
Targeting BRAF Activation as Acquired Resistance Mechanism to EGFR Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small-Cell Lung Cancer


Abstract
:1. Introduction
2. BRAF-Mutant NSCLC
3. BRAF Alterations as ARMs to Osimertinib in EGFR-Mutant NSCLC
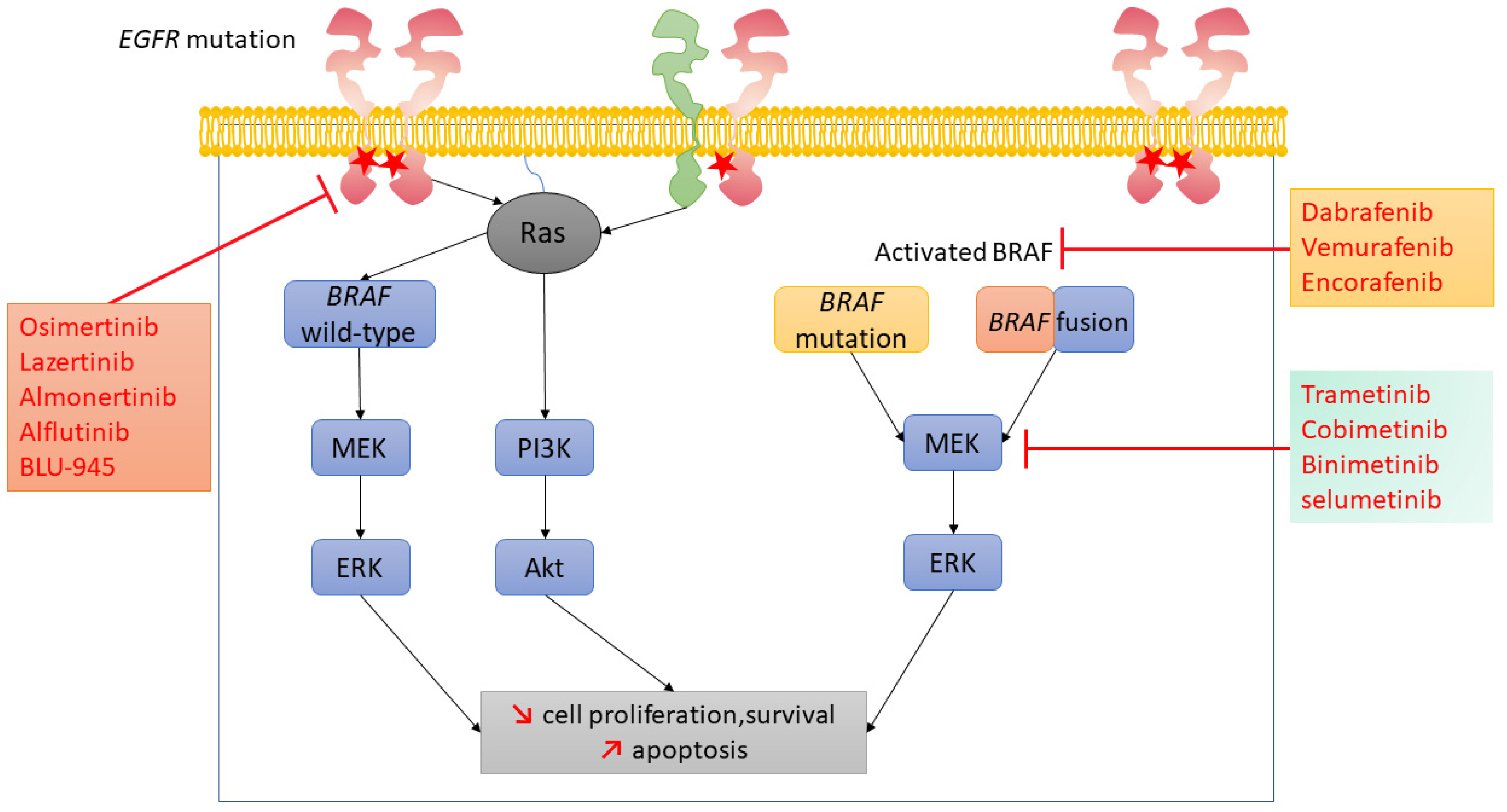
4. Targeting EGFR/BRAF/MEK Pathway in NSCLC with BRAF Activation as an ARM to Osimertinib
4.1. BRAF V600 Mutation
4.2. Non-BRAF V600 Mutations and BRAF Rearrangements
5. Discussion and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar]
- Houston, K.A.; Henley, S.J.; Li, J.; White, M.C.; Richards, T.B. Patterns in lung cancer incidence rates and trends by histologic type in the United States, 2004–2009. Lung Cancer 2014, 86, 22–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlesi, F.; Mazieres, J.; Merlio, J.P.; Debieuvre, D.; Mosser, J.; Lena, H.; Ouafik, L.; Besse, B.; Rouquette, I.; Westeel, V.; et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: Results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016, 387, 1415–1426. [Google Scholar] [CrossRef]
- Couraud, S.; Souquet, P.J.; Paris, C.; Do, P.; Doubre, H.; Pichon, E.; Dixmier, A.; Monnet, I.; Etienne-Mastroianni, B.; Vincent, M.; et al. BioCAST/IFCT-1002: Epidemiological and molecular features of lung cancer in never-smokers. Eur. Respir. J. 2015, 45, 1403–1414. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Alexander, R.E.; MacLennan, G.T.; Cummings, O.W.; Montironi, R.; Lopez-Beltran, A.; Cramer, H.M.; Davidson, D.D.; Zhang, S. Molecular pathology of lung cancer: Key to personalized medicine. Mod. Pathol. 2012, 25, 347–369. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2019, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 378, 113–125. [Google Scholar] [CrossRef]
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 2019, 121, 725–737. [Google Scholar] [CrossRef]
- Schalm, S.S.; Dineen, T.; Lim, S.M.; Park, C.W.; Hsieh, J.; Woessner, R.; Zhang, Z.; Wilson, K.; Eno, M.; Wilson, D.; et al. 384P BLU-945, a highly potent and selective 4th generation EGFR TKI for the treatment of EGFR T790M/C797S resistant NSCLC. Ann. Oncol. 2020, 31, S1391. [Google Scholar] [CrossRef]
