
Klotho and Mesenchymal Stem Cells: A Review on Cell and Gene Therapy for Chronic Kidney Disease and Acute Kidney Disease


Abstract
:1. Introduction
2. Klotho and Chronic Kidney Disease
2.1. Chronic Kidney Disease
2.2. Klotho in Chronic Kidney Disease
2.2.1. Klotho and FGF-23
2.2.2. Klotho/FGF/PTH Axis
2.2.3. CKD and Cardiovascular Disease
2.2.4. Klotho and Inflammation
2.2.5. Klotho and Fibrosis
3. Acute Kidney Injury
3.1. Klotho in Acute Kidney Injury
3.1.1. Klotho, Inflammation and AKI
3.1.2. Klotho and Non-Inflammatory Mechanisms in AKI
4. Therapeutic Potential of Klotho in Acute and Chronic Kidney Diseases
- (a).
- DNA methyltransferase inhibitor: In regard to the DNA methyltransferase inhibitor azacytidine, it has been observed that Klotho’s promoter is located in a region rich in cytosine and guanine—a CpG island [206,207] that lacks sequences for classic regulatory elements in this region [206]. Azacytidine, in turn, is able to promote an augmentation in the promoter activity of the Klotho gene, leading to a rise in the levels of this protein in cells. The use of this compound in vivo, though, is difficult and regards future research due to the variety of possible activities presented by this compound [2].
- (b).
- Agonists for PPAR-γ: Moving on to troglitazone and ciglitazone, these drugs are classified as thiazolidinediones and they act as agonists of peroxisome proliferator-activated receptor gamma (PPAR-γ) [208]. One study indicated that, in cells from medullary collecting ducts, proximal tubules and distal tubules, there is an induction of the expression of Klotho genes by these compounds, both in a time- and dose-dependent manner. The same study proposed that the activation of this receptor is, then, a potential mechanism for the effect observed, since a selective PPAR-γ antagonist abolished the process [208]. In regard to the clinical use of these molecules, the challenges involve edema, weight gain and osteoporosis, among others [209].
- (c).
- Histone deacetylase inhibitors: The other approach discussed in Figure 4 is the use of histone deacetylase inhibitors, such as trichostatin A and valproic acid. Data indicate that in cells treated with these compounds, the reduced expression of Klotho induced by TWEAK or TNF-α is prevented [110]. Likewise, an increase in Klotho’s gene expression in some cell lineages has been reported [206]. In spite of the fact that these inhibitors are being evaluated in clinical trials involving the treatment of cancer, for example, their use in vivo is still difficult, because of adverse reactions, especially cardio-toxicity [210].
- (d).
- RAAS inhibitors: Regarding the use of RAAS inhibitors, in turn, it has been demonstrated in a rodent model of chronic nephropathy that the inhibition of angiotensin II type 1 receptor with the use of losartan results in an augmentation in Klotho expression, along with a reduction in kidney histological damage [211]. A meta-analysis of randomized controlled trials and systematic reviews, however, has shown that in order to improve some functional parameters in CKD patients, such as blood pressure control and the reduction of proteinuria, dual blockade of RAAS is better than monotherapy [212]. Thus, although the previously discussed alternative represents a potential strategy for the treatment of CKD, the approach itself does not diminish the development of ESKD, for example, so it does not cause a long-term amelioration in the treatment of CKD, as reviewed by that group [212].
- (e).
- Paricalcitol: As illustrated in Figure 4, other compounds are interesting for the elevation of Klotho levels, such as vitamin derivatives, such as paricalcitol. In a study with uremic rats, conducted by Ritter, it was demonstrated that paricalcitol blocks the reduction of Klotho mRNA and protein levels in renal tubules [213]. In another study, on the other hand, an increase in Klotho levels in the kidneys was not observed [214]. Currently, there is a lack of information in the literature in regard to the influence of paricalcitol and other agonists for vitamin D receptors on CKD progression and cardiovascular risk [215]. Furthermore, a meta-analysis highlighted the necessity of future randomized trials to assess the effects of paricalcitol on ESKD progression and mortality in individuals, although some data point out that this approach is effective in the reduction of proteinuria [216]. Moreover, the authors reported that there was a trend towards hypocalcemia in patients [216]. Thus, the clinical feasibility of this approach still needs further studies.
- (f).
- Intermedin: In regard to intermedin, a study with rats with CKD has shown that the decrease in Klotho protein levels was overturned by intermedin in the kidneys, plasma and calcified aorta [217]. This compound also diminished vascular calcification in this animal model [217]. Intermedin, then, could be a promising strategy to increase Klotho levels. However, the lack of information in literature about this compound is a downside of it. Only two experimental papers were found when we searched for information about intermedin and Klotho in Pubmed [217,218]. Thus, further studies are needed in order to shed light on the efficacy and safety of intermedin and on how exactly it is related to Klotho levels.
- (g).
- Statins: Statins have also been proposed as a promising approach to promote Klotho overexpression. Studies with rodents demonstrated that these drugs led both to an increase in renal Klotho levels and to the attenuation of the reduction of Klotho in nephropathy [219]. Furthermore, there was an improvement in the resistance to oxidative stress in CKD [219]. Although the use of statins for patients with correct medical recommendations is considered acceptable because of their benefits [220], a review has pointed out that genetic testing of transporter genes which affect the internalization or efflux of statins would be interesting, considering the existent polymorphisms that can affect both the safety and efficacy of statins, since this genetic background might interfere in the incidence of myopathy and statins results in patients [221].
- (h).
- Recombinant protein: The administration of recombinant Klotho, in turn, is a potential strategy to increase its levels [48]. Studies with rodent models for kidney diseases have shown the attenuation of renal fibrosis [177], the avoidance of progression from AKI to CKD, cardiac remodeling and an improvement in renal and cardiac parameters [177]. Researchers have also observed the reduction of kidney damage and recovery from AKI [46]. However, the Klotho protein’s instability in urine and blood [222] might demand alternatives to an effective recombinant protein administration [223]. Timing for treatment should also be analyzed so that there would not be an impaired recovery of the kidneys [223].
- (i).
- Adenoviral delivery: Lastly, as described in Figure 4, adenoviral delivery of Klotho leads to an increase in this protein level. Studies with rodents with reduced Klotho expression have indicated the mitigation of renal damage promoted by adenoviral Klotho delivery, seen, for example, with a decrease in tubular atrophy [224]. Regarding this approach, there are also other studies involving rodent models for diabetic kidney disease (DKD) that show the amelioration of the disease after the delivery of Klotho, which might be associated with the inhibition of RAAS activation and the Wnt/β-catenin pathway [225] and the improvement of creatinine clearance, proteinuria and tubulointerstitial damage in animals with Ang-II infusion [226]. Moreover, in rodent models for AKI studies have demonstrated the mitigation of histological damage and apoptosis and the improvement of serum creatinine levels post-injury after Klotho delivery [183]. Although promising results have been achieved so far, it is important to mention that the adeno-viral delivery of genes requires caution, due to possible mutagenesis or immunogenicity [227].
- (j).
- Mesenchymal stem cells and extracellular vesicles: Taking into account all the approaches discussed previously, we decided to explore the potential of MSCs due to their applicability in regenerative medicine. Currently, these are the most studied type of stem cells [228] and they are also the most commonly used ones [229]. They are seen both as a therapeutic approach themselves, through their direct administration to patients—either genetically modified or not—and as a way to grow organoids in culture, for instance, which can also be administered to patients later [230]. These cells present potential for the treatment of different diseases, due to their immune modulation and differentiation properties, along with their tropism for injured tissues and their rich secretome, as it will be addressed in this review [229]. Hence, considering these characteristics of MSCs, alongside Klotho’s anti-inflammatory potential, we speculate that these cells can act like a “Trojan Horse’’ in vivo, delivering Klotho to the renal tissue, and that they could therefore modify and improve the microenvironment conditions for Klotho. This protein, in turn, would be able to ameliorate the surroundings conditions for MSCs as well, and consequently increase their efficacy in vivo. Therefore, we propose that MSCs and Klotho would act in a synergic way, contributing to the improvement of kidney conditions in CKD and AKI, as illustrated in Figure 5.
4.1. Mesenchymal Stem Cells
4.1.1. Properties and Characterization of Mesenchymal Stem Cells
4.1.2. Efficacy and Safety of Mesenchymal Stem Cells
4.2. Crosstalk between Klotho and Mesenchymal Stem Cells
4.3. Perspectives on MSC and Gene Therapy for Chronic and Acute Kidney Disease
4.3.1. MSC-Derived Extracellular Vesicles
4.3.2. MicroRNAs
4.3.3. MSCs Combined with Sodium-Glucose Co-Transporter-2 Inhibitors
4.3.4. Klotho, microRNAs and Genetically Modified MSCs in Chronic and Acute Kidney Disease
4.3.5. Challenges for the Clinical Application of MSCs
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kuro-O, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, S.; Combet, E.; Stenvinkel, P.; Shiels, P.G. Klotho, Aging, and the Failing Kidney. Front. Endocrinol. 2020, 11, 560. [Google Scholar] [CrossRef]
- Lanzani, C.; Citterio, L.; Vezzoli, G. Klotho: A link between cardiovascular and non-cardiovascular mortality. Clin. Kidney J. 2020, 13, 926–932. [Google Scholar] [CrossRef]
- Bloch, L.; Sineshchekova, O.; Reichenbach, D.; Reiss, K.; Saftig, P.; Kuro-O, M.; Kaether, C. Klotho is a substrate for α-, β- and γ-secretase. FEBS Lett. 2009, 583, 3221–3224. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-D.; Podvin, S.; Gillespie, E.; Leeman, S.E.; Abraham, C.R. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc. Natl. Acad. Sci. USA 2007, 104, 19796–19801. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-D.; Tung, T.Y.; Liang, J.; Zeldich, E.; Zhou, T.B.T.; Turk, B.E.; Abraham, C.R. Identification of Cleavage Sites Leading to the Shed Form of the Anti-Aging Protein Klotho. Biochemistry 2014, 53, 5579–5587. [Google Scholar] [CrossRef] [Green Version]
- Zou, D.; Wu, W.; He, Y.; Ma, S.; Gao, J. The role of klotho in chronic kidney disease. BMC Nephrol. 2018, 19, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuro-O, M. The Klotho proteins in health and disease. Nat. Rev. Nephrol. 2018, 15, 27–44. [Google Scholar] [CrossRef]
- Imura, A.; Iwano, A.; Tohyama, O.; Tsuji, Y.; Nozaki, K.; Hashimoto, N.; Fujimori, T.; Nabeshima, Y.-I. Secreted Klotho protein in sera and CSF: Implication for post-translational cleavage in release of Klotho protein from cell membrane. FEBS Lett. 2004, 565, 143–147. [Google Scholar] [CrossRef]
- Mencke, R.; Harms, G.; Moser, J.; Van Meurs, M.; Diepstra, A.; Leuvenink, H.; Hillebrands, J.-L. Human alternative Klotho mRNA is a nonsense-mediated mRNA decay target inefficiently spliced in renal disease. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.; Groen, A.J.; Molostvov, G.; Lu, T.; Lilley, K.S.; Snead, D.; James, S.; Wilkinson, I.B.; Ting, S.; Hsiao, L.-L.; et al. α-Klotho Expression in Human Tissues. J. Clin. Endocrinol. Metab. 2015, 100, E1308–E1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.C.; Kuro-O, M.; Moe, O.W. Renal and Extrarenal Actions of Klotho. Semin. Nephrol. 2013, 33, 118–129. [Google Scholar] [CrossRef] [Green Version]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of Aging in Mice by the Hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akimoto, T.; Yoshizawa, H.; Watanabe, Y.; Numata, A.; Yamazaki, T.; Takeshima, E.; Iwazu, K.; Komada, T.; Otani, N.; Morishita, Y.; et al. Characteristics of urinary and serum soluble Klotho protein in patients with different degrees of chronic kidney disease. BMC Nephrol. 2012, 13, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semba, R.D.; Moghekar, A.R.; Hu, J.; Sun, K.; Turner, R.; Ferrucci, L.; O’Brien, R. Klotho in the cerebrospinal fluid of adults with and without Alzheimer’s disease. Neurosci. Lett. 2014, 558, 37–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, R.; Nakada, Y.; Hu, M.C.; Kurosu, H.; Wang, L.; Nakatani, T.; Shi, M.; Eliseenkova, A.V.; Razzaque, M.S.; Moe, O.W.; et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc. Natl. Acad. Sci. USA 2010, 107, 407–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urakawa, I.; Yamazaki, Y.; Shimada, T.; Iijima, K.; Hasegawa, H.; Okawa, K.; Fujita, T.; Fukumoto, S.; Yamashita, T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nat. Cell Biol. 2006, 444, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H.; Ogawa, Y.; Miyoshi, M.; Yamamoto, M.; Nandi, A.; Rosenblatt, K.P.; Baum, M.G.; Schiavi, S.; Hu, M.-C.; Moe, O.W.; et al. Regulation of Fibroblast Growth Factor-23 Signaling by Klotho. J. Biol. Chem. 2006, 281, 6120–6123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.C.; Shi, M.; Zhang, J.; Pastor, J.; Nakatani, T.; Lanske, B.; Razzaque, M.S.; Rosenblatt, K.P.; Baum, M.G.; Kuro-O, M.; et al. Klotho: A novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 2010, 24, 3438–3450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Q.; Hoefs, S.; van der Kemp, A.W.; Topala, C.N.; Bindels, R.J.; Hoenderop, J.G. The ß-Glucuronidase Klotho Hydrolyzes and Activates the TRPV5 Channel. Science 2005, 310, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.-K.; Ortega, B.; Kurosu, H.; Rosenblatt, K.P.; Kuro-O, M.; Huang, C.-L. Removal of sialic acid involving Klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc. Natl. Acad. Sci. USA 2008, 105, 9805–9810. [Google Scholar] [CrossRef] [Green Version]
- Erben, R.G.; Andrukhova, O. FGF23-Klotho signaling axis in the kidney. Bone 2017, 100, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Saar-Kovrov, V.; Donners, M.M.P.C.; van der Vorst, E.P.C. Shedding of Klotho: Functional Implications in Chronic Kidney Disease and Associated Vascular Disease. Front. Cardiovasc. Med. 2021, 7, 617842. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Kuro-O, M.; Moe, O.W. Secreted Klotho and Chronic Kidney Disease. Adv. Exp. Med. Biol. 2012, 728, 126–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangos, S.; Amaral, A.P.; Faul, C.; Jüppner, H.; Reiser, J.; Wolf, M. Expression of fgf23 and klotho in developing embryonic tissues and adult kidney of the zebrafish, Danio rerio. Off. Publ. Eur. Dial. Transpl. Assoc.—Eur. Ren. Assoc. 2012, 27, 4314–4322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.-A.; Watanabe, M.; Yamada, H.; Nagai, A.; Kinuta, M.; Takei, K. Immunohistochemical Localization of Klotho Protein in Brain, Kidney, and Reproductive Organs of Mice. Cell Struct. Funct. 2004, 29, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Ohyama, Y.; Kurabayashi, M.; Masuda, H.; Nakamura, T.; Aihara, Y.; Kaname, T.; Suga, T.; Arai, M.; Aizawa, H.; Matsumura, Y.; et al. Molecular Cloning of RatklothocDNA: Markedly Decreased Expression ofklothoby Acute Inflammatory Stress. Biochem. Biophys. Res. Commun. 1998, 251, 920–925. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Cappola, A.R.; Sun, K.; Bandinelli, S.; Dalal, M.; Crasto, C.; Guralnik, J.M.; Ferrucci, L. Plasma Klotho and Mortality Risk in Older Community-Dwelling Adults. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2011, 66, 794–800. [Google Scholar] [CrossRef] [Green Version]
- Pavik, I.; Jaeger, P.; Ebner, L.; Wagner, C.A.; Petzold, K.; Spichtig, D.; Poster, D.; Wüthrich, R.P.; Russmann, S.; Serra, A.L. Secreted Klotho and FGF23 in chronic kidney disease Stage 1 to 5: A sequence suggested from a cross-sectional study. Off. Publ. Eur. Dial. Transpl. Assoc.—Eur. Ren. Assoc. 2013, 28, 352–359. [Google Scholar] [CrossRef] [Green Version]
- De Borst, M.; Vervloet, M.G.; Ter Wee, P.M.; Navis, G. Cross Talk Between the Renin-Angiotensin-Aldosterone System and Vitamin D-FGF-23-klotho in Chronic Kidney Disease: Figure 1. J. Am. Soc. Nephrol. 2011, 22, 1603–1609. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Fernandez, B.; Izquierdo, M.C.; Valiño-Rivas, L.; Nastou, D.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Albumin downregulates Klotho in tubular cells. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2018, 33, 1712–1722. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, M.; Rotundo, S.; Chiodini, P.; Gagliardi, I.; Michael, A.; Angotti, E.; Borrelli, S.; Serra, R.; Foti, D.; De Sarro, G.; et al. Contribution of Predictive and Prognostic Biomarkers to Clinical Research on Chronic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 5846. [Google Scholar] [CrossRef] [PubMed]
- Lousa, I.; Reis, F.; Beirão, I.; Alves, R.; Belo, L.; Santos-Silva, A. New Potential Biomarkers for Chronic Kidney Disease Management—A Review of the Literature. Int. J. Mol. Sci. 2020, 22, 43. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Su, W.; Shen, Z.; Wang, R. Correlation between Soluble α-Klotho and Renal Function in Patients with Chronic Kidney Disease: A Review and Meta-Analysis. BioMed Res. Int. 2018, 2018, 9481475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- US Government. Centers for Disease Control and Prevention. Chronic Kidney Disease Initiative. Chronic Kidney Disease Basics. Available online: https://www.Cdc.Gov/kidneydisease/basics.Html (accessed on 9 May 2021).
