRNA-298 | BACE1 1 | Repression of APP 1, BACE1 1, Aβ 1 and some Tau forms | [134] |
| APP 1 | |||
| Tau under study | |||
| miRNA-326 | VAV1 1 | Inhibition of Aβ 1 deposition, apoptosis, Tau phosphorylation | [135] |
| miRNA-328 | BACE1 1 | Regulation of BACE1 1 expression | [136] |
| miRNA-339-5p | BACE1 1 | Inhibition of BACE1 1 expression | [137] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riscado, M.; Baptista, B.; Sousa, F. New RNA-Based Breakthroughs in Alzheimer’s Disease Diagnosis and Therapeutics. Pharmaceutics 2021, 13, 1397. https://doi.org/10.3390/pharmaceutics13091397
Riscado M, Baptista B, Sousa F. New RNA-Based Breakthroughs in Alzheimer’s Disease Diagnosis and Therapeutics. Pharmaceutics. 2021; 13(9):1397. https://doi.org/10.3390/pharmaceutics13091397
Chicago/Turabian StyleRiscado, Micaela, Bruno Baptista, and Fani Sousa. 2021. "New RNA-Based Breakthroughs in Alzheimer’s Disease Diagnosis and Therapeutics" Pharmaceutics 13, no. 9: 1397. https://doi.org/10.3390/pharmaceutics13091397