- Schalm, S.S.; Dineen, T.; Lim, S.M.; Park, C.W.; Hsieh, J.; Woessner, R.; Zhang, Z.; Wilson, K.; Eno, M.; Wilson, D.; et al. 1296P BLU-945, a highly potent and selective 4th generation EGFR TKI for the treatment of EGFR T790M/C797S resistant NSCLC. Ann. Oncol. 2020, 31, S839. [Google Scholar] [CrossRef]
- Wang, S.; Song, Y.; Liu, D. EAI045: The fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. 2017, 385, 51–54. [Google Scholar] [CrossRef]
- Dankort, D.; Filenova, E.; Collado, M.; Serrano, M.; Jones, K.; McMahon, M. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev. 2007, 21, 379–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, P.T.C.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Project, C.G.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; et al. Mechanism of Activation of the RAF-ERK Signaling Pathway by Oncogenic Mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Yao, Z.; Torres, N.M.; Tao, A.; Gao, Y.; Luo, L.; Li, Q.; de Stanchina, E.; Abdel-Wahab, O.; Solit, D.B.; Poulikakos, P.I.; et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell 2015, 28, 370–383. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and Clinicopathologic Associations of Oncogenic BRAF in Metastatic Melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef]
- Villaruz, L.C.; Socinski, M.A.; Abberbock, S.; Berry, L.D.; Johnson, B.E.; Kwiatkowski, D.J.; Iafrate, A.J.; Varella-Garcia, M.; Franklin, W.A.; Camidge, D.R.; et al. Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the Lung Cancer Mutation Consortium. Cancer 2015, 121, 448–456. [Google Scholar] [CrossRef] [Green Version]
- Kris, M.G.; Johnson, B.E.; Berry, L.D.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Varella-Garcia, M.; Franklin, W.A.; Aronson, S.L.; Su, P.-F.; et al. Using Multiplexed Assays of Oncogenic Drivers in Lung Cancers to Select Targeted Drugs. JAMA 2014, 311, 1998–2006. [Google Scholar] [CrossRef]
- Skoulidis, F.; Heymach, J.V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 2019, 19, 495–509. [Google Scholar] [CrossRef]
- In The Cancer Genome Atlas Database. Available online: http://www.cbioportal.org/ (accessed on 20 August 2021).