- Lu, X.; Hu, M.C. Klotho/FGF23 Axis in Chronic Kidney Disease and Cardiovascular Disease. Kidney Dis. 2017, 3, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Shabaka, A.; Cases-Corona, C.; Fernandez-Juarez, G. Therapeutic Insights in Chronic Kidney Disease Progression. Front. Med. 2021, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Kuro-O, M.; Moe, O.W. Klotho and Chronic Kidney Disease. Contrib. Nephrol. 2013, 180, 47–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zewinger, S.; Rauen, T.; Rudnicki, M.; Federico, G.; Wagner, M.; Triem, S.; Schunk, S.J.; Petrakis, I.; Schmit, D.; Wagenpfeil, S.; et al. Dickkopf-3 (DKK3) in Urine Identifies Patients with Short-Term Risk of eGFR Loss. J. Am. Soc. Nephrol. 2018, 29, 2722–2733. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.R.; Lee, D.; Cai, M.M.; Tomlinson, L.; Ford, M.L.; McMahon, L.P.; Holt, S. Urinary neutrophil gelatinase-associated lipocalin may aid prediction of renal decline in patients with non-proteinuric Stages 3 and 4 chronic kidney disease (CKD). Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2013, 28, 1569–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Castañeda, J.R.; Rodelo-Haad, C.; De Mier, M.V.P.-R.; Martin-Malo, A.; Santamaria, R.; Rodriguez, M. Klotho/FGF23 and Wnt Signaling as Important Players in the Comorbidities Associated with Chronic Kidney Disease. Toxins 2020, 12, 185. [Google Scholar] [CrossRef] [Green Version]
- Gagliardini, E.; Benigni, A. Role of anti-TGF-β antibodies in the treatment of renal injury. Cytokine Growth Factor Rev. 2006, 17, 89–96. [Google Scholar] [CrossRef]
- Guan, Q.; Li, S.; Gao, S.; Chen, H.; Nguan, C.Y.C.; Du, C. Reduction of chronic rejection of renal allografts by anti-transforming growth factor-β antibody therapy in a rat model. Am. J. Physiol. Physiol. 2013, 305, F199–F207. [Google Scholar] [CrossRef] [Green Version]
- Ziyadeh, F.N.; Hoffman, B.B.; Han, D.C.; Iglesias-de la Cruz, M.C.; Hong, S.; Isono, M.; Chen, S.; McGowan, T.A.; Sharma, K. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-β antibody in db/db diabetic mice. Proc. Natl. Acad. Sci. USA 2000, 97, 8015–8020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Border, W.A.; Noble, N.A. Interactions of Transforming Growth Factor-β and Angiotensin II in Renal Fibrosis. Hypertension 1998, 31, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.-C.; Shi, M.; Zhang, J.; Quiñones, H.; Kuro-O, M.; Moe, O.W. Klotho deficiency is an early biomarker of renal ischemia–reperfusion injury and its replacement is protective. Kidney Int. 2010, 78, 1240–1251. [Google Scholar] [CrossRef] [Green Version]
- Haruna, Y.; Kashihara, N.; Satoh, M.; Tomita, N.; Namikoshi, T.; Sasaki, T.; Fujimori, T.; Xie, P.; Kanwar, Y.S. Amelioration of progressive renal injury by genetic manipulation of Klotho gene. Proc. Natl. Acad. Sci. USA 2007, 104, 2331–2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neyra, J.A.; Hu, M.C. Potential application of klotho in human chronic kidney disease. Bone 2017, 100, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Farrow, E.G.; Davis, S.I.; Summers, L.J.; White, K.E. Initial FGF23-Mediated Signaling Occurs in the Distal Convoluted Tubule. J. Am. Soc. Nephrol. 2009, 20, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Farrow, E.G.; Summers, L.J.; Schiavi, S.C.; McCormick, J.A.; Ellison, D.H.; White, E.K. Altered renal FGF23-mediated activity involving MAPK and Wnt: Effects of the Hyp mutation. J. Endocrinol. 2010, 207, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.C.; Shiizaki, K.; Kuro-O, M.; Moe, O.W. Fibroblast Growth Factor 23 and Klotho: Physiology and Pathophysiology of an Endocrine Network of Mineral Metabolism. Annu. Rev. Physiol. 2013, 75, 503–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.C.; Bian, A.; Neyra, J.; Zhan, M. Klotho, stem cells, and aging. Clin. Interv. Aging 2015, 10, 1233–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuro-O, M. A potential link between phosphate and aging—Lessons from Klotho-deficient mice. Mech. Ageing Dev. 2010, 131, 270–275. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, O.; Isakova, T.; Rhee, E.; Shah, A.; Holmes, J.; Collerone, G.; Jüppner, H.; Wolf, M. Fibroblast Growth Factor-23 Mitigates Hyperphosphatemia but Accentuates Calcitriol Deficiency in Chronic Kidney Disease. J. Am. Soc. Nephrol. 2005, 16, 2205–2215. [Google Scholar] [CrossRef] [PubMed]
- John, G.B.; Cheng, C.-Y.; Kuro-O, M. Role of Klotho in Aging, Phosphate Metabolism, and CKD. Am. J. Kidney Dis. 2011, 58, 127–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonelli, M.; Sacks, F.; Pfeffer, M.; Gao, Z.; Curhan, G. Relation Between Serum Phosphate Level and Cardiovascular Event Rate in People with Coronary Disease. Circulation 2005, 112, 2627–2633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesh, S.K.; Stack, A.G.; Levin, N.W.; Hulbert-Shearon, T.; Port, F.K. Association of Elevated Serum PO4, Ca × PO4Product, and Parathyroid Hormone with Cardiac Mortality Risk in Chronic Hemodialysis Patients. J. Am. Soc. Nephrol. 2001, 12, 2131–2138. [Google Scholar] [CrossRef] [PubMed]
- Dusso, A.S.; Brown, A.J.; Slatopolsky, E. Vitamin D. Am. J. Physiol. Ren. Physiol. 2005, 289, 8–28. [Google Scholar] [CrossRef] [PubMed]
- Berndt, T.; Kumar, R. Novel mechanisms in the regulation of phosphorus homeostasis. Physiology 2009, 24, 17–25. [Google Scholar] [CrossRef]
- Canalejo, R.; Canalejo, A.; Martinez-Moreno, J.M.; Rodriguez-Ortiz, M.E.; Estepa, J.C.; Mendoza, F.J.; Munoz-Castaneda, J.R.; Shalhoub, V.; Almaden, Y.; Rodriguez, M. FGF23 Fails to Inhibit Uremic Parathyroid Glands. J. Am. Soc. Nephrol. 2010, 21, 1125–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isakova, T.; Wahl, P.; Vargas, G.S.; Gutiérrez, O.M.; Scialla, J.; Xie, H.; Appleby, D.; Nessel, L.; Bellovich, K.; Chen, J.; et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011, 79, 1370–1378. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, H.; Nagano, N.; Urakawa, I.; Yamazaki, Y.; Iijima, K.; Fujita, T.; Yamashita, T.; Fukumoto, S.; Shimada, T. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int. 2010, 78, 975–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumock, G.T.; Andress, D.L.; Marx, S.E.; Sterz, R.; Joyce, A.T.; Kalantar-Zadeh, K. Association of Secondary Hyperparathyroidism with CKD Progression, Health Care Costs and Survival in Diabetic Predialysis CKD Patients. Nephron Clin. Pract. 2009, 113, c54–c61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.-C.; Sloan, A.; Isakova, T.; Gutierrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef] [Green Version]
- Komaba, H.; Goto, S.; Fujii, H.; Hamada, Y.; Kobayashi, A.; Shibuya, K.; Tominaga, Y.; Otsuki, N.; Nibu, K.-I.; Nakagawa, K.; et al. Depressed expression of Klotho and FGF receptor 1 in hyperplastic parathyroid glands from uremic patients. Kidney Int. 2010, 77, 232–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofman-Bang, J.; Martus, G.; Santini, M.A.; Olgaard, K.; Lewin, E. Increased parathyroid expression of klotho in uremic rats. Kidney Int. 2010, 78, 1119–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imura, A.; Tsuji, Y.; Murata, M.; Maeda, R.; Kubota, K.; Iwano, A.; Obuse, C.; Togashi, K.; Tominaga, M.; Kita, N.; et al. α-Klotho as a Regulator of Calcium Homeostasis. Science 2007, 316, 1615–1618. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Urakawa, I.; Isakova, T.; Yamazaki, Y.; Epstein, M.; Wesseling-Perry, K.; Wolf, M.; Salusky, I.B.; Jüppner, H. Circulating Fibroblast Growth Factor 23 in Patients with End-Stage Renal Disease Treated by Peritoneal Dialysis Is Intact and Biologically Active. J. Clin. Endocrinol. Metab. 2010, 95, 578–585. [Google Scholar] [CrossRef] [Green Version]
- Kumata, C.; Mizobuchi, M.; Ogata, H.; Koiwa, F.; Nakazawa, A.; Kondo, F.; Kadokura, Y.; Kinugasa, E.; Akizawa, T. Involvement of Alpha-Klotho and Fibroblast Growth Factor Receptor in the Development of Secondary Hyperparathyroidism. Am. J. Nephrol. 2010, 31, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Krajisnik, T.; Olauson, H.; Mirza, M.A.; Hellman, P.; Åkerström, G.; Westin, G.; Larsson, T.E.; Björklund, P. Parathyroid Klotho and FGF-receptor 1 expression decline with renal function in hyperparathyroid patients with chronic kidney disease and kidney transplant recipients. Kidney Int. 2010, 78, 1024–1032. [Google Scholar] [CrossRef] [Green Version]
- Lafage-Proust, M.-H. Does the downregulation of the FGF23 signaling pathway in hyperplastic parathyroid glands contribute to refractory secondary hyperparathyroidism in CKD patients? Kidney Int. 2010, 77, 390–392. [Google Scholar] [CrossRef] [Green Version]
- Razzaque, M.S.; Sitara, D.; Taguchi, T.; St-Arnaud, R.; Lanske, B. Premature aging-like phenotype in fibroblast growth factor 23 null mice is a vitamin D-mediated process. FASEB J. 2006, 20, 720–722. [Google Scholar] [CrossRef] [Green Version]
- Tsujikawa, H.; Kurotaki, Y.; Fujimori, T.; Fukuda, K.; Nabeshima, Y.-I. Klotho, a Gene Related to a Syndrome Resembling Human Premature Aging, Functions in a Negative Regulatory Circuit of Vitamin D Endocrine System. Mol. Endocrinol. 2003, 17, 2393–2403. [Google Scholar] [CrossRef]
- Stubbs, J.R.; Liu, S.; Tang, W.; Zhou, J.; Wang, Y.; Yao, X.; Quarles, L.D. Role of Hyperphosphatemia and 1,25-Dihydroxyvitamin D in Vascular Calcification and Mortality in Fibroblastic Growth Factor 23 Null Mice. J. Am. Soc. Nephrol. 2007, 18, 2116–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesse, M.; Fröhlich, L.F.; Zeitz, U.; Lanske, B.; Erben, R.G. Ablation of vitamin D signaling rescues bone, mineral, and glucose homeostasis in Fgf-23 deficient mice. Matrix Biol. 2007, 26, 75–84. [Google Scholar] [CrossRef]
- Ohnishi, M.; Nakatani, T.; Lanske, B.; Razzaque, M.S. In Vivo Genetic Evidence for Suppressing Vascular and Soft-Tissue Calcification Through the Reduction of Serum Phosphate Levels, Even in the Presence of High Serum Calcium and 1,25-Dihydroxyvitamin D Levels. Circ. Cardiovasc. Genet. 2009, 2, 583–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Qureshi, A.R.; Ripsweden, J.; Wennberg, L.; Heimburger, O.; Lindholm, B.; Barany, P.; Haarhaus, M.; Brismar, T.B.; Stenvinkel, P. Vertebral bone density associates with coronary artery calcification and is an independent predictor of poor outcome in end-stage renal disease patients. Bone 2016, 92, 50–57. [Google Scholar] [CrossRef]
- Jiang, Y.; Shen, Z.; Zhang, J.; Xing, C.; Zha, X.; Shen, C.; Zeng, M.; Yang, G.; Mao, H.; Zhang, B.; et al. Parathyroidectomy Increases Heart Rate Variability and Leptin Levels in Patients with Stage 5 Chronic Kidney Disease. Am. J. Nephrol. 2016, 44, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Lee, M.J.; Ryu, G.W.; Jhee, J.H.; Kim, H.W.; Park, S.; Jung, S.-Y.; Oh, H.J.; Park, J.T.; Han, S.H.; et al. Low serum intact parathyroid hormone level is an independent risk factor for overall mortality and major adverse cardiac and cerebrovascular events in incident dialysis patients. Osteoporos. Int. 2016, 27, 2717–2726. [Google Scholar] [CrossRef]
- Seifert, M.; Fuentes, L.D.L.; Rothstein, M.; Dietzen, D.J.; Bierhals, A.J.; Cheng, S.C.; Ross, W.; Windus, D.; Dávila-Román, V.G.; Hruska, K.A. Effects of Phosphate Binder Therapy on Vascular Stiffness in Early-Stage Chronic Kidney Disease. Am. J. Nephrol. 2013, 38, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Shoji, T.; Marubayashi, S.; Shigematsu, T.; Iseki, K.; Tsubakihara, Y. Use of Vitamin D Receptor Activator, Incident Cardiovascular Disease and Death in a Cohort of Hemodialysis Patients. Ther. Apher. Dial. 2014, 19, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, S. FGF23-FGF Receptor/Klotho Pathway as a New Drug Target for Disorders of Bone and Mineral Metabolism. Calcif. Tissue Int. 2015, 98, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, T.; Perkovic, V.; De Galan, B.E.; Zoungas, S.; Pillai, A.; Jardine, M.; Patel, A.; Cass, A.; Neal, B.; Poulter, N.; et al. Albuminuria and Kidney Function Independently Predict Cardiovascular and Renal Outcomes in Diabetes. J. Am. Soc. Nephrol. 2009, 20, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Memmos, E.; Sarafidis, P.; Pateinakis, P.; Tsiantoulas, A.; Faitatzidou, D.; Giamalis, P.; Vasilikos, V.; Papagianni, A. Soluble Klotho is associated with mortality and cardiovascular events in hemodialysis. BMC Nephrol. 2019, 20, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karalliedde, J.; Maltese, G.; Hill, B.; Viberti, G.; Gnudi, L. Effect of Renin-Angiotensin System Blockade on Soluble Klotho in Patients with Type 2 Diabetes, Systolic Hypertension, and Albuminuria. Clin. J. Am. Soc. Nephrol. 2013, 8, 1899–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, Y.; Nakamura, T.; Ohyama, Y.; Suzuki, T.; Iida, A.; Shiraki-Iida, T.; Kuro-O, M.; Nabeshima, Y.-I.; Kurabayashi, M.; Nagai, R. In Vivo klotho Gene Delivery Protects against Endothelial Dysfunction in Multiple Risk Factor Syndrome. Biochem. Biophys. Res. Commun. 2000, 276, 767–772. [Google Scholar] [CrossRef]
- Xie, J.; Yoon, J.; An, S.-W.; Kuro-O, M.; Huang, C.-L. Soluble Klotho Protects against Uremic Cardiomyopathy Independently of Fibroblast Growth Factor 23 and Phosphate. J. Am. Soc. Nephrol. 2014, 26, 1150–1160. [Google Scholar] [CrossRef]
- Ding, J.; Tang, Q.; Luo, B.; Zhang, L.; Lin, L.; Han, L.; Hao, M.; Li, M.; Yu, L.; Li, M. Klotho inhibits angiotensin II-induced cardiac hypertrophy, fibrosis, and dysfunction in mice through suppression of transforming growth factor-β1 signaling pathway. Eur. J. Pharmacol. 2019, 859, 172549. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, V.M.; Kleber, M.E.; Vervloet, M.G.; Larsson, T.E.; Tomaschitz, A.; Pilz, S.; Stojakovic, T.; Delgado, G.; Grammer, T.B.; Marx, N.; et al. Soluble klotho and mortality: The Ludwigshafen Risk and Cardiovascular Health Study. Atherosclerosis 2015, 242, 483–489. [Google Scholar] [CrossRef]
- Pan, H.-C.; Chou, K.-M.; Lee, C.-C.; Yang, N.-I.; Sun, C.-Y. Circulating Klotho levels can predict long-term macrovascular outcomes in type 2 diabetic patients. Atherosclerosis 2018, 276, 83–90. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, O.M.; Januzzi, J.L.; Isakova, T.; Laliberte, K.; Smith, K.; Collerone, G.; Sarwar, A.; Hoffmann, U.; Coglianese, E.; Christenson, R.; et al. Fibroblast Growth Factor 23 and Left Ventricular Hypertrophy in Chronic Kidney Disease. Circulation 2009, 119, 2545–2552. [Google Scholar] [CrossRef] [Green Version]
- Feldman, H.I.; Appel, L.J.; Chertow, G.M.; Cifelli, D.; Cizman, B.; Daugirdas, J.; Fink, J.; Franklin-Becker, E.D.; Go, A.S.; Hamm, L.L.; et al. The Chronic Renal Insufficiency Cohort (CRIC) Study: Design and Methods. J. Am. Soc. Nephrol. 2003, 14, S148–S153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Ibrahimi, O.A.; Olsen, S.; Umemori, H.; Mohammadi, M.; Ornitz, D.M. Receptor Specificity of the Fibroblast Growth Factor Family. J. Biol. Chem. 2006, 281, 15694–15700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Ibrahimi, O.A.; Goetz, R.; Zhang, F.; Davis, S.I.; Garringer, H.J.; Linhardt, R.J.; Ornitz, D.; Mohammadi, M.; White, K.E. Analysis of the Biochemical Mechanisms for the Endocrine Actions of Fibroblast Growth Factor-23. Endocrinology 2005, 146, 4647–4656. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, G.S.; Reuter, S.; Kentrup, D.; Grabner, A.; Amaral, A.P.; Fobker, M.; Stypmann, J.; Pavenstädt, H.; Wolf, M.; Faul, C.; et al. Treatment of established left ventricular hypertrophy with fibroblast growth factor receptor blockade in an animal model of CKD. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.—Eur. Ren. Assoc. 2014, 29, 2028–2035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhao, M.-M.; Cai, Y.; Zheng, M.-F.; Sun, W.-L.; Zhang, S.-Y.; Kong, W.; Gu, J.; Wang, X.; Xu, M.-J. Mammalian target of rapamycin signaling inhibition ameliorates vascular calcification via Klotho upregulation. Kidney Int. 2015, 88, 711–721. [Google Scholar] [CrossRef] [Green Version]
- Kestenbaum, B.; Sampson, J.N.; Rudser, K.D.; Patterson, D.J.; Seliger, S.L.; Young, B.; Sherrard, D.J.; Andress, D.L. Serum Phosphate Levels and Mortality Risk among People with Chronic Kidney Disease. J. Am. Soc. Nephrol. 2004, 16, 520–528. [Google Scholar] [CrossRef] [Green Version]
- Neyra, J.; Hu, M. αKlotho and Chronic Kidney Disease. Vitam. Horm. 2016, 101, 257–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, J.; Huang, J.; Luo, C.; Ye, H.; Ling, X.; Wu, Q.; Shen, W.; Zhou, L. Klotho retards renal fibrosis through targeting mitochondrial dysfunction and cellular senescence in renal tubular cells. Physiol. Rep. 2021, 9, e14696. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Mo, H.; Miao, J.; Zhou, D.; Tan, R.J.; Hou, F.F.; Liu, Y. Klotho Ameliorates Kidney Injury and Fibrosis and Normalizes Blood Pressure by Targeting the Renin-Angiotensin System. Am. J. Pathol. 2015, 185, 3211–3223. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Liu, Y. Wnt/β-catenin signaling and renin–angiotensin system in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2016, 25, 100–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Li, Y.; Hao, S.; Zhou, D.; Tan, R.J.; Nie, J.; Hou, F.F.; Kahn, M.; Liu, Y. Multiple Genes of the Renin-Angiotensin System Are Novel Targets of Wnt/β-Catenin Signaling. J. Am. Soc. Nephrol. 2015, 26, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Stenvinkel, P.; Larsson, T.E. Chronic Kidney Disease: A Clinical Model of Premature Aging. Am. J. Kidney Dis. 2013, 62, 339–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecoits-Filho, R.; Heimbürger, O.; Bárány, P.; Suliman, M.; Fehrman-Ekholm, I.; Lindholm, B.; Stenvinkel, P. Associations between circulating inflammatory markers and residual renal function in CRF patients. Am. J. Kidney Dis. 2003, 41, 1212–1218. [Google Scholar] [CrossRef]
- Buendía, P.; Ramírez, R.; Aljama, P.; Carracedo, J. Klotho Prevents Translocation of NFκB; Academic Press: Cambridge, MA, USA, 2016; Volume 101, pp. 119–150. [Google Scholar]
- Maekawa, Y.; Ishikawa, K.; Yasuda, O.; Oguro, R.; Hanasaki, H.; Kida, I.; Takemura, Y.; Ohishi, M.; Katsuya, T.; Rakugi, H. Klotho suppresses TNF-α-induced expression of adhesion molecules in the endothelium and attenuates NF-κB activation. Endocrinology 2009, 35, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, J.-L.; Schneider, P.; Tschopp, J. The molecular architecture of the TNF superfamily. Trends Biochem. Sci. 2002, 27, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Ucero, A.; Berzal, S.; Ocaña-Salceda, C.; Sancho, M.; Orzaez, M.; Messeguer, A.; Ruiz-Ortega, M.; Egido, J.; Vicent, M.J.; Ortiz, A.; et al. A Polymeric Nanomedicine Diminishes Inflammatory Events in Renal Tubular Cells. PLoS ONE 2013, 8, e51992. [Google Scholar] [CrossRef] [Green Version]
- Sanz, A.B.; Justo, P.; Sanchez-Niño, M.D.; Blanco-Colio, L.M.; Winkles, J.A.; Kreztler, M.; Jakubowski, A.; Blanco, J.; Egido, J.; Ruiz-Ortega, M.; et al. The Cytokine TWEAK Modulates Renal Tubulointerstitial Inflammation. J. Am. Soc. Nephrol. 2008, 19, 695–703. [Google Scholar] [CrossRef] [Green Version]
- Moreno, J.A.; Izquierdo, M.C.; Sanchez-Niño, M.D.; Suárez-Alvarez, B.; Lopez-Larrea, C.; Jakubowski, A.; Blanco, J.; Ramirez, R.; Selgas, R.; Ruiz-Ortega, M.; et al. The Inflammatory Cytokines TWEAK and TNFα Reduce Renal Klotho Expression through NFκB. J. Am. Soc. Nephrol. 2011, 22, 1315–1325. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Kuro-O, M.; Sun, Z. Klotho gene delivery suppresses Nox2 expression and attenuates oxidative stress in rat aortic smooth muscle cells via the cAMP-PKA pathway. Aging Cell 2012, 11, 410–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.-Y.; Pan, S.-Y.; Li, B.; Chen, Y.-M.; Lin, S.-L. Rejuvenation: Turning back the clock of aging kidney. J. Formos. Med. Assoc. 2020, 119, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Hsia, C.C.; Ravikumar, P.; Ye, J. Acute lung injury complicating acute kidney injury: A model of endogenous αKlotho deficiency and distant organ dysfunction. Bone 2017, 100, 100–109. [Google Scholar] [CrossRef]
- Ologunde, R.; Zhao, H.; Lu, K.; Ma, D. Organ cross talk and remote organ damage following acute kidney injury. Int. Urol. Nephrol. 2014, 46, 2337–2345. [Google Scholar] [CrossRef] [PubMed]
- Andrade, L.; Rodrigues, C.; Gomes, S.A.; Noronha, I.L. Acute Kidney Injury as a Condition of Renal Senescence. Cell Transplant. 2018, 27, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Khan, I.; Simpson, W.; Prescott, G.; Townend, J.; Smith, W.; MacLeod, A. Incidence and Outcomes in Acute Kidney Injury: A Comprehensive Population-Based Study. J. Am. Soc. Nephrol. 2007, 18, 1292–1298. [Google Scholar] [CrossRef] [Green Version]
- Chawla, L.S.; Bellomo, R.; Bihorac, A.; Goldstein, S.L.; Siew, E.D.; Bagshaw, S.M.; Bittleman, D.; Cruz, D.; Endre, Z.; Fitzgerald, R.L.; et al. Acute kidney disease and renal recovery: Consensus report of the Acute Disease Quality Initiative (ADQI) 16 Workgroup. Nat. Rev. Nephrol. 2017, 13, 241–257. [Google Scholar] [CrossRef] [Green Version]
- Waikar, S.S.; Liu, K.; Chertow, G.M. Diagnosis, Epidemiology and Outcomes of Acute Kidney Injury. Clin. J. Am. Soc. Nephrol. 2008, 3, 844–861. [Google Scholar] [CrossRef] [PubMed]
- Megyesi, J.; Udvarhelyi, N.; Safirstein, R.L.; Price, P.M. The p53-independent activation of transcription of p21 WAF1/CIP1/SDI1 after acute renal failure. Am. J. Physiol. Physiol. 1996, 271, F1211–F1216. [Google Scholar] [CrossRef] [PubMed]
- Megyesi, J.; Safirstein, R.L.; Price, P.M. Induction of p21WAF1/CIP1/SDI1 in kidney tubule cells affects the course of cisplatin-induced acute renal failure. J. Clin. Investig. 1998, 101, 777–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellomo, R.; Kellum, A.J.; Ronco, C. Acute kidney injury. Lancet 2012, 380, 756–766. [Google Scholar] [CrossRef]
- Wen, Y.; Jiang, L.; Xu, Y.; Qian, C.-Y.; Li, S.-S.; Qin, T.-H.; Chen, E.-Z.; Lin, J.-D.; Ai, Y.-H.; Wu, D.-W.; et al. Prevalence, risk factors, clinical course, and outcome of acute kidney injury in Chinese intensive care units: A prospective cohort study. Chin. Med. J. 2013, 126, 4409–4416. [Google Scholar] [PubMed]