- Paik, P.K.; Arcila, M.E.; Fara, M.; Sima, C.S.; Miller, V.A.; Kris, M.G.; Ladanyi, M.; Riely, G.J. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 2046–2051. [Google Scholar] [CrossRef] [Green Version]
- Pratilas, C.A.; Hanrahan, A.J.; Halilovic, E.; Persaud, Y.; Soh, J.; Chitale, D.; Shigematsu, H.; Yamamoto, H.; Sawai, A.; Janakiraman, M.; et al. Genetic Predictors of MEK Dependence in Non–Small Cell Lung Cancer. Cancer Res. 2008, 68, 9375–9383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.W.; Hutter, B.; Jäger, N.; Korshunov, A.; Kool, M.; Warnatz, H.-J.; Zichner, T.; Lambert, S.R.; Ryzhova, M.; Quang, D.A.K.; et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat. Genet. 2013, 45, 927–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberlein, C.A.; Stetson, D.; Markovets, A.A.; Al-Kadhimi, K.J.; Lai, Z.; Fisher, P.R.; Meador, C.B.; Spitzler, P.; Ichihara, E.; Ross, S.J.; et al. Acquired Resistance to the Mutant-Selective EGFR Inhibitor AZD9291 Is Associated with Increased Dependence on RAS Signaling in Preclinical Models. Cancer Res. 2015, 75, 2489–2500. [Google Scholar] [CrossRef] [Green Version]
- Mauclet, C.; Collard, P.; Ghaye, B.; Hoton, D.; Nana, F.A. Tumor response to EGFR/BRAF/MEK co-inhibition in a patient with EGFR mutated lung adenocarcinoma developing a BRAFV600 mutation as an acquired resistance mechanism. Lung Cancer 2021, 159, 42–44. [Google Scholar] [CrossRef] [PubMed]
- Vojnic, M.; Kubota, D.; Kurzatkowski, C.; Offin, M.; Suzawa, K.; Benayed, R.; Schoenfeld, A.J.; Plodkowski, A.J.; Poirier, J.T.; Rudin, C.M.; et al. Acquired BRAF Rearrangements Induce Secondary Resistance to EGFR therapy in EGFR-Mutated Lung Cancers. J. Thorac. Oncol. 2019, 14, 802–815. [Google Scholar] [CrossRef] [PubMed]
- Vojnic, M.; Kurzatkowski, C.; Kubota, D.; Suzawa, K.; Liu, Z.; Mattar, M.; Khodos, I.; Poirier, J.T.; Stanchina, E.d.; Rudin, C.M.; et al. Acquired BRAF fusions as a mechanism of resistance to EGFR therapy. J. Clin. Oncol. 2018, 36, 12122. [Google Scholar] [CrossRef]
- Roper, N.; Brown, A.-L.; Wei, J.S.; Pack, S.; Trindade, C.; Kim, C.; Restifo, O.; Gao, S.; Sindiri, S.; Mehrabadi, F.; et al. Clonal Evolution and Heterogeneity of Osimertinib Acquired Resistance Mechanisms in EGFR Mutant Lung Cancer. Cell Rep. Med. 2020, 1, 100007. [Google Scholar] [CrossRef] [PubMed]
- La Monica, S.; Minari, R.; Cretella, D.; Bonelli, M.; Fumarola, C.; Cavazzoni, A.; Galetti, M.; Digiacomo, G.; Riccardi, F.; Petronini, P.G.; et al. Acquired BRAF G469A Mutation as a Resistance Mechanism to First-Line Osimertinib Treatment in NSCLC Cell Lines Harboring an EGFR Exon 19 Deletion. Target. Oncol. 2019, 14, 619–626. [Google Scholar] [CrossRef]
- Ho, C.-C.; Liao, W.-Y.; Lin, C.-A.; Shih, J.-Y.; Yu, C.-J.; Chih-Hsin Yang, J. Acquired BRAF V600E Mutation as Resistant Mechanism after Treatment with Osimertinib. J. Target. Oncol. 2017, 12, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Sequist, L.V.; Han, J.Y.; Ahn, M.J.; Cho, B.C.; Yu, H.; Kim, S.W.; Yang, J.C.; Lee, J.S.; Su, W.C.; Kowalski, D.; et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: Interim results from a multicentre, open-label, phase 1b study. Lancet. Oncol. 2020, 21, 373–386. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Cantarini, M.; Frewer, P.; Hawkins, G.; Peters, J.; Howarth, P.; Ahmed, G.F.; Sahota, T.; Hartmaier, R.; Li-Sucholeiki, X.; et al. SAVANNAH: A Phase II trial of osimertinib plus savolitinib for patients (pts) with EGFR-mutant, MET-driven (MET+), locally advanced or metastatic non-small cell lung cancer (NSCLC), following disease progression on osimertinib. J. Clin. Oncol. 2019, 37, TPS9119. [Google Scholar] [CrossRef]
- Yu, H.; Goldberg, S.; Le, X.; Piotrowska, Z.; Smith, P.; Mensi, I.; Kirova, B.; Chmielecki, J.; Li-Sucholeicki, X.; Szekeres, P.; et al. P2.01-22 ORCHARD: A Phase II Platform Study in Patients with Advanced NSCLC Who Have Progressed on First-Line Osimertinib Therapy. J. Target. Oncol. 2019, 14, S647. [Google Scholar] [CrossRef]