- Silver, S.A.; Harel, Z.; McArthur, E.; Nash, D.M.; Acedillo, R.; Kitchlu, A.; Garg, A.X.; Chertow, G.M.; Bell, C.M.; Wald, R. Causes of Death after a Hospitalization with AKI. J. Am. Soc. Nephrol. 2018, 29, 1001–1010. [Google Scholar] [CrossRef] [Green Version]
- Bucaloiu, I.D.; Kirchner, H.L.; Norfolk, E.R.; Hartle, J.E.; Perkins, R.M. Increased risk of death and de novo chronic kidney disease following reversible acute kidney injury. Kidney Int. 2012, 81, 477–485. [Google Scholar] [CrossRef] [Green Version]
- Wu, V.-C.; Wu, C.-H.; Huang, T.-M.; Wang, C.-Y.; Lai, C.-F.; Shiao, C.-C.; Chang, C.-H.; Lin, S.-L.; Chen, Y.-Y.; Chen, Y.-M.; et al. Long-Term Risk of Coronary Events after AKI. J. Am. Soc. Nephrol. 2014, 25, 595–605. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.-Y. Yes, AKI Truly Leads to CKD. J. Am. Soc. Nephrol. 2012, 23, 967–969. [Google Scholar] [CrossRef] [Green Version]
- Cerdá, J.; Lameire, N.; Eggers, P.; Pannu, N.; Uchino, S.; Wang, H.; Bagga, A.; Levin, A. Epidemiology of Acute Kidney Injury. Clin. J. Am. Soc. Nephrol. 2008, 3, 881–886. [Google Scholar] [CrossRef] [Green Version]
- Case, J.; Khan, S.; Khalid, R.; Khan, A. Epidemiology of Acute Kidney Injury in the Intensive Care Unit. Crit. Care Res. Pract. 2013, 2013, 479730. [Google Scholar] [CrossRef] [Green Version]
- Grams, M.E.; Estrella, M.M.; Coresh, J.; Brower, R.G.; Liu, K. Fluid Balance, Diuretic Use, and Mortality in Acute Kidney Injury. Clin. J. Am. Soc. Nephrol. 2011, 6, 966–973. [Google Scholar] [CrossRef]
- Nash, K.; Hafeez, A.; Hou, S. Hospital-acquired renal insufficiency. Am. J. Kidney Dis. 2002, 39, 930–936. [Google Scholar] [CrossRef]
- Mehta, R.L.; Chertow, G.M.; Masereeuw, R.; Notenboom, S.; Smeets, P.H.E.; Wouterse, A.C.; Russel, F.G.M. Acute Renal Failure Definitions and Classification: Time for Change? J. Am. Soc. Nephrol. 2003, 14, 2178–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feest, T.G.; Round, A.; Hamad, S. Incidence of severe acute renal failure in adults: Results of a community based study. BMJ 1993, 306, 481–483. [Google Scholar] [CrossRef] [Green Version]
- Uchino, S.; Kellum, J.A.; Bellomo, R.; Doig, G.S.; Morimatsu, H.; Morgera, S.; Schetz, M.; Tan, I.; Bouman, C.; Macedo, E.; et al. Acute Renal Failure in Critically Ill PatientsA Multinational, Multicenter Study. JAMA 2005, 294, 813–818. [Google Scholar] [CrossRef] [Green Version]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; James, M.T. Acute Kidney Injury. Ann. Intern. Med. 2017, 167, ITC66–ITC80. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.L.; Starfield, B.; Anderson, G. Prevalence, Expenditures, and Complications of Multiple Chronic Conditions in the Elderly. Arch. Intern. Med. 2002, 162, 2269–2276. [Google Scholar] [CrossRef] [PubMed]
- Khwaja, A. KDIGO Clinical Practice Guidelines for Acute Kidney Injury. Nephron. Clin. Pract. 2012, 120, c179–c184. [Google Scholar] [CrossRef] [PubMed]
- Sanz, A.B.; Sánchez-Niño, M.D.; Martín-Cleary, C.; Ortiz, A.; Ramos, A.M.; Arduan, A.O. Progress in the development of animal models of acute kidney injury and its impact on drug discovery. Expert Opin. Drug Discov. 2013, 8, 879–895. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Ricksten, S.-E.; Bragadottir, G.; Redfors, B.; Nordquist, L. Renal oxygenation and haemodynamics in acute kidney injury and chronic kidney disease. Clin. Exp. Pharmacol. Physiol. 2012, 40, 138–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tampe, B.; Steinle, U.; Tampe, D.; Carstens, J.; Korsten, P.; Zeisberg, E.M.; Müller, G.A.; Kalluri, R.; Zeisberg, M. Low-dose hydralazine prevents fibrosis in a murine model of acute kidney injury–to–chronic kidney disease progression. Kidney Int. 2017, 91, 157–176. [Google Scholar] [CrossRef]
- Molitoris, B.A. Therapeutic translation in acute kidney injury: The epithelial/endothelial axis. J. Clin. Investig. 2014, 124, 2355–2363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akcay, A.; Nguyen, Q.; Edelstein, C.L. Mediators of Inflammation in Acute Kidney Injury. Mediat. Inflamm. 2009, 2009, 137072. [Google Scholar] [CrossRef]
- Dellepiane, S.; Marengo, M.; Cantaluppi, V. Detrimental cross-talk between sepsis and acute kidney injury: New pathogenic mechanisms, early biomarkers and targeted therapies. Crit. Care 2016, 20, 1. [Google Scholar] [CrossRef] [Green Version]
- Coelho, S.; Cabral, M.D.G.; Lopes, J.A.; Jacinto, A. Renal regeneration after acute kidney injury. Nephrology 2018, 23, 805–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Chen, W.; Chen, Y.; Zhou, Q.; Xiao, P.; Tang, R.; Xue, J. Neferine Attenuates Acute Kidney Injury by Inhibiting NF-κB Signaling and Upregulating Klotho Expression. Front. Pharmacol. 2019, 10, 1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holditch, S.J.; Brown, C.N.; Lombardi, A.M.; Nguyen, K.N.; Edelstein, C.L. Recent Advances in Models, Mechanisms, Biomarkers, and Interventions in Cisplatin-Induced Acute Kidney Injury. Int. J. Mol. Sci. 2019, 20, 3011. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef] [Green Version]
- Ferenbach, D.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Basile, D.P.; Bonventre, J.V.; Mehta, R.; Nangaku, M.; Unwin, R.; Rosner, M.H.; Kellum, J.A.; Ronco, C. Progression after AKI: Understanding Maladaptive Repair Processes to Predict and Identify Therapeutic Treatments. J. Am. Soc. Nephrol. 2016, 27, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Duffield, J.S. Macrophages and Immunologic Inflammation of the Kidney. Semin. Nephrol. 2010, 30, 234–254. [Google Scholar] [CrossRef] [Green Version]
- Ko, G.J.; Boo, C.-S.; Jo, S.-K.; Cho, W.Y.; Kim, H.K. Macrophages contribute to the development of renal fibrosis following ischaemia/reperfusion-induced acute kidney injury. Nephrol. Dial. Transplant. 2007, 23, 842–852. [Google Scholar] [CrossRef] [Green Version]
- Guijarro, C.; Egido, J. Transcription factor-κB (NF-κB) and renal disease. Kidney Int. 2001, 59, 415–424. [Google Scholar] [CrossRef] [Green Version]
- Sanz, A.B.; Sanchez-Niño, M.D.; Ramos, A.M.; Moreno, J.A.; Santamaria, B.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. NF-κB in Renal Inflammation. J. Am. Soc. Nephrol. 2010, 21, 1254–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panah, F.; Ghorbanihaghjo, A.; Argani, H.; Zarmehri, M.A.; Ahmad, S.N.S. Ischemic acute kidney injury and klotho in renal transplantation. Clin. Biochem. 2018, 55, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Pannu, N. Bidirectional relationships between acute kidney injury and chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2013, 22, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Polichnowski, A.J.; Lan, R.; Geng, H.; Griffin, K.A.; Venkatachalam, M.A.; Bidani, A.K. Severe Renal Mass Reduction Impairs Recovery and Promotes Fibrosis after AKI. J. Am. Soc. Nephrol. 2014, 25, 1496–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chawla, L.S.; Eggers, P.W.; Star, R.A.; Kimmel, P.L. Acute Kidney Injury and Chronic Kidney Disease as Interconnected Syndromes. N. Engl. J. Med. 2014, 371, 58–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chawla, L.; Kimmel, P.L. Acute kidney injury and chronic kidney disease: An integrated clinical syndrome. Kidney Int. 2012, 82, 516–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Shi, M.; Maique, J.; Shaffer, J.; Yan, S.; Moe, O.W.; Hu, M.C. Beclin 1/Bcl-2 complex-dependent autophagy activity modulates renal susceptibility to ischemia-reperfusion injury and mediates renoprotection by Klotho. Am. J. Physiol. Physiol. 2020, 318, F772–F792. [Google Scholar] [CrossRef]
- Endre, Z.H.; Kellum, J.A.; Di Somma, S.; Doi, K.; Goldstein, S.L.; Koyner, J.L.; Macedo, E.; Mehta, R.L.; Murray, P.T. Differential Diagnosis of AKI in Clinical Practice by Functional and Damage Biomarkers: Workgroup Statements from the Tenth Acute Dialysis Quality Initiative Consensus Conference. Contrib. Nephrol. 2013, 182, 30–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francoz, C.; Nadim, M.K.; Durand, F. Kidney biomarkers in cirrhosis. J. Hepatol. 2016, 65, 809–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, W.; Chen, J.; Jiang, Y.; Wu, J.; Shou, Z.; He, Q.; Wang, Y.; Chen, Y.; Wang, H. Urinary fractalkine is a marker of acute rejection. Kidney Int. 2008, 74, 1454–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, K.; Coelho, S.; Rodrigues, B.; Martins, H.; Frade, F.; Lopes, S.; Cunha, L.; Papoila, A.L.; Devarajan, P. Cystatin C as a Marker of Acute Kidney Injury in the Emergency Department. Clin. J. Am. Soc. Nephrol. 2010, 5, 1745–1754. [Google Scholar] [CrossRef] [Green Version]
- Chang-Panesso, M.; Shi, M.; Cho, H.J.; Paek, J.; Ye, J.; Moe, O.W.; Hu, M.C. Klotho has dual protective effects on cisplatin-induced acute kidney injury. Kidney Int. 2014, 85, 855–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.C.; Kuro-O, M.; Moe, O.W. The emerging role of Klotho in clinical nephrology. Nephrol. Dial. Transplant. 2012, 27, 2650–2657. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Torreggiani, M.; Post, J.B.; Zheng, F.; Uribarri, J.; Striker, G.E. Role of oxidants/inflammation in declining renal function in chronic kidney disease and normal aging. Kidney Int. 2009, 76, S3–S11. [Google Scholar] [CrossRef] [Green Version]
- Mitobe, M.; Yoshida, T.; Sugiura, H.; Shirota, S.; Tsuchiya, K.; Nihei, H. Oxidative Stress Decreases Klotho Expression in a Mouse Kidney Cell Line. Nephron Exp. Nephrol. 2005, 101, e67–e74. [Google Scholar] [CrossRef]
- Maltese, G.; Psefteli, P.-M.; Rizzo, B.; Srivastava, S.; Gnudi, L.; Mann, G.E.; Siow, R.C. The anti-ageing hormone klotho induces Nrf2-mediated antioxidant defences in human aortic smooth muscle cells. J. Cell. Mol. Med. 2016, 21, 621–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeldich, E.; Chen, C.-D.; Colvin, T.A.; Bove-Fenderson, E.A.; Liang, J.; Zhou, T.B.T.; Harris, D.; Abraham, C.R. The Neuroprotective Effect of Klotho is Mediated via Regulation of Members of the Redox System. J. Biol. Chem. 2014, 289, 24700–24715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, H.J.; Nam, B.Y.; Lee, M.J.; Kim, C.H.; Koo, H.M.; Doh, F.M.; Han, J.H.; Kim, E.J.; Han, J.S.; Park, J.T.; et al. Decreased Circulating Klotho Levels in Patients Undergoing Dialysis and Relationship to Oxidative Stress and Inflammation. Perit. Dial. Int. 2015, 35, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.-C.; Shi, M.; Cho, H.J.; Zhang, J.; Pavlenco, A.; Liu, S.; Sidhu, S.; Huang, L.J.-S.; Moe, O.W. The erythropoietin receptor is a downstream effector of Klotho-induced cytoprotection. Kidney Int. 2013, 84, 468–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.; Fan, H.; Le, J.; Chen, G.; Chen, H.; Li, J.; Zhu, J. Protective effects of Klotho protein on acute kidney injury in septic mice and its mechanism. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2019, 31, 160–164. [Google Scholar]
- Bi, F.; Chen, F.; Li, Y.; Wei, A.; Cao, W. Klotho preservation by Rhein promotes toll-like receptor 4 proteolysis and attenuates lipopolysaccharide-induced acute kidney injury. J. Mol. Med. 2018, 96, 915–927. [Google Scholar] [CrossRef]
- Lin, W.; Wu, X.; Wen, J.; Fei, Y.; Wu, J.; Li, X.; Zhang, Q.; Dong, Y.; Xu, T.; Fan, Y.; et al. Nicotinamide retains Klotho expression and ameliorates rhabdomyolysis-induced acute kidney injury. Nutrition 2021, 91–92, 111376. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Li, Y.; Zhou, D.; Tan, R.J.; Liu, Y. Loss of Klotho Contributes to Kidney Injury by Derepression of Wnt/β-Catenin Signaling. J. Am. Soc. Nephrol. 2013, 24, 771–785. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, H.; Yoshida, T.; Shiohira, S.; Kohei, J.; Mitobe, M.; Kurosu, H.; Kuro-O, M.; Nitta, K.; Tsuchiya, K. Reduced Klotho expression level in kidney aggravates renal interstitial fibrosis. Am. J. Physiol. Ren. Physiol. 2012, 302, F1252–F1264. [Google Scholar] [CrossRef] [PubMed]
- Doi, S.; Zou, Y.; Togao, O.; Pastor, J.V.; John, G.B.; Wang, L.; Shiizaki, K.; Gotschall, R.; Schiavi, S.; Yorioka, N.; et al. Klotho Inhibits Transforming Growth Factor-β1 (TGF-β1) Signaling and Suppresses Renal Fibrosis and Cancer Metastasis in Mice. J. Biol. Chem. 2011, 286, 8655–8665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, K.; Yin, Z.; Xie, Y. Roles of the kidney in the formation, remodeling and repair of bone. J. Nephrol. 2016, 29, 349–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marumo, T.; Hishikawa, K.; Yoshikawa, M.; Fujita, T. Epigenetic Regulation of BMP7 in the Regenerative Response to Ischemia. J. Am. Soc. Nephrol. 2008, 19, 1311–1320. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Li, Y.; Chen, F.; Yin, S.; Liu, Z.; Cao, W. Klotho preservation via histone deacetylase inhibition attenuates chronic kidney disease-associated bone injury in mice. Sci. Rep. 2017, 7, 46195. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, Y.; Ohishi, M.; Ikushima, M.; Yamamoto, K.; Yasuda, O.; Oguro, R.; Yamamoto-Hanasaki, H.; Tatara, Y.; Takeya, Y.; Rakugi, H. Klotho protein diminishes endothelial apoptosis and senescence via a mitogen-activated kinase pathway. Geriatr. Gerontol. Int. 2011, 11, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Fergusson, M.M.; Castilho, R.M.; Liu, J.; Cao, L.; Chen, J.; Malide, D.; Rovira, I.I.; Schimel, D.; Kuo, C.J.; et al. Augmented Wnt Signaling in a Mammalian Model of Accelerated Aging. Science 2007, 317, 803–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiura, H.; Yoshida, T.; Mitobe, M.; Yoshida, S.; Shiohira, S.; Nitta, K.; Tsuchiya, K. Klotho reduces apoptosis in experimental ischaemic acute kidney injury via HSP-70. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2009, 25, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Kim, S.L.; Lee, S.-Y.; Koo, J.K.; Wang, Z.; Choi, M.E. Autophagy Regulates TGF-β Expression and Suppresses Kidney Fibrosis Induced by Unilateral Ureteral Obstruction. J. Am. Soc. Nephrol. 2014, 25, 2835–2846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Zepeda-Orozco, D.; Black, R.; Lin, F. Autophagy Is a Component of Epithelial Cell Fate in Obstructive Uropathy. Am. J. Pathol. 2010, 176, 1767–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Wei, Q.; Dong, G.; Komatsu, M.; Su, Y.; Dong, Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012, 82, 1271–1283. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, M.; Urushido, M.; Hamada, K.; Matsumoto, T.; Shimamura, Y.; Ogata, K.; Inoue, K.; Taniguchi, Y.; Horino, T.; Fujieda, M.; et al. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am. J. Physiol. Physiol. 2013, 305, F495–F509. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, G.P. Autophagy protects proximal tubular cells from injury and apoptosis. Kidney Int. 2012, 82, 1250–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Periyasamy-Thandavan, S.; Jiang, M.; Wei, Q.; Smith, R.; Yin, X.-M.; Dong, Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int. 2008, 74, 631–640. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, A.; Kimura, T.; Takabatake, Y.; Namba, T.; Kaimori, J.; Kitamura, H.; Matsui, I.; Niimura, F.; Matsusaka, T.; Fujita, N.; et al. Autophagy Guards Against Cisplatin-Induced Acute Kidney Injury. Am. J. Pathol. 2012, 180, 517–525. [Google Scholar] [CrossRef]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Kaimori, J.-Y.; Matsui, I.; Namba, T.; Kitamura, H.; Niimura, F.; Matsusaka, T.; Soga, T.; et al. Autophagy Protects the Proximal Tubule from Degeneration and Acute Ischemic Injury. J. Am. Soc. Nephrol. 2011, 22, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Isaka, Y.; Kimura, T.; Takabatake, Y. The protective role of autophagy against aging and acute ischemic injury in kidney proximal tubular cells. Autophagy 2011, 7, 1085–1087. [Google Scholar] [CrossRef] [Green Version]
- Livingston, M.J.; Dong, Z. Autophagy in Acute Kidney Injury. Semin. Nephrol. 2014, 34, 17–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Fan, X.; Lawson, W.E.; Paueksakon, P.; Harris, R.C. Telomerase deficiency delays renal recovery in mice after ischemia–reperfusion injury by impairing autophagy. Kidney Int. 2015, 88, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G.J.C. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, M.; Neilson, E.G. Mechanisms of Tubulointerstitial Fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1819–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drüeke, T.B.; Massy, Z.A. Phosphate Binders in CKD: Bad News or Good News? J. Am. Soc. Nephrol. 2012, 23, 1277–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohab, N.; Fujimori, T.; Nishiguchib, S.; Tamorib, A.; Shiomib, S.; Nakatanic, T.; Sugimurac, K.; Kishimotoc, T.; Kinoshitaad, S.; Kurokib, T.; et al. Severely Reduced Production of Klotho in Human Chronic Renal Failure Kidney. Biochem. Biophys. Res. Commun. 2001, 280, 1015–1020. [Google Scholar] [CrossRef]
- Lin, Y.; Sun, Z. In Vivo Pancreatic β-Cell–Specific Expression of Antiaging GeneKlotho: A Novel Approach for Preserving β-Cells in Type 2 Diabetes. Diabetes 2014, 64, 1444–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iida, R.-H.; Kanko, S.; Suga, T.; Morito, M.; Yamane, A. Autophagic-lysosomal pathway functions in the masseter and tongue muscles in the klotho mouse, a mouse model for aging. Mol. Cell. Biochem. 2010, 348, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Shiozaki, M.; Yoshimura, K.; Shibata, M.; Koike, M.; Matsuura, N.; Uchiyama, Y.; Gotow, T. Morphological and biochemical signs of age-related neurodegenerative changes in klotho mutant mice. Neuroscience 2008, 152, 924–941. [Google Scholar] [CrossRef] [PubMed]
- Shu, G.; Xie, B.; Ren, F.; Liu, D.-C.; Zhou, J.; Li, Q.; Chen, J.; Yuan, L.; Zhou, J. Restoration of klotho expression induces apoptosis and autophagy in hepatocellular carcinoma cells. Cell. Oncol. 2013, 36, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Flores, B.; Gillings, N.; Bian, A.; Cho, H.J.; Yan, S.; Liu, Y.; Levine, B.; Moe, O.W.; Hu, M.C. αKlotho Mitigates Progression of AKI to CKD through Activation of Autophagy. J. Am. Soc. Nephrol. 2016, 27, 2331–2345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Livingston, M.J.; Dong, Z. Autophagy in Acute Kidney Injury and Repair. Nephron Clin. Pract. 2014, 127, 56–60. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.I.; Na, H.-J.; Ding, Y.; Wang, Z.; Lee, S.-J.; Choi, M.E. Autophagy Promotes Intracellular Degradation of Type I Collagen Induced by Transforming Growth Factor (TGF)-β1. J. Biol. Chem. 2012, 287, 11677–11688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azuma, M.; Koyama, D.; Kikuchi, J.; Yoshizawa, H.; Thasinas, D.; Shiizaki, K.; Kuro-O, M.; Furukawa, Y.; Kusano, E. Promoter methylation confers kidney-specific expression of the Klotho gene. FASEB J. 2012, 26, 4264–4274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, D.; Jones, P.A. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl. Acad. Sci. USA 2002, 99, 3740–3745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Li, Y.; Fan, Y.; Wu, J.; Zhao, B.; Guan, Y.; Chien, S.; Wang, N. Klotho is a target gene of PPAR-γ. Kidney Int. 2008, 74, 732–739. [Google Scholar] [CrossRef] [Green Version]
- Feingold, K.R. Oral and Injectable (Non-Insulin) Pharmacological Agents for the Treatment of Type 2 Diabetes; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Benedetti, R.; Conte, M.; Altucci, L. Targeting Histone Deacetylases in Diseases: Where Are We? Antioxid. Redox Signal. 2015, 23, 99–126. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.E.; Ghee, J.Y.; Piao, S.; Song, J.-H.; Han, D.H.; Kim, S.; Ohashi, N.; Kobori, H.; Kuro-O, M.; Yang, C.W. Angiotensin II blockade upregulates the expression of Klotho, the anti-ageing gene, in an experimental model of chronic cyclosporine nephropathy. Nephrol. Dial. Transplant. 2010, 26, 800–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Huang, R.; Kavanagh, J.; Li, L.; Zeng, X.; Li, Y.; Fu, P. Efficacy and Safety of Dual Blockade of the Renin–Angiotensin–Aldosterone System in Diabetic Kidney Disease: A Meta-Analysis. Am. J. Cardiovasc. Drugs 2019, 19, 259–286. [Google Scholar] [CrossRef] [PubMed]
- Ritter, C.S.; Zhang, S.; Delmez, J.; Finch, J.L.; Slatopolsky, E. Differential expression and regulation of Klotho by paricalcitol in the kidney, parathyroid, and aorta of uremic rats. Kidney Int. 2015, 87, 1141–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, W.L.; Leaf, E.M.; Hu, M.C.; Takeno, M.; Kuro-O, M.; Moe, O.W.; Giachelli, C.M. Vitamin D receptor agonists increase klotho and osteopontin while decreasing aortic calcification in mice with chronic kidney disease fed a high phosphate diet. Kidney Int. 2012, 82, 1261–1270. [Google Scholar] [CrossRef] [Green Version]
- Dyer, C.A. Safety and tolerability of paricalcitol in patients with chronic kidney disease. Expert Opin. Drug Saf. 2013, 12, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Zhang, W.; Zhang, X.; Li, X.; Chen, J. Efficacy and Safety of Paricalcitol Therapy for Chronic Kidney Disease: A Meta-Analysis. Clin. J. Am. Soc. Nephrol. 2012, 7, 391–400. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.R.; Guo, J.; Wang, Y.; Hou, Y.L.; Lu, W.W.; Zhang, J.S.; Yu, Y.R.; Xu, M.J.; Liu, X.Y.; Wang, X.J.; et al. Intermedin1–53 attenuates vascular calcification in rats with chronic kidney disease by upregulation of α-Klotho. Kidney Int. 2016, 89, 586–600. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.-S.; Liu, Y.; Chen, Y.; Ren, J.-L.; Zhang, Y.-R.; Yu, Y.-R.; Jia, M.-Z.; Ning, Z.-P.; Du, J.; Tang, C.-S.; et al. Intermedin alleviates pathological cardiac remodeling by upregulating klotho. Pharmacol. Res. 2020, 159, 104926. [Google Scholar] [CrossRef]
- Yoon, H.E.; Lim, S.W.; Piao, S.G.; Song, J.-H.; Kim, J.; Yang, C.W. Statin Upregulates the Expression of Klotho, an Anti-Aging Gene, in Experimental Cyclosporine Nephropathy. Nephron Dial. Transplant. 2012, 120, e123–e133. [Google Scholar] [CrossRef] [PubMed]