- Cho, B.C.; Lee, K.H.; Cho, E.K.; Kim, D.W.; Lee, J.S.; Han, J.Y.; Kim, S.W.; Spira, A.; Haura, E.B.; Sabari, J.K.; et al. 1258O Amivantamab (JNJ-61186372), an EGFR-MET bispecific antibody, in combination with lazertinib, a 3rd-generation tyrosine kinase inhibitor (TKI), in advanced EGFR NSCLC. Ann. Oncol. 2020, 31, S813. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.M.E.; et al. Osimertinib or Platinum–Pemetrexed in EGFR T790M–Positive Lung Cancer. N. Engl. J. Med. 2016, 376, 629–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planchard, D.; Smit, E.F.; Groen, H.J.M.; Mazieres, J.; Besse, B.; Helland, Å.; Giannone, V.; D’Amelio, A.M., Jr.; Zhang, P.; Mookerjee, B.; et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: An open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1307–1316. [Google Scholar] [CrossRef]
- Planchard, D.; Besse, B.; Kim, T.M.; Quoix, E.A.; Souquet, P.J.; Mazieres, J.; Barlesi, F.; Groen, H.J.M.; Smit, E.F.; Baik, C.S.; et al. Updated survival of patients (pts) with previously treated BRAF V600E–mutant advanced non-small cell lung cancer (NSCLC) who received dabrafenib (D) or D + trametinib (T) in the phase II BRF113928 study. J. Clin. Oncol. 2017, 35, 9075. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined Vemurafenib and Cobimetinib in BRAF-Mutated Melanoma. J. Clin. Oncol. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [Green Version]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Tsuboi, M.; He, J.; John, T.; Grohe, C.; Majem, M.; Goldman, J.W.; Laktionov, K.; Kim, S.-W.; Kato, T.; et al. Osimertinib in Resected EGFR-Mutated Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Meng, P.; Koopman, B.; Kok, K.; ter Elst, A.; Schuuring, E.; van Kempen, L.C.; Timens, W.; Hiltermann, T.J.N.; Groen, H.J.M.; van den Berg, A.; et al. Combined osimertinib, dabrafenib and trametinib treatment for advanced non-small-cell lung cancer patients with an osimertinib-induced BRAFV600E mutation. Lung Cancer 2020, 146, 358–361. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cancer Types | BRAF Alterations | Proportions | References |
|---|---|---|---|
| NSCLC | BRAF mutations | 2–6% of NSCLC | [3,18,19] |
| V600E | 50–65% of BRAF mutations | [19,20,21] | |
| Non-V600E | 35–50% of BRAF mutations | [19,20,21,22] | |
| In never smokers | 19–23% of BRAF mutations | [16,19] | |
| In former smokers | 69–72% of BRAF mutations | [16,19] | |
| In current smokers | 5–13% of BRAF mutations | [16,19] | |
| BRAF rearrangements | 0 (not reported) | [19] | |
| Pilocytic astrocytoma | BRAF rearrangements | 78.13% | [19,23] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aboubakar Nana, F.; Ocak, S. Targeting BRAF Activation as Acquired Resistance Mechanism to EGFR Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small-Cell Lung Cancer. Pharmaceutics 2021, 13, 1478. https://doi.org/10.3390/pharmaceutics13091478
Aboubakar Nana F, Ocak S. Targeting BRAF Activation as Acquired Resistance Mechanism to EGFR Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small-Cell Lung Cancer. Pharmaceutics. 2021; 13(9):1478. https://doi.org/10.3390/pharmaceutics13091478
Chicago/Turabian StyleAboubakar Nana, Frank, and Sebahat Ocak. 2021. "Targeting BRAF Activation as Acquired Resistance Mechanism to EGFR Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small-Cell Lung Cancer" Pharmaceutics 13, no. 9: 1478. https://doi.org/10.3390/pharmaceutics13091478
APA StyleAboubakar Nana, F., & Ocak, S. (2021). Targeting BRAF Activation as Acquired Resistance Mechanism to EGFR Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small-Cell Lung Cancer. Pharmaceutics, 13(9), 1478. https://doi.org/10.3390/pharmaceutics13091478