- Newman, C.B.; Preiss, D.; Tobert, J.A.; Jacobson, T.A.; Page, I.R.L.; Goldstein, L.B.; Chin, C.; Tannock, L.R.; Miller, M.; Raghuveer, G.; et al. Statin Safety and Associated Adverse Events: A Scientific Statement from the American Heart Association. Arter. Thromb. Vasc. Biol. 2019, 39, e38–e81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Z.W.; Wu, K.R.; Li, R.; Yin, Y.; Li, X.L.; Zhang, S.F.; Li, Y. Pharmacogenetics of statins treatment: Efficacy and safety. J. Clin. Pharm. Ther. 2019, 44, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Adema, A.Y.; Vervloet, M.G.; Blankenstein, M.A.; Heijboer, A.C. α-Klotho is unstable in human urine. Kidney Int. 2015, 88, 1442–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheikhi, A.; Barchowsky, A.; Sahu, A.; Shinde, S.N.; Pius, A.; Clemens, Z.J.; Li, H.; Kennedy, C.A.; Hoeck, J.; Franti, M.; et al. Klotho: An Elephant in Aging Research. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2019, 74, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, Z. Klotho Gene Delivery Prevents the Progression of Spontaneous Hypertension and Renal Damage. Hypertension 2009, 54, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ren, D.; Li, Y.; Xu, G. Klotho attenuates diabetic nephropathy in db/db mice and ameliorates high glucose-induced injury of human renal glomerular endothelial cells. Cell Cycle 2019, 18, 696–707. [Google Scholar] [CrossRef] [Green Version]
- Mitani, H.; Ishizaka, N.; Aizawa, T.; Ohno, M.; Usui, S.-I.; Suzuki, T.; Amaki, T.; Mori, I.; Nakamura, Y.; Sato, M.; et al. In Vivo klotho Gene Transfer Ameliorates Angiotensin II-Induced Renal Damage. Hypertension 2002, 39, 838–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lien, Y.H.; Lai, L.W. Gene therapy for renal diseases. Kidney Int. Suppl. 1997, 61, 85–88. [Google Scholar]
- Kim, N.; Cho, S.-G. Clinical applications of mesenchymal stem cells. Korean J. Intern. Med. 2013, 28, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Squillaro, T.; Peluso, G.; Galderisi, U. Clinical Trials with Mesenchymal Stem Cells: An Update. Cell Transplant. 2016, 25, 829–848. [Google Scholar] [CrossRef] [Green Version]
- Bochon, B.; Kozubska, M.; Surygała, G.; Witkowska, A.; Kuźniewicz, R.; Grzeszczak, W.; Wystrychowski, G. Mesenchymal Stem Cells—Potential Applications in Kidney Diseases. Int. J. Mol. Sci. 2019, 20, 2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulini, J.; Higuti, E.; Bastos, R.M.C.; Gomes, S.A.; Rangel, É.B. Mesenchymal Stem Cells as Therapeutic Candidates for Halting the Progression of Diabetic Nephropathy. Stem Cells Int. 2016, 2016, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- MohammadAlipour, A.; Dumbali, S.P.; Wenzel, P.L. Mitochondrial Transfer and Regulators of Mesenchymal Stromal Cell Function and Therapeutic Efficacy. Front. Cell Dev. Biol. 2020, 8, 603292. [Google Scholar] [CrossRef] [PubMed]
- Vizoso, F.J.; Eiro, N.; Cid, S.; Schneider, J.; Perez-Fernandez, R. Mesenchymal Stem Cell Secretome: Toward Cell-Free Therapeutic Strategies in Regenerative Medicine. Int. J. Mol. Sci. 2017, 18, 1852. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Yang, L. Mesenchymal stem cells and extracellular vesicles in therapy against kidney diseases. Stem Cell Res. Ther. 2021, 12, 219. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, K.; Kitamura, S.; Wada, J. Immunomodulatory and Regenerative Effects of Mesenchymal Stem Cell-Derived Extracellular Vesicles in Renal Diseases. Int. J. Mol. Sci. 2020, 21, 756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grange, C.; Papadimitriou, E.; Dimuccio, V.; Pastorino, C.; Molina, J.; O’Kelly, R.; Niedernhofer, L.J.; Robbins, P.D.; Camussi, G.; Bussolati, B. Urinary Extracellular Vesicles Carrying Klotho Improve the Recovery of Renal Function in an Acute Tubular Injury Model. Mol. Ther. 2020, 28, 490–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, S.; Grange, C.; Deregibus, M.C.; Calogero, R.A.; Saviozzi, S.; Collino, F.; Morando, L.; Busca, A.; Falda, M.; Bussolati, B.; et al. Mesenchymal Stem Cell-Derived Microvesicles Protect Against Acute Tubular Injury. J. Am. Soc. Nephrol. 2009, 20, 1053–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vakhshiteh, F.; Atyabi, F.; Ostad, S.N. Mesenchymal stem cell exosomes: A two-edged sword in cancer therapy. Int. J. Nanomed. 2019, 14, 2847–2859. [Google Scholar] [CrossRef] [Green Version]
- Watson, D.C.; Yung, B.C.; Bergamaschi, C.; Chowdhury, B.; Bear, J.; Stellas, D.; Morales-Kastresana, A.; Jones, J.C.; Felber, B.K.; Chen, X.; et al. Scalable, cGMP-compatible purification of extracellular vesicles carrying bioactive human heterodimeric IL-15/lactadherin complexes. J. Extracell. Vesicles 2018, 7, 1442088. [Google Scholar] [CrossRef] [PubMed]
- Nikfarjam, S.; Rezaie, J.; Zolbanin, N.M.; Jafari, R. Mesenchymal stem cell derived-exosomes: A modern approach in translational medicine. J. Transl. Med. 2020, 18, 449. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.J.; Prockop, D.M.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Galipeau, J.; Krampera, M.; Barrett, J.; Dazzi, F.; Deans, R.J.; DeBruijn, J.; Dominici, M.; Fibbe, W.E.; Gee, A.P.; Gimble, J.M.; et al. International Society for Cellular Therapy perspective on immune functional assays for mesenchymal stromal cells as potency release criterion for advanced phase clinical trials. Cytotherapy 2016, 18, 151–159. [Google Scholar] [CrossRef] [Green Version]
- Chinnadurai, R.; Rajan, D.; Qayed, M.; Arafat, D.; Garcia, M.; Liu, Y.; Kugathasan, S.; Anderson, L.J.; Gibson, G.; Galipeau, J. Potency Analysis of Mesenchymal Stromal Cells Using a Combinatorial Assay Matrix Approach. Cell Rep. 2018, 22, 2504–2517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimarino, A.M.; Caplan, A.I.; Bonfield, T.L. Mesenchymal Stem Cells in Tissue Repair. Front. Immunol. 2013, 4, 201. [Google Scholar] [CrossRef] [Green Version]
- Kabat, M.; Bobkov, I.; Kumar, S.; Grumet, M. Trends in mesenchymal stem cell clinical trials 2004–2018: Is efficacy optimal in a narrow dose range? Stem Cells Transl. Med. 2020, 9, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keating, A. Mesenchymal Stromal Cells: New Directions. Cell Stem Cell 2012, 10, 709–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardo, M.E.; Fibbe, W.E. Mesenchymal Stromal Cells: Sensors and Switchers of Inflammation. Cell Stem Cell 2013, 13, 392–402. [Google Scholar] [CrossRef] [Green Version]
- El-Badawy, A.; El-Badri, N. Clinical Efficacy of Stem Cell Therapy for Diabetes Mellitus: A Meta-Analysis. PLoS ONE 2016, 11, e0151938. [Google Scholar] [CrossRef] [Green Version]
- Cramer, C.; Freisinger, E.; Jones, R.K.; Slakey, U.P.; Dupin, C.L.; Newsome, E.R.; Alt, E.U.; Izadpanah, R. Persistent High Glucose Concentrations Alter the Regenerative Potential of Mesenchymal Stem Cells. Stem Cells Dev. 2010, 19, 1875–1884. [Google Scholar] [CrossRef]
- Koci, Z.; Turnovcova, K.; Dubský, M.; Baranovicova, L.; Holan, V.; Chudíčková, M.; Syková, E.; Kubinová, Š. Characterization of human adipose tissue-derived stromal cells isolated from diabetic patient’s distal limbs with critical ischemia. Cell Biochem. Funct. 2014, 32, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.; Alm, J.J.; Heldring, N.; Moll, G.; Gavin, C.; Batsis, I.; Qian, H.; Sigvardsson, M.; Nilsson, B.; Kyllonen, L.E.; et al. Type 1 Diabetes Mellitus Donor Mesenchymal Stromal Cells Exhibit Comparable Potency to Healthy Controls In Vitro. Stem Cells Transl. Med. 2016, 5, 1485–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Rhijn-Brouwer, F.C.C.; Gremmels, H.; Fledderus, J.O.; Verhaar, M.C. Mesenchymal Stromal Cell Characteristics and Regenerative Potential in Cardiovascular Disease. Cell Transplant. 2018, 27, 765–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hare, J.M.; Fishman, J.E.; Gerstenblith, G.; Velazquez, D.L.D.; Zambrano, J.P.; Suncion, V.Y.; Tracy, M.; Ghersin, E.; Johnston, P.V.; Brinker, J.A.; et al. Comparison of Allogeneic vs Autologous Bone Marrow–Derived Mesenchymal Stem Cells Delivered by Transendocardial Injection in Patients with Ischemic Cardiomyopathy. JAMA 2012, 308, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Huang, Z.; Han, F.; Zhao, M.; Cao, R.; Zhao, D.; Hong, L.; Na, N.; Li, H.; Miao, B.; et al. Allogeneic mesenchymal stem cells as induction therapy are safe and feasible in renal allografts: Pilot results of a multicenter randomized controlled trial. J. Transl. Med. 2018, 16, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sávio-Silva, C.; Beyerstedt, S.; Soinski-Sousa, P.E.; Casaro, E.B.; Balby-Rocha, M.T.A.; Simplício-Filho, A.; Alves-Silva, J.; Rangel, É.B. Mesenchymal Stem Cell Therapy for Diabetic Kidney Disease: A Review of the Studies Using Syngeneic, Autologous, Allogeneic, and Xenogeneic Cells. Stem Cells Int. 2020, 2020, 1–28. [Google Scholar] [CrossRef]
- Jimenez-Puerta, G.J.; Marchal, J.A.; Ruiz, E.L.; Gálvez-Martín, P. Role of Mesenchymal Stromal Cells as Therapeutic Agents: Potential Mechanisms of Action and Implications in Their Clinical Use. J. Clin. Med. 2020, 9, 445. [Google Scholar] [CrossRef] [Green Version]
- Bhat, S.; Viswanathan, P.; Chandanala, S.; Prasanna, S.J.; Seetharam, R.N. Expansion and characterization of bone marrow derived human mesenchymal stromal cells in serum-free conditions. Sci. Rep. 2021, 11, 3403. [Google Scholar] [CrossRef]
- Jing-Li, Z.; Wu, X.-Y.; Tong, J.-B.; Yang, X.-X.; Zhao, J.-L.; Zheng, Q.-F.; Zhao, G.-B.; Ma, Z.-J. Comparative analysis of human mesenchymal stem cells from bone marrow and adipose tissue under xeno-free conditions for cell therapy. Stem Cell Res. Ther. 2015, 6, 55. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Omar, O.; Vazirisani, F.; Thomsen, P.; Ekström, K. Mesenchymal stem cell-derived exosomes have altered microRNA profiles and induce osteogenic differentiation depending on the stage of differentiation. PLoS ONE 2018, 13, e0193059. [Google Scholar] [CrossRef] [PubMed]
- Kehl, D.; Generali, M.; Mallone, A.; Heller, M.; Uldry, A.-C.; Cheng, P.; Gantenbein, B.; Hoerstrup, S.P.; Weber, B. Proteomic analysis of human mesenchymal stromal cell secretomes: A systematic comparison of the angiogenic potential. Regen. Med. 2019, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Sávio-Silva, C.; Soinski-Sousa, P.E.; Balby-Rocha, M.A.T.; Lira, Á.D.O.; Rangel, É.B. Mesenchymal stem cell therapy in acute kidney injury (AKI): Review and perspectives. Rev. Da Assoc. Méd. Bras. 2020, 66, s45–s54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sávio-Silva, C.; Soinski-Sousa, P.; Simplício-Filho, A.; Bastos, R.; Beyerstedt, S.; Rangel, É.B. Therapeutic Potential of Mesenchymal Stem Cells in a Pre-Clinical Model of Diabetic Kidney Disease and Obesity. Int. J. Mol. Sci. 2021, 22, 1546. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Zhang, B.; Guo, W.; Liu, N.; Bai, Y.; Miao, J.; Zhao, G.; Liu, B.; Wang, S.; Ma, L.; et al. Effects of Mesenchymal Stem Cells Transplanted at Different Time Points in a Rat Remnant Kidney Model. Am. J. Nephrol. 2014, 39, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.; Lira, R.; Oliveira, M.; Martins, M.; Azevedo, Y.; Silva, K.; Carvalho, S.; Cortez, E.; Stumbo, A.C.; Carvalho, L.; et al. Bone Marrow-Derived Mesenchymal Stem Cells Transplantation Ameliorates Renal Injury through Anti-Fibrotic and Anti-Inflammatory Effects in Chronic Experimental Renovascular Disease. Biomed. J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Fang, Y.; Xie, L.; Liu, X.; Shan, W.; Zeng, R.; Liu, J.; Liu, X. Adipose-Derived Mesenchymal Stem Cells Transplantation Alleviates Renal Injury in Streptozotocin-Induced Diabetic Nephropathy. J. Histochem. Cytochem. 2015, 63, 842–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Li, L. Preconditioning influences mesenchymal stem cell properties in vitro and in vivo. J. Cell. Mol. Med. 2018, 22, 1428–1442. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Zhu, M.; Dangelmajer, S.; Lee, Y.M.; Wijesekera, O.; Castellanos, C.X.; Denduluri, A.; Chaichana, K.L.; Li, Q.; Zhang, H.; et al. Hypoxia-cultured human adipose-derived mesenchymal stem cells are non-oncogenic and have enhanced viability, motility, and tropism to brain cancer. Cell Death Dis. 2014, 5, e1567. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yoon, Y.M.; Lee, S.H. Hypoxic Preconditioning Promotes the Bioactivities of Mesenchymal Stem Cells via the HIF-1α-GRP78-Akt Axis. Int. J. Mol. Sci. 2017, 18, 1320. [Google Scholar] [CrossRef] [PubMed]
- Szabó, E.; Fajka-Boja, R.; Kriston-Pál, É.; Hornung, Á.; Makra, I.; Kudlik, G.; Uher, F.; Katona, R.L.; Monostori, É.; Czibula, Á. Licensing by Inflammatory Cytokines Abolishes Heterogeneity of Immunosuppressive Function of Mesenchymal Stem Cell Population. Stem Cells Dev. 2015, 24, 2171–2180. [Google Scholar] [CrossRef] [Green Version]
- Oses, C.; Olivares, B.; Ezquer, M.; Acosta, C.; Bosch, P.; Donoso, M.; Léniz, P.; Ezquer, F. Preconditioning of adipose tissue-derived mesenchymal stem cells with deferoxamine increases the production of pro-angiogenic, neuroprotective and anti-inflammatory factors: Potential application in the treatment of diabetic neuropathy. PLoS ONE 2017, 12, e0178011. [Google Scholar] [CrossRef]
- Azar, Y.M.; Green, R.; Niesler, C.U.; van de Vyver, M. Antioxidant Preconditioning Improves the Paracrine Responsiveness of Mouse Bone Marrow Mesenchymal Stem Cells to Diabetic Wound Fluid. Stem Cells Dev. 2018, 27, 1646–1657. [Google Scholar] [CrossRef]
- Capilla-González, V.; López-Beas, J.; Escacena, N.; Aguilera, Y.; de la Cuesta, A.; Ruiz-Salmerón, R.; Martin, F.; Hmadcha, A.; Soria, B. PDGF Restores the Defective Phenotype of Adipose-Derived Mesenchymal Stromal Cells from Diabetic Patients. Mol. Ther. 2018, 26, 2696–2709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagaishi, K.; Mizue, Y.; Chikenji, T.; Otani, M.; Nakano, M.; Saijo, Y.; Tsuchida, H.; Ishioka, S.; Nishikawa, A.; Saito, T.; et al. Umbilical cord extracts improve diabetic abnormalities in bone marrow-derived mesenchymal stem cells and increase their therapeutic effects on diabetic nephropathy. Sci. Rep. 2017, 7, 8484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.; Russell, P.J.; Martiniello-Wilks, R.; Rasko, J.E.J.; Khatri, A. Concise review: Nanoparticles and cellular carriers-allies in cancer imaging and cellular gene therapy? Stem Cells 2010, 28, 1686–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Lu, C.; Dai, Q.; Sheng, J.; Xu, M. SIRT3 Facilitates Amniotic Fluid Stem Cells to Repair Diabetic Nephropathy Through Protecting Mitochondrial Homeostasis by Modulation of Mitophagy. Cell. Physiol. Biochem. 2018, 46, 1508–1524. [Google Scholar] [CrossRef] [Green Version]
- Kaundal, U.; Bagai, U.; Rakha, A. Immunomodulatory plasticity of mesenchymal stem cells: A potential key to successful solid organ transplantation. J. Transl. Med. 2018, 16, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namgung, R.; Singha, K.; Yu, M.K.; Jon, S.; Kim, Y.S.; Ahn, Y.; Park, I.-K.; Kim, W.J. Hybrid superparamagnetic iron oxide nanoparticle-branched polyethylenimine magnetoplexes for gene transfection of vascular endothelial cells. Biomaterials 2010, 31, 4204–4213. [Google Scholar] [CrossRef] [PubMed]
- Tögel, F.; Isaac, J.; Hu, Z.; Weiss, K.; Westenfelder, C. Renal SDF-1 signals mobilization and homing of CXCR4-positive cells to the kidney after ischemic injury. Kidney Int. 2005, 67, 1772–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Tian, J.; Cheng, J.; Zhang, J. Migration of CXCR4 gene-modified bone marrow-derived mesenchymal stem cells to the acute injured kidney. J. Cell. Biochem. 2013, 114, 2677–2689. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Ma, R.; Cai, W.; Huang, W.; Paul, C.; Liang, J.; Wang, Y.; Zhao, T.; Kim, H.W.; Xu, M.; et al. Exosomes Secreted from CXCR4 Overexpressing Mesenchymal Stem Cells Promote Cardioprotection via Akt Signaling Pathway following Myocardial Infarction. Stem Cells Int. 2015, 2015, 659890. [Google Scholar] [CrossRef] [Green Version]
- Russell, A.L.; Lefavor, R.C.; Zubair, A.C. Characterization and cost-benefit analysis of automated bioreactor-expanded mesenchymal stem cells for clinical applications. Transfussion 2018, 58, 2374–2382. [Google Scholar] [CrossRef] [PubMed]
- Haack-Sørensen, M.; Juhl, M.; Follin, B.; Søndergaard, R.H.; Kirchhoff, M.; Kastrup, J.; Ekblond, A. Development of large-scale manufacturing of adipose-derived stromal cells for clinical applications using bioreactors and human platelet lysate. Scand. J. Clin. Lab. Investig. 2018, 78, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Phan, J.; Kumar, P.; Hao, D.; Gao, K.; Farmer, D.; Wang, A. Engineering mesenchymal stem cells to improve their exosome efficacy and yield for cell-free therapy. J. Extracell. Vesicles 2018, 7, 1522236. [Google Scholar] [CrossRef] [PubMed]
- Amable, P.R.; Teixeira, M.V.T.; Carias, R.B.V.; Granjeiro, J.M.; Borojevic, R. Protein synthesis and secretion in human mesenchymal cells derived from bone marrow, adipose tissue and Wharton’s jelly. Stem Cell Res. Ther. 2014, 5, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driscoll, J.; Patel, T. The mesenchymal stem cell secretome as an acellular regenerative therapy for liver disease. J. Gastroenterol. 2019, 54, 763–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, W.; Zhang, Y.; Yin, Z. The protective mechanism of Klotho gene-modified bone marrow mesenchymal stem cells on acute kidney injury induced by rhabdomyolysis. Regen. Ther. 2021, 18, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xue, D.; Hu, D.; Xie, T.; Tao, Y.; Zhu, T.; Chen, E.; Pan, Z. Secreted klotho protein attenuates osteogenic differentiation of human bone marrow mesenchymal stem cellsin vitrovia inactivation of the FGFR1/ERK signaling pathway. Growth Factors 2015, 33, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wan, X.; Cao, Y.-Z.; Sun, D.; Cao, C.-C. Klotho gene-modified BMSCs showed elevated antifibrotic effects by inhibiting the Wnt/β-catenin pathway in kidneys after acute injury. Cell Biol. Int. 2018, 42, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.; Sun, Z. Klotho Deficiency Accelerates Stem Cells Aging by Impairing Telomerase Activity. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2018, 74, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.-B.; Chen, X.; Chen, B.; Wang, X.-D.; Jiang, R.; Lu, Y.-P. Protective effect of bone marrow mesenchymal stem cells modified with klotho on renal ischemia-reperfusion injury. Ren. Fail. 2019, 41, 175–182. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Su, B.; Ni, H.; Li, L.; Chen, X.; You, X.; Zhang, H. microRNA-199a may be involved in the pathogenesis of lupus nephritis via modulating the activation of NF-κB by targeting Klotho. Mol. Immunol. 2018, 103, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Lee, S.H. Potential and Therapeutic Efficacy of Cell-based Therapy Using Mesenchymal Stem Cells for Acute/chronic Kidney Disease. Int. J. Mol. Sci. 2019, 20, 1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Bernal, D.; García-Arranz, M.; Yáñez, R.M.; Hervás-Salcedo, R.; Cortés, A.; Fernández-García, M.; Hernando-Rodríguez, M.; Quintana-Bustamante, Ó.; Bueren, J.A.; García-Olmo, D.; et al. The Current Status of Mesenchymal Stromal Cells: Controversies, Unresolved Issues and Some Promising Solutions to Improve Their Therapeutic Efficacy. Front. Cell Dev. Biol. 2021, 9, 609. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.-Z.; Lin, Y.-H.; Su, L.-J.; Wu, M.-S.; Jeng, H.-Y.; Chang, H.-C.; Huang, Y.-H.; Ling, T.-Y. Mesenchymal stem/stromal cell-based therapy: Mechanism, systemic safety and biodistribution for precision clinical applications. J. Biomed. Sci. 2021, 28, 28. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.-Y.; Wang, B.; Tang, T.-T.; Wen, Y.; Li, Z.-L.; Feng, S.-T.; Wu, M.; Liu, D.; Yin, D.; Ma, K.-L.; et al. Exosomal miR-125b-5p deriving from mesenchymal stem cells promotes tubular repair by suppression of p53 in ischemic acute kidney injury. Theranostics 2021, 11, 5248–5266. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-J.; Jiang, K.; Ferguson, C.M.; Tang, H.; Zhu, X.; Lerman, A.; Lerman, L.O. Augmented efficacy of exogenous extracellular vesicles targeted to injured kidneys. Signal. Transduct. Target. Ther. 2020, 5, 199. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-K.; Yang, C.; Su, Y.; Luo, J.-C.; Luo, M.-H.; Huang, D.-L.; Tu, G.-W.; Luo, Z. Mesenchymal Stem Cell-Derived Extracellular Vesicles: A Potential Therapeutic Strategy for Acute Kidney Injury. Front. Immunol. 2021, 12, 2124. [Google Scholar] [CrossRef] [PubMed]
- Collino, F.; Bruno, S.; Incarnato, D.; Dettori, D.; Neri, F.; Provero, P.; Pomatto, M.; Oliviero, S.; Tetta, C.; Quesenberry, P.J.; et al. AKI Recovery Induced by Mesenchymal Stromal Cell-Derived Extracellular Vesicles Carrying MicroRNAs. J. Am. Soc. Nephrol. 2015, 26, 2349–2360. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Li, H.-Y.; Yang, Q.; Chen, G.; Lin, S.; Liao, C.; Zhou, T. Administration of mesenchymal stem cells in diabetic kidney disease: A systematic review and meta-analysis. Stem Cell Res. Ther. 2021, 12, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Nowak, N.; Yamanouchi, M.; Satake, E. The Nephroprotective Properties of Extracellular Vesicles in Experimental Models of Chronic Kidney Disease: A Systematic Review. Stem Cell Rev. Rep. 2021, 1–31. [Google Scholar] [CrossRef]
- Khatri, R.; Mazurek, S.; Petry, S.F.; Linn, T. Mesenchymal stem cells promote pancreatic β-cell regeneration through downregulation of FoxO1 pathway. Stem Cell Res. Ther. 2020, 11, 497. [Google Scholar] [CrossRef]
- Sun, Y.; Shi, H.; Yin, S.; Ji, C.; Zhang, X.; Zhang, B.; Wu, P.; Shi, Y.; Mao, F.; Yan, Y.; et al. Human Mesenchymal Stem Cell Derived Exosomes Alleviate Type 2 Diabetes Mellitus by Reversing Peripheral Insulin Resistance and Relieving β-Cell Destruction. ACS Nano 2018, 12, 7613–7628. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragni, E.; Banfi, F.; Barilani, M.; Cherubini, A.; Parazzi, V.; Larghi, P.; Dolo, V.; Bollati, V.; Lazzari, L. Extracellular Vesicle-Shuttled mRNA in Mesenchymal Stem Cell Communication. Stem Cells 2017, 35, 1093–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Guo, Z.-H. Downregulation of lncRNA NEAT1 Ameliorates LPS-Induced Inflammatory Responses by Promoting Macrophage M2 Polarization via miR-125a-5p/TRAF6/TAK1 Axis. Inflammation 2020, 43, 1548–1560. [Google Scholar] [CrossRef]
- Yang, C.; Yang, C.; Huang, Z.; Zhang, J.; Chen, N.; Guo, Y.; Zahoor, A.; Deng, G. Reduced expression of MiR-125a-5p aggravates LPS-induced experimental acute kidney injury pathology by targeting TRAF6. Life Sci. 2021, 288, 119657. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pan, X.; Fu, X.; Yang, Y.; Chen, J.; Lin, W. MicroRNA-26a: An Emerging Regulator of Renal Biology and Disease. Kidney Blood Press. Res. 2019, 44, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Koga, K.; Yokoi, H.; Mori, K.; Kasahara, M.; Kuwabara, T.; Imamaki, H.; Ishii, A.; Mori, K.P.; Kato, Y.; Ohno, S.; et al. MicroRNA-26a inhibits TGF-β-induced extracellular matrix protein expression in podocytes by targeting CTGF and is downregulated in diabetic nephropathy. Diabetology 2015, 58, 2169–2180. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Wang, H.; Wang, B.; Yuan, Y.; Klein, J.D.; Wang, X.H. Exogenous miR-26a suppresses muscle wasting and renal fibrosis in obstructive kidney disease. FASEB J. 2019, 33, 13590–13601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, N.; Bera, A.; Das, F.; Ghosh-Choudhury, N.; Kasinath, B.S.; Choudhury, G.G. High glucose enhances microRNA-26a to activate mTORC1 for mesangial cell hypertrophy and matrix protein expression. Cell. Signal. 2015, 27, 1276–1285. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Wang, G.; Peng, Z. MicroRNA-191-5p diminished sepsis-induced acute kidney injury through targeting oxidative stress responsive 1 in rat models. Biosci. Rep. 2019, 39, 20190548. [Google Scholar] [CrossRef] [Green Version]
- Berillo, O.; Huo, K.-G.; Fraulob-Aquino, J.C.; Richer, C.; Briet, M.; Boutouyrie, P.; Lipman, M.L.; Sinnett, D.; Paradis, P.; Schiffrin, E.L. Circulating let-7g-5p and miR-191-5p Are Independent Predictors of Chronic Kidney Disease in Hypertensive Patients. Am. J. Hypertens. 2020, 33, 505–513. [Google Scholar] [CrossRef]
- An, X.; Liao, G.; Chen, Y.; Luo, A.; Liu, J.; Yuan, Y.; Li, L.; Yang, L.; Wang, H.; Liu, F.; et al. Intervention for early diabetic nephropathy by mesenchymal stem cells in a preclinical nonhuman primate model. Stem Cell Res. Ther. 2019, 10, 363. [Google Scholar] [CrossRef]
- Kluger, A.Y.; Tecson, K.M.; Lee, A.Y.; Lerma, E.; Rangaswami, J.; Lepor, N.E.; Cobble, M.E.; McCullough, P.A. Class effects of SGLT2 inhibitors on cardiorenal outcomes. Cardiovasc. Diabetol. 2019, 18, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.-F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Neuen, B.L.; Young, T.; Heerspink, H.J.L.; Neal, B.; Perkovic, V.; Billot, L.; Mahaffey, K.W.; Charytan, D.M.; Wheeler, D.C.; Arnott, C.; et al. SGLT2 inhibitors for the prevention of kidney failure in patients with type 2 diabetes: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2019, 7, 845–854. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, J.; Qu, D.; Wang, L.; Luo, J.-Y.; Lau, C.W.; Liu, P.; Gao, Z.; Tipoe, G.L.; Lee, H.K.; et al. Inhibition of miR-200c Restores Endothelial Function in Diabetic Mice Through Suppression of COX-2. Diabetes 2016, 65, 1196–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morii, K.; Yamasaki, S.; Doi, S.; Irifuku, T.; Sasaki, K.; Doi, T.; Nakashima, A.; Arihiro, K.; Masaki, T. microRNA-200c regulates KLOTHO expression in human kidney cells under oxidative stress. PLoS ONE 2019, 14, e0218468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Bi, X.; Xiong, J.; Han, W.; Xiao, T.; Xu, X.; Yang, K.; Liu, C.; Jiang, W.; He, T.; et al. MicroRNA-34a Promotes Renal Fibrosis by Downregulation of Klotho in Tubular Epithelial Cells. Mol. Ther. 2019, 27, 1051–1065. [Google Scholar] [CrossRef] [PubMed]
- Mehi, S.J.; Maltare, A.; Abraham, C.R.; King, G.D. MicroRNA-339 and microRNA-556 regulate Klotho expression in vitro. AGE 2014, 36, 141–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, W.-L.; Xu, G.-S. Atrasentan increased the expression of klotho by mediating miR-199b-5p and prevented renal tubular injury in diabetic nephropathy. Sci. Rep. 2016, 6, 19979. [Google Scholar] [CrossRef]
- Ortega, F.J.; Moreno-Navarrete, J.M.; Pardo, G.; Sabater-Masdeu, M.; Hummel, M.; Ferrer, A.; Rodriguez-Hermosa, J.I.; Ruiz, B.; Ricart, W.; Peral, B.; et al. MiRNA Expression Profile of Human Subcutaneous Adipose and during Adipocyte Differentiation. PLoS ONE 2010, 5, e9022. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Guo, Y.F.; Fu, G.P.; Guan, C.; Zhang, X.; Yang, D.G.; Shi, Y.C. Protective effect of miRNA-containing extracellular vesicles derived from mesenchymal stromal cells of old rats on renal function in chronic kidney disease. Stem Cell Res. Ther. 2020, 11, 274. [Google Scholar] [CrossRef]
- Meng, Y.; Eirin, A.; Zhu, X.-Y.; O’Brien, D.R.; Lerman, A.; Van Wijnen, A.J.; Lerman, L.O. The metabolic syndrome modifies the mRNA expression profile of extracellular vesicles derived from porcine mesenchymal stem cells. Diabetol. Metab. Syndr. 2018, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Froelich, K.; Mickler, J.; Steusloff, G.; Technau, A.; Tirado, M.R.; Scherzed, A.; Hackenberg, S.; Radeloff, A.; Hagen, R.; Kleinsasser, N. Chromosomal aberrations and deoxyribonucleic acid single-strand breaks in adipose-derived stem cells during long-term expansion in vitro. Cytotherapy 2013, 15, 767–781. [Google Scholar] [CrossRef] [PubMed]
- Neri, S.; Bourin, P.; Peyrafitte, J.-A.; Cattini, L.; Facchini, A.; Mariani, E. Human Adipose Stromal Cells (ASC) for the Regeneration of Injured Cartilage Display Genetic Stability after In Vitro Culture Expansion. PLoS ONE 2013, 8, e77895. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Huang, H.; Xu, X. Biological characteristics and karyotiping of a new isolation method for human adipose mesenchymal stem cells in vitro. Tissue Cell 2017, 49, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Trounson, A.; McDonald, C. Stem Cell Therapies in Clinical Trials: Progress and Challenges. Cell Stem Cell 2015, 17, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdi, R.; Fiorina, P.; Adra, C.N.; Atkinson, M.; Sayegh, M.H. Immunomodulation by Mesenchymal Stem Cells: A Potential Therapeutic Strategy for Type 1 Diabetes. Diabetes 2008, 57, 1759–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casiraghi, F.; Remuzzi, G.; Abbate, M.; Perico, N. Multipotent Mesenchymal Stromal Cell Therapy and Risk of Malignancies. Stem Cell Rev. Rep. 2013, 9, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Skyler, J.S.; Fonseca, V.A.; Segal, K.R.; Rosenstock, J. Allogeneic Mesenchymal Precursor Cells in Type 2 Diabetes: A Randomized, Placebo-Controlled, Dose-Escalation Safety and Tolerability Pilot Study. Diabetes Care 2015, 38, 1742–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packham, D.K.; Fraser, I.R.; Kerr, P.G.; Segal, K.R. Allogeneic Mesenchymal Precursor Cells (MPC) in Diabetic Nephropathy: A Randomized, Placebo-controlled, Dose Escalation Study. EBioMedicine 2016, 12, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Fisher, S.A.; Doree, C.; Mathur, A.; Martin-Rendon, E. Meta-Analysis of Cell Therapy Trials for Patients with Heart Failure. Circ. Res. 2015, 116, 1361–1377. [Google Scholar] [CrossRef] [PubMed]
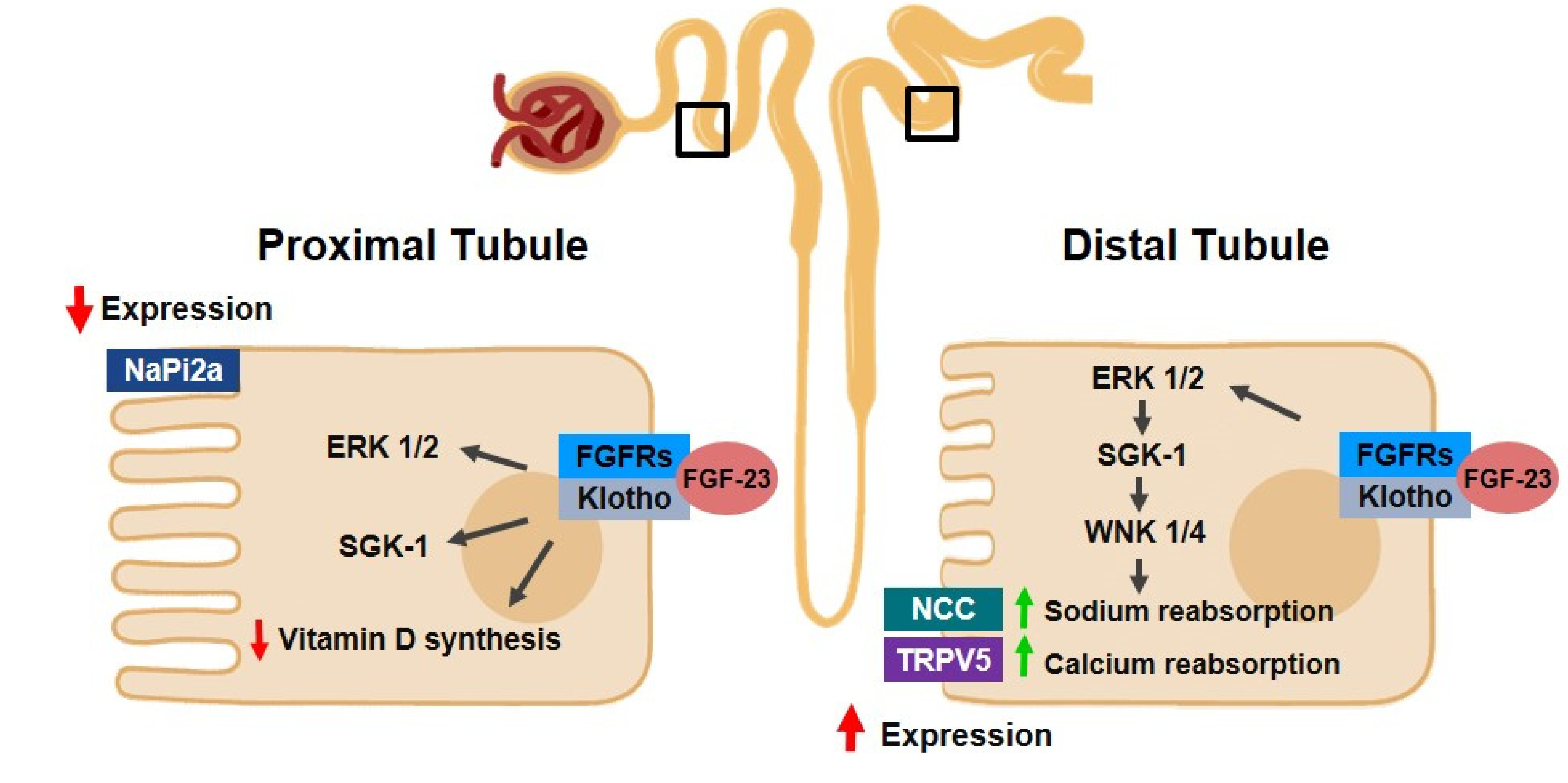


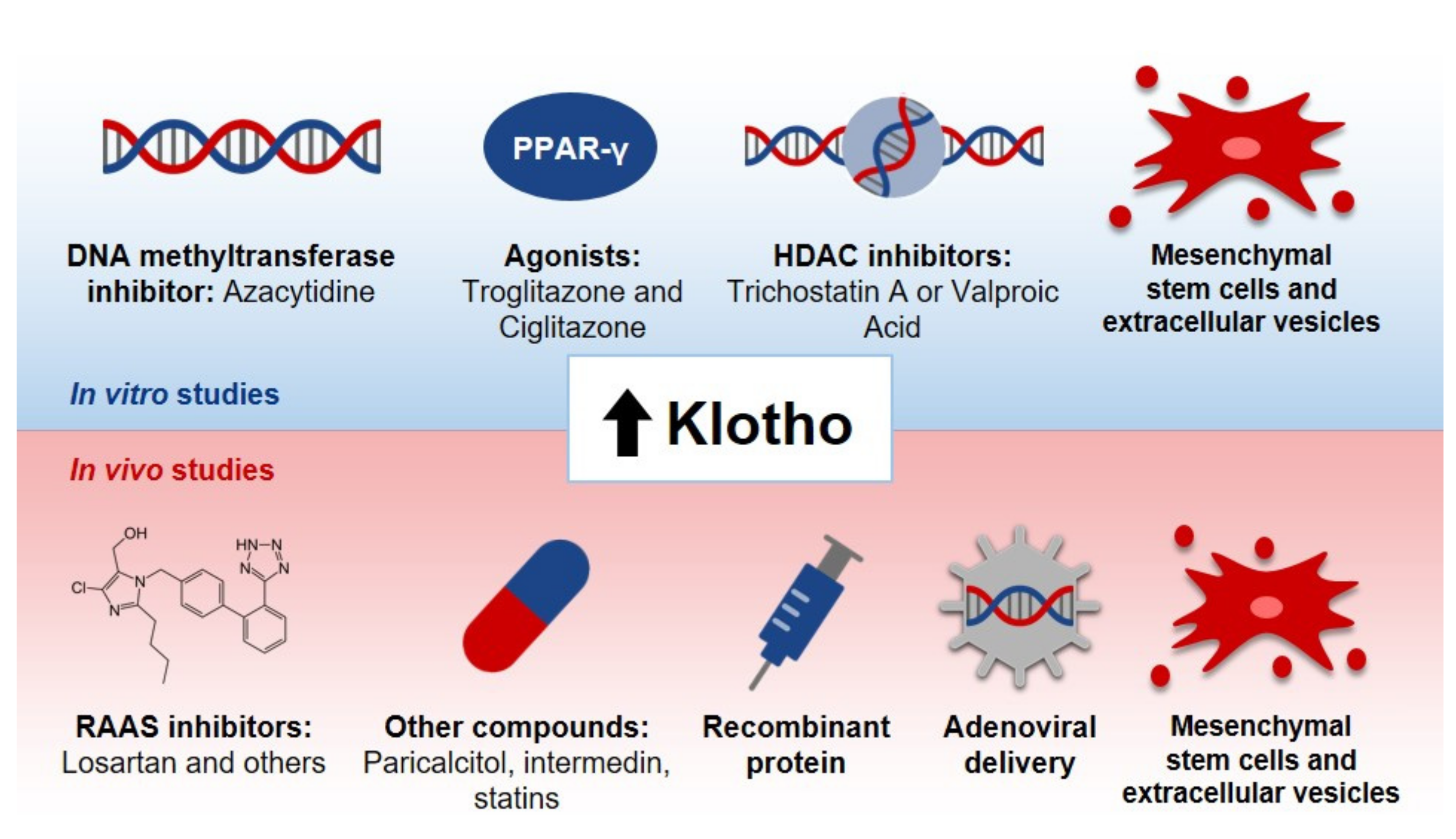


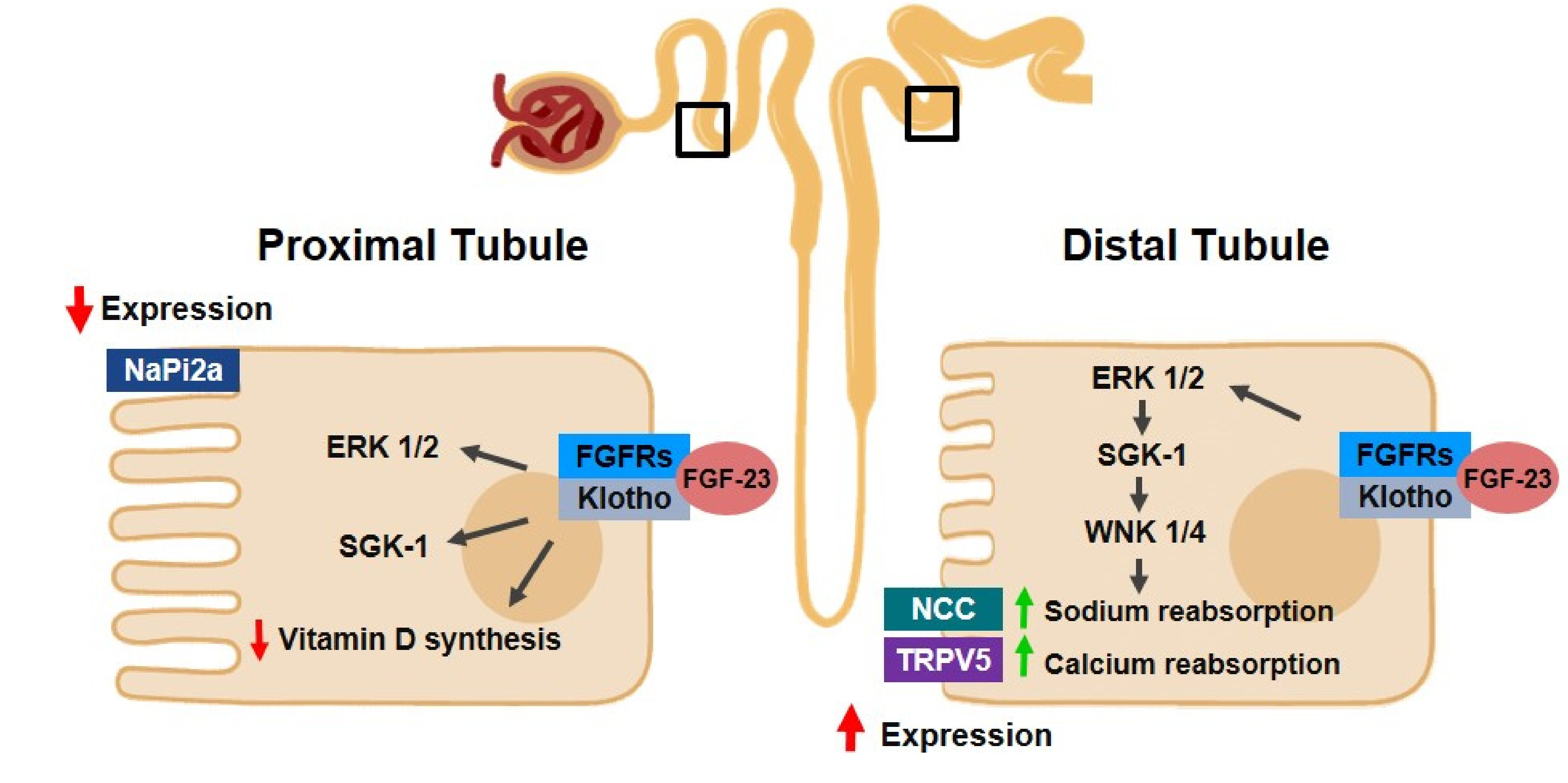{kind=link}
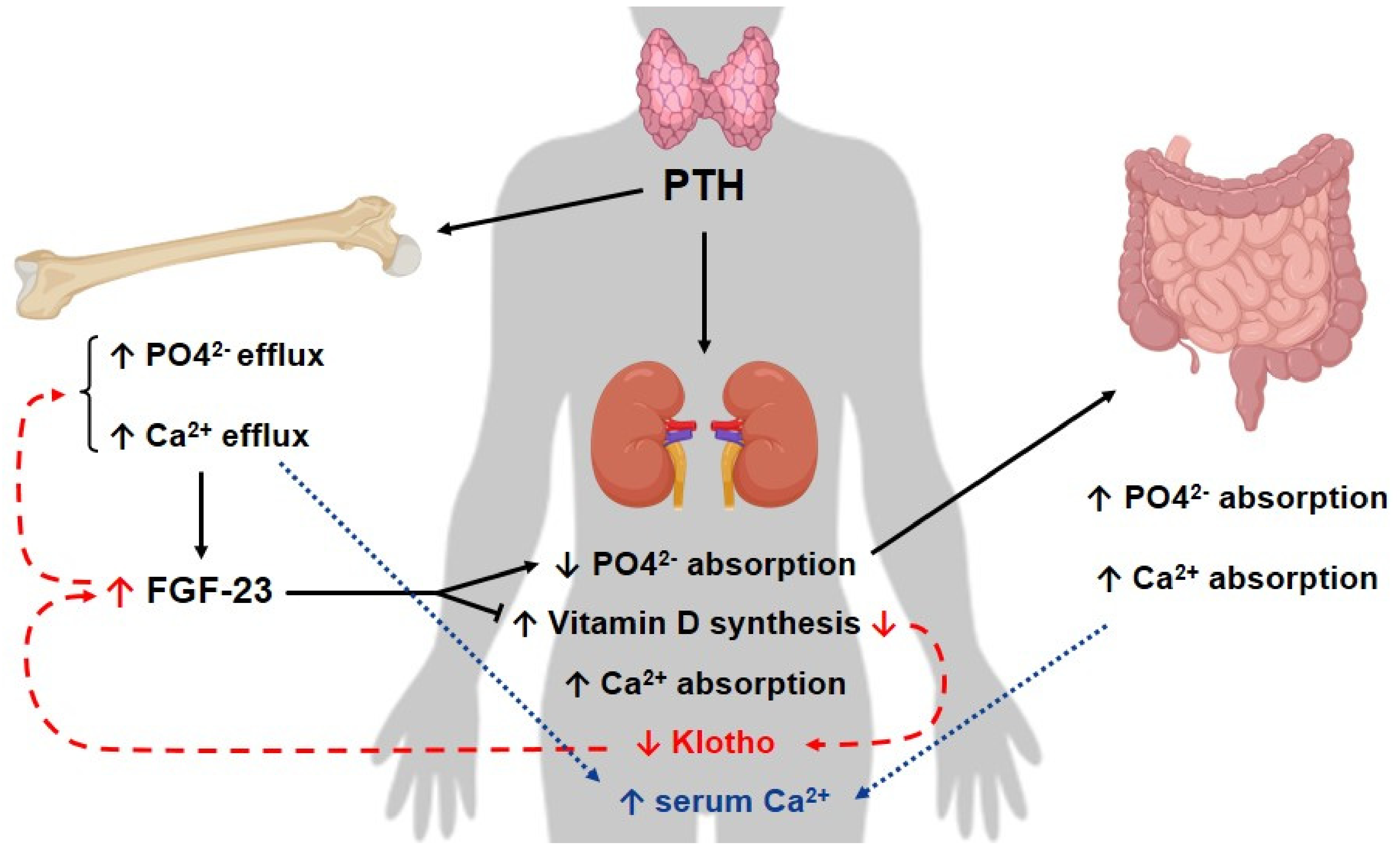{kind=link}
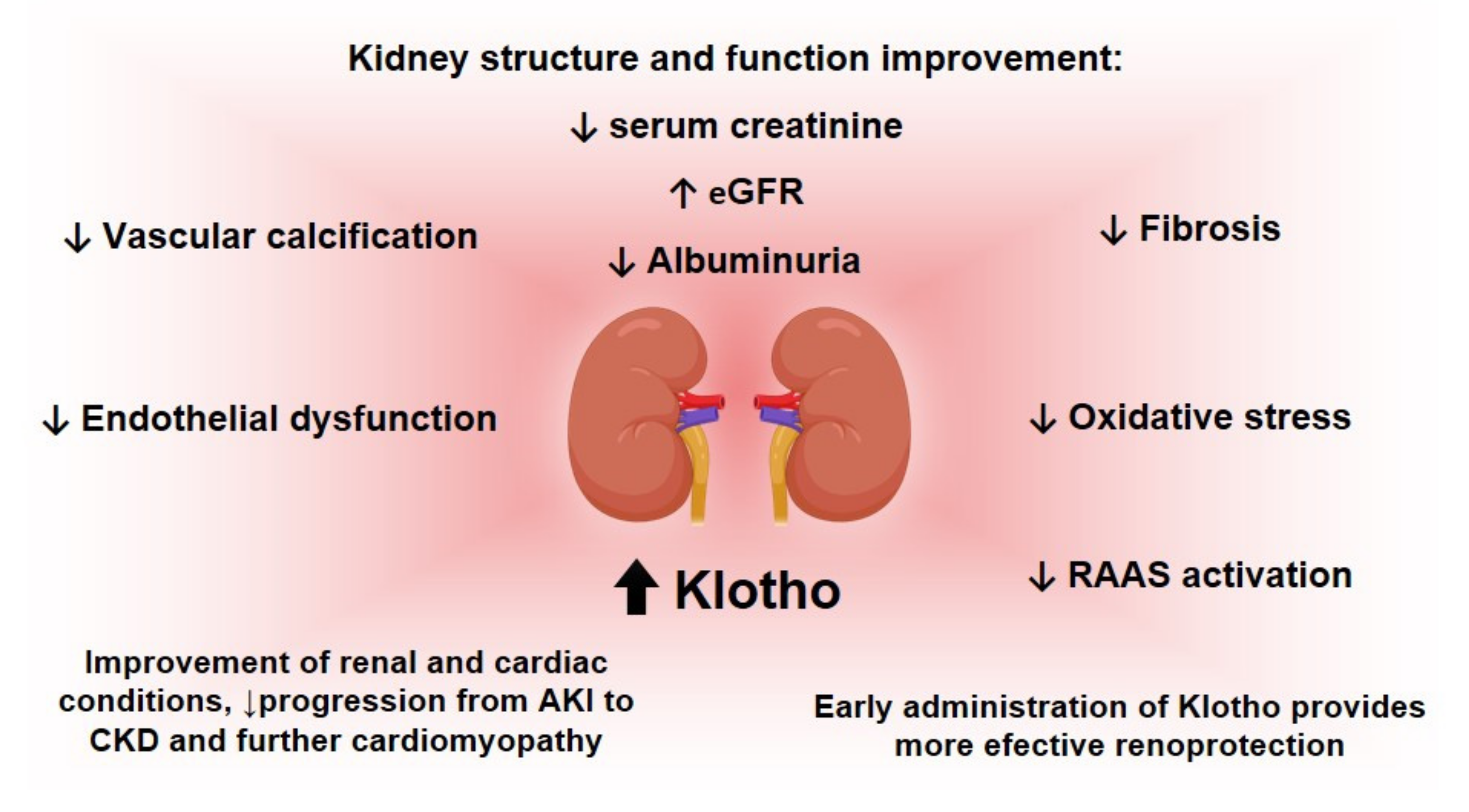{kind=link}
{kind=link}
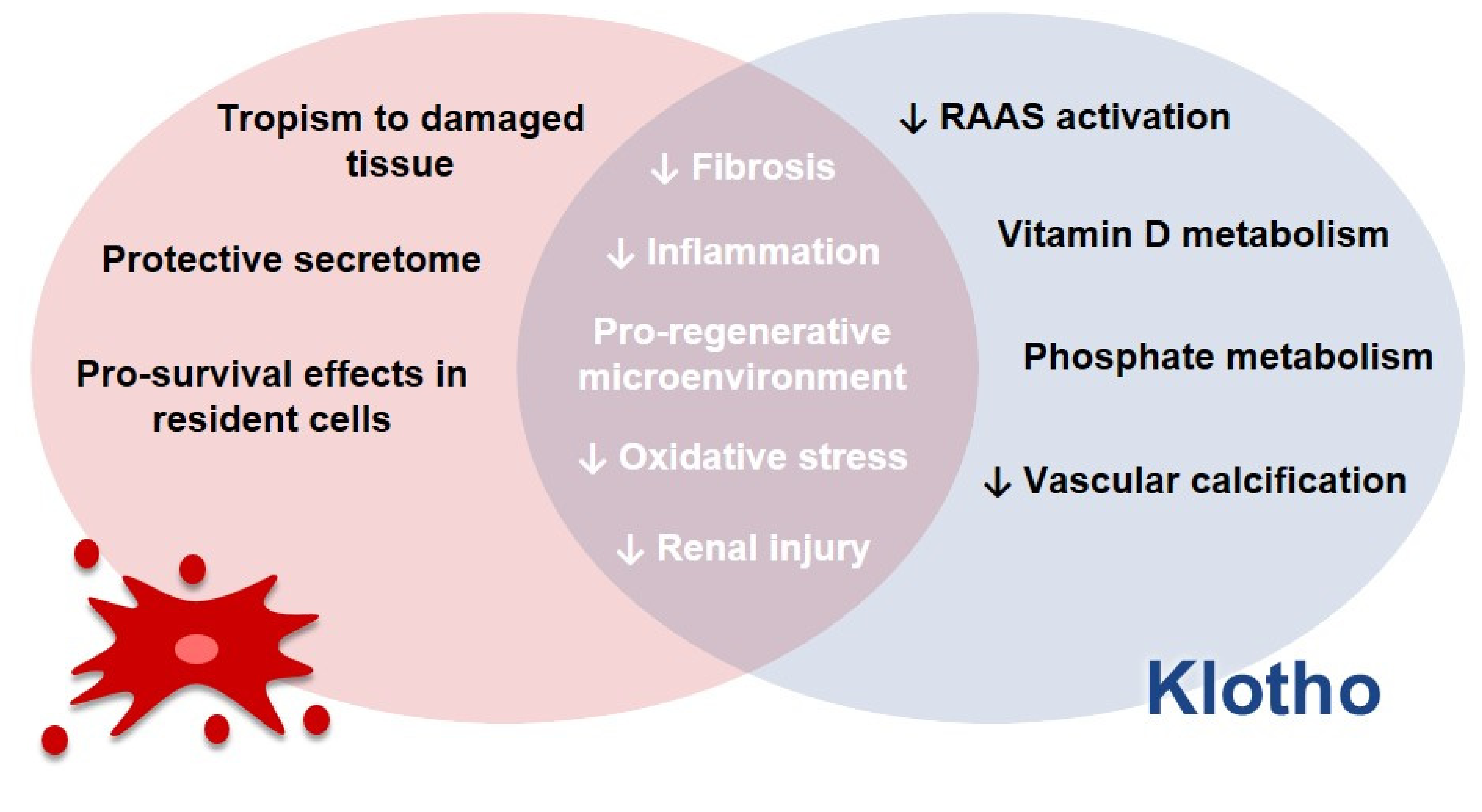{kind=link}
| Author/Year | Model Used | Study Design | Conclusion |
|---|---|---|---|
| Karalliedde, J., et al., 2013 [85] | Patients with diabetes type 2, presenting systolic hypertension and albuminuria | Single-Center, Double-Blind Randomized Controlled Trial | Inhibition of RAAS led to an increase in soluble Klotho levels. |
| Saito, Y., et al., 2000 [86] | Rats with atherosclerosis | Preclinical Study | Klotho adenoviral delivery resulted in the mitigation of vascular endothelial dysfunction and reduction of blood pressure values |
| Kuro-o, M., et al.,1997 [1] | Klotho-deficient mice | Preclinical Study | The animals presented artery calcification, cardiac fibrosis and hypertrophy. Klotho might participate in the signaling pathways involved in these processes. |
| Xie, J., et al., 2015 [87] | Klotho-deficient mice | Preclinical Study | The increase in soluble Klotho levels attenuated cardiac remodeling in CKD animals. Decrease in this protein level is proposed to be an independent factor for cardiomyopathy in CKD. |
| Ding, et al., 2019 [88] | Mice with angiotensin-II infusion | Preclinical Study | Klotho was related to the decrease of cardiac FGF-23 expression in vitro and in vivo; moreover, it prevented cardiac remodeling and dysfunction in this model. |
| Memmos, et al., 2019 [84] | 79 patients on dialysis | Prospective Cohort Study | Low levels of Klotho are correlated with an increased risk of cardiovascular disease and reduced overall survival in these patients. It might contribute to cardiovascular disease in individuals with CKD. |
| Brandenburg, V.M., et al., 2015 [89] | 2948 patients | Multicenter Longitudinal Study | In individuals with normal kidney function, Klotho does not act as a predictive marker of cardiovascular and mortality risk. |
| Pan, H.C., et al., 2018 [90] | 168 patients with diabetes type 2 | Prospective Study | Low levels of Klotho are associated with cardiovascular outcomes, such as coronary disease. In these patients, Klotho level is a predictor for vascular events. |
| Gutierrez, O.M., et al., 2009 [91] | 162 patients with CKD | Cross-Sectional Study | FGF-23 is correlated with vascular dysfunction, such as left ventricular mass index and hypertrophy in these individuals. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco, M.L.; Beyerstedt, S.; Rangel, É.B. Klotho and Mesenchymal Stem Cells: A Review on Cell and Gene Therapy for Chronic Kidney Disease and Acute Kidney Disease. Pharmaceutics 2022, 14, 11. https://doi.org/10.3390/pharmaceutics14010011
Franco ML, Beyerstedt S, Rangel ÉB. Klotho and Mesenchymal Stem Cells: A Review on Cell and Gene Therapy for Chronic Kidney Disease and Acute Kidney Disease. Pharmaceutics. 2022; 14(1):11. https://doi.org/10.3390/pharmaceutics14010011
Chicago/Turabian StyleFranco, Marcella Liciani, Stephany Beyerstedt, and Érika Bevilaqua Rangel. 2022. "Klotho and Mesenchymal Stem Cells: A Review on Cell and Gene Therapy for Chronic Kidney Disease and Acute Kidney Disease" Pharmaceutics 14, no. 1: 11. https://doi.org/10.3390/pharmaceutics14010011
APA StyleFranco, M. L., Beyerstedt, S., & Rangel, É. B. (2022). Klotho and Mesenchymal Stem Cells: A Review on Cell and Gene Therapy for Chronic Kidney Disease and Acute Kidney Disease. Pharmaceutics, 14(1), 11. https://doi.org/10.3390/pharmaceutics14